
Abstract
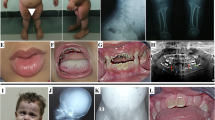
Opsismodysplasia is an autosomal recessive skeletal disorder characterized by facial dysmorphism, micromelia, platyspondyly and retarded bone maturation. Recently, mutations in the gene encoding inositol polyphosphate phosphatase-like 1 (INPPL1) are found in several families with opsismodysplasia by a homozygosity mapping, followed by whole genome sequencing. We performed an exome sequencing in two unrelated Japanese families with opsismodysplasia and identified a novel INPPL1 mutation, c.1960_1962delGAG, in one family. The mutation is predicted to result in an in-frame deletion (p.E654del) within the central catalytic 5-phosphate domain. Our results further support that INPPL1 is the disease gene for opsismodysplasia and that opsismodysplasia has genetic heterogeneity.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Cormier-Daire, V., Delezoide, A. L., Philip, N., Marcorelles, P., Casas, K., Hillion, Y. et al. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J. Med. Genet. 40, 195–200 (2003).
Beemer, F. A. & Kozlowski, K. S Additional case of opsismodysplasia supporting autosomal recessive inheritance. Am. J. Med. Genet. 49, 344–347 (1994).
Santos, H. G. & Saraiva, J. M Opsismodysplasia: another case and literature review. Clin. Dysmorphol. 4, 222–226 (1995).
Tyler, K., Sarioglu, N. & Kunze, J Five familial cases of opsismodysplasia substantiate the hypothesis of autosomal recessive inheritance. Am. J. Med. Genet. 83, 47–52 (1999).
Below, J. E., Earl, D. L., Shively, K. M., McMillin, M. J., Smith, J. D., Turner, E. H. et al. Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 92, 137–143 (2013).
Miyake, N., Elcioglu, N. H., Iida, A., Isguven, P., Dai, J., Murakami, N. et al. PAPSS2 mutations cause autosomal recessive brachyolmia. J. Med. Genet. 49, 533–538 (2012).
Ohnishi, Y., Tanaka, T., Ozaki, K., Yamada, R., Suzuki, H. & Nakamura, Y A high-throughput SNP typing system for genome-wide association studies. J. Hum. Genet. 46, 471–477 (2001).
Huber, C., Faqeih, E. A., Bartholdi, D., Bole-Feysot, C., Borochowitz, Z., Cavalcanti, D. P. et al. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am. J. Hum. Genet. 92, 144–149 (2013).
Dyson, J. M., Kong, A. M., Wiradjaja, F., Astle, M. V., Gurung, R. & Mitchell, C. A The SH2 domain containing inositol polyphosphate 5-phosphatase-2: SHIP2. Int. J. Biochem. Cell. Biol. 37, 2260–2265 (2005).
Suwa, A., Kurama, T. & Shimokawa, T. SHIP2 and its involvement in various diseases. Expert Opin. Ther. Targets 14, 727–737 (2010).
Hejna, J. A., Saito, H., Merkens, L. S., Tittle, T. V., Jakobs, P. M., Whitney, M. A. et al. Cloning and characterization of a human cDNA (INPPL1) sharing homology with inositol polyphosphate phosphatases. Genomics 29, 285–287 (1995).
Pesesse, X., Deleu, S., De Smedt, F., Drayer, L. & Erneux, C Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem. Biophys. Res. Commun. 239, 697–700 (1997).
Sleeman, M. W., Wortley, K. E., Lai, K. M., Gowen, L. C., Kintner, J., Kline, W. O. et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 11, 199–205 (2005).
Acknowledgements
We thank the patients and their family for their help to the study. We also thank the Japanese Skeletal Dysplasia Consortium. This study is supported by research grants from the Ministry of Health, Labor and Welfare (23300101 to SI and NMat.; 23300201 to SI), by Grants-in-Aid for Young Scientists (23689052 to NMiy.) from the Japan Society for the Promotion of Science; by Research on intractable diseases, Health and Labour Sciences Research Grants, H23-Nanchi-Ippan-123 (SI) and by grants from the Japan Science and Technology Agency, the Strategic Research Program for Brain Sciences (11105137 to NMat.), a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12024421 to NMat.), a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (12020465 to NMat.) and the Takeda Science Foundation (to N Miy. and N Mat.). We thank Professors Debora Krakow and Michael Bamshad for their comments on the patients’ phenotypes. We also thank Ms Tomoko Kusadokoro for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Iida, A., Okamoto, N., Miyake, N. et al. Exome sequencing identifies a novel INPPL1 mutation in opsismodysplasia. J Hum Genet 58, 391–394 (2013). https://doi.org/10.1038/jhg.2013.25
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2013.25
Keywords
This article is cited by
-
INPPL1 gene mutations in opsismodysplasia
Journal of Human Genetics (2017)
-
Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex
Orphanet Journal of Rare Diseases (2014)
