
Abstract
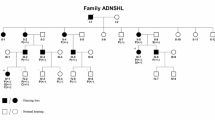
There are 68 sex-linked syndromes that include hearing loss as one feature and five sex-linked nonsyndromic deafness loci listed in the OMIM database. The possibility of additional such sex-linked loci was explored by ascertaining three unrelated Pakistani families (PKDF536, PKDF1132 and PKDF740) segregating X-linked recessive deafness. Sequence analysis of POU3F4 (DFN3) in affected members of families PKDF536 and PKDF1132 revealed two novel nonsense mutations, p.Q136X and p.W114X, respectively. Family PKDF740 is segregating congenital blindness, mild-to-profound progressive hearing loss that is characteristic of Norrie disease (MIM#310600). Sequence analysis of NDP among affected members of this family revealed a novel single nucleotide deletion c.49delG causing a frameshift and premature truncation (p.V17fsX1) of the encoded protein. These mutations were not found in 150 normal DNA samples. Identification of pathogenic alleles causing X-linked recessive deafness will improve molecular diagnosis, genetic counseling and molecular epidemiology of hearing loss among Pakistanis.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Cohen, M. M. & Gorlin, R. J. Epidemiology Etiology and Genetic Patterns (Oxford University Press, Oxford, 1995).
Petersen, M. B., Wang, Q. & Willems, P. J. Sex-linked deafness. Clin. Genet. 73, 14–23 (2008).
de Kok, Y. J., van der Maarel, S. M., Bitner-Glindzicz, M., Huber, I., Monaco, A. P., Malcolm, S. et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 267, 685–688 (1995).
de Brouwer, A. P., van Bokhoven, H., Nabuurs, S. B., Arts, W. F., Christodoulou, J. & Duley, J. PRPS1 mutations: four distinct syndromes and potential treatment. Am. J. Hum. Genet. 86, 506–518 (2010).
Mathis, J. M., Simmons, D. M., He, X., Swanson, L. W. & Rosenfeld, M. G. Brain 4: a novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J. 11, 2551–2561 (1992).
Li, J., Cheng, J., Lu, Y., Chen, A., Sun, Y., Kang, D. et al. Identification of a novel mutation in POU3F4 for prenatal diagnosis in a Chinese family with X-linked nonsyndromic hearing loss. J. Genet. Genomics 37, 787–793 (2010).
Bleeker-Wagemakers, L. M., Friedrich, U., Gal, A., Wienker, T. F., Warburg, M. & Ropers, H. H. Close linkage between Norrie disease, a cloned DNA sequence from the proximal short arm, and the centromere of the X chromosome. Hum. Genet. 71, 211–214 (1985).
Berger, W., Meindl, A., van de Pol, T. J., Cremers, F. P., Ropers, H. H., Doerner, C. et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nat. Genet. 2, 84 (1992).
Meindl, A., Berger, W., Meitinger, T., van de Pol, D., Achatz, H., Dorner, C. et al. Norrie disease is caused by mutations in an extracellular protein resembling C-terminal globular domain of mucins. Nat. Genet. 2, 139–143 (1992).
Chen, Z. Y., Battinelli, E. M., Fielder, A., Bundey, S., Sims, K., Breakefield, X. O. et al. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 5, 180–183 (1993).
Ahmed, Z. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z., Khan, S., Griffith, A. J. et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 69, 25–34 (2001).
Wang, Q. J., Li, Q. Z., Rao, S. Q., Zhao, Y. L., Yuan, H., Yang, W. Y. et al. A novel mutation of POU3F4 causes congenital profound sensorineural hearing loss in a large Chinese family. Laryngoscope 116, 944–950 (2006).
Vore, A. P., Chang, E. H., Hoppe, J. E., Butler, M. G., Forrester, S., Schneider, M. C. et al. Deletion of and novel missense mutation in POU3F4 in 2 families segregating X-linked nonsyndromic deafness. Arch. Otolaryngol. Head Neck Surg. 131, 1057–1063 (2005).
Lee, H. K., Lee, S. H., Lee, K. Y., Lim, E. J., Choi, S. Y., Park, R. K. et al. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants. Clin. Genet. 75, 572–575 (2009).
de Kok, Y. J., Merkx, G. F., van der Maarel, S. M., Huber, I., Malcolm, S., Ropers, H. H. et al. A duplication/paracentric inversion associated with familial X-linked deafness (DFN3) suggests the presence of a regulatory element more than 400 kb upstream of the POU3F4 gene. Hum. Mol. Genet. 4, 2145–2150 (1995).
Bitner-Glindzicz, M., Turnpenny, P., Hoglund, P., Kaariainen, H., Sankila, E. M., van der Maarel, S. M. et al. Further mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked deafness DFN3. Hum. Mol. Genet. 4, 1467–1469 (1995).
Collin, R. W., Chellappa, R., Pauw, R. J., Vriend, G., Oostrik, J., van Drunen, W. et al. Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum. Mutat. 29, 545–554 (2008).
Phelps, P. D., Reardon, W., Pembrey, M., Bellman, S. & Luxom, L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology 33, 326–330 (1991).
Song, M. H., Lee, H. K., Choi, J. Y., Kim, S., Bok, J. & Kim, U. K. Clinical evaluation of DFN3 patients with deletions in the POU3F4 locus and detection of carrier female using MLPA. Clin. Genet. 78, 524–532 (2010).
Marlin, S., Moizard, M. P., David, A., Chaissang, N., Raynaud, M., Jonard, L. et al. Phenotype and genotype in females with POU3F4 mutations. Clin. Genet. 76, 558–563 (2009).
Samadi, D. S., Saunders, J. C. & Crenshaw, E. B. III Mutation of the POU-domain gene Brn4/Pou3f4 affects middle-ear sound conduction in the mouse. Hear. Res. 199, 11–21 (2005).
Cremers, C. W., Hombergen, G. C. & Wentges, R. T. Perilymphatic gusher and stapes surgery. A predictable complication? Clin. Otolaryngol. Allied Sci. 8, 235–240 (1983).
Staropoli, J. F., Xin, W. & Sims, K. B. Co-segregation of Norrie disease and idiopathic pulmonary hypertension in a family with a microdeletion of the NDP region at Xp11.3–p11.4. J. Med. Genet. 47, 786–790 (2010).
Gal, A., Wieringa, B., Smeets, D. F., Bleeker-Wagemakers, L. & Ropers, H. H. Submicroscopic interstitial deletion of the X chromosome explains a complex genetic syndrome dominated by Norrie disease. Cytogenet. Cell Genet. 42, 219–224 (1986).
Sims, K. B., Ozelius, L., Corey, T., Rinehart, W. B., Liberfarb, R., Haines, J. et al. Norrie disease gene is distinct from the monoamine oxidase genes. Am. J. Hum. Genet. 45, 424–434 (1989).
Zhu, D. P., Antonarakis, S. E., Schmeckpeper, B. J., Diergaarde, P. J., Greb, A. E. & Maumenee, I. H. Microdeletion in the X-chromosome and prenatal diagnosis in a family with Norrie disease. Am. J. Med. Genet. 33, 485–488 (1989).
Collins, F. A., Murphy, D. L., Reiss, A. L., Sims, K. B., Lewis, J. G., Freund, L. et al. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am. J. Med. Genet. 42, 127–134 (1992).
Rodriguez-Revenga, L., Madrigal, I., Alkhalidi, L. S., Armengol, L., Gonzalez, E., Badenas, C. et al. Contiguous deletion of the NDP, MAOA, MAOB, and EFHC2 genes in a patient with Norrie disease, severe psychomotor retardation and myoclonic epilepsy. Am. J. Med. Genet. A 143A, 916–920 (2007).
Chen, Z. Y., Hendriks, R. W., Jobling, M. A., Powell, J. F., Breakefield, X. O., Sims, K. B. et al. Isolation and characterization of a candidate gene for Norrie disease. Nat. Genet. 1, 204–208 (1992).
Royer, G., Hanein, S., Raclin, V., Gigarel, N., Rozet, J. M., Munnich, A. et al. NDP gene mutations in 14 French families with Norrie disease. Hum. Mutat. 22, 499 (2003).
Schuback, D. E., Chen, Z. Y., Craig, I. W., Breakefield, X. O. & Sims, K. B. Mutations in the Norrie disease gene. Hum. Mutat. 5, 285–292 (1995).
Wu, W. C., Drenser, K., Trese, M., Capone, A. Jr & Dailey, W. Retinal phenotype-genotype correlation of pediatric patients expressing mutations in the Norrie disease gene. Arch. Ophthalmol. 125, 225–230 (2007).
Meire, F. M., Lafaut, B. A., Speleman, F. & Hanssens, M. Isolated Norrie disease in a female caused by a balanced translocation t(X,6). Ophthalmic. Genet. 19, 203–207 (1998).
Sims, K. B., Irvine, A. R. & Good, W. V. Norrie disease in a family with a manifesting female carrier. Arch. Ophthalmol. 115, 517–519 (1997).
Yamada, K., Limprasert, P., Ratanasukon, M., Tengtrisorn, S., Yingchareonpukdee, J., Vasiknanonte, P. et al. Two Thai families with Norrie disease (ND): association of two novel missense mutations with severe ND phenotype, seizures, and a manifesting carrier. Am. J. Med. Genet. 100, 52–55 (2001).
Criswick, V. G. & Schepens, C. L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 68, 578–594 (1969).
de Crecchio, G., Simonelli, F., Nunziata, G., Mazzeo, S., Greco, G. M., Rinaldi, E. et al. Autosomal recessive familial exudative vitreoretinopathy: evidence for genetic heterogeneity. Clin. Genet. 54, 315–320 (1998).
Robitaille, J., MacDonald, M. L., Kaykas, A., Sheldahl, L. C., Zeisler, J., Dube, M. P. et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 32, 326–330 (2002).
Toomes, C., Bottomley, H. M., Jackson, R. M., Towns, K. V., Scott, S., Mackey, D. A. et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 74, 721–730 (2004).
Ye, X., Wang, Y. & Nathans, J. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med. 16, 417–425 (2010).
Huebner, A. K., Gandia, M., Frommolt, P., Maak, A., Wicklein, E. M., Thiele, H. et al. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am. J. Hum. Genet. 88, 621–627 (2011).
Schraders, M., Haas, S. A., Weegerink, N. J. D., Oostrik, J., Hu, H., Hoefsloot, L. H. et al. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am. J. Hum. Genet 88, 628–634 (2011).
Acknowledgements
We are grateful to the families who made this research possible. This work was supported by Cincinnati Children's Hospital Research Foundation (CCHMC) intramural research funds to Sheikh Riazuddin and ZMA, the National Institute on Deafness and Other Communication Disorders (NIDCD/NIH) research Grant R00-DC009287-03, a Career Development Award from RPB to ZMA. Work in Pakistan was supported by the Higher Education Commission to Sheikh Riazuddin, Islamabad; EMRO/WHO-COMSTECH and Ministry of Science and Technology (MoST) to Sheikh Riazuddin, Islamabad; the International Center for Genetic Engineering and Biotechnology, Trieste, Italy under project CRP/PAK08-01 Contract no. 08/009 to Sheikh Riazuddin. Part of this study carried out in the USA was supported by intramural funds from the NIDCD/NIH (Z01-DC00039-14) to TBF.
Note : While this manuscript was in press, two independent studies published in the May 2011 issue of American Journal of Human Genetics,41, 42 reported mutations of SMPX as another cause of X-linked nonsyndromic hearing loss in humans.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Waryah, A., Ahmed, Z., Binder, M. et al. Molecular and clinical studies of X-linked deafness among Pakistani families. J Hum Genet 56, 534–540 (2011). https://doi.org/10.1038/jhg.2011.55
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2011.55
Keywords
This article is cited by
-
Molecular genetic landscape of hereditary hearing loss in Pakistan
Human Genetics (2022)
-
Clinical and molecular characterization of POU3F4 mutations in multiple DFNX2 Chinese families
BMC Medical Genetics (2018)
-
Genetic Testing of Non-familial Deaf Patients for CIB2 and GJB2 Mutations: Phenotype and Genetic Counselling
Biochemical Genetics (2017)
