
Abstract
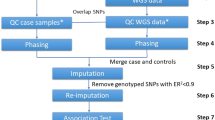
Genome-wide association studies (GWAS) have successfully identified many common genetic variants associated with complex diseases over the past decade. The ‘gold standard’ method for validating the top single nucleotide polymorphisms (SNPs) identified in GWAS is to independently replicate the findings in similar or diverse large-scale external cohorts. However, for rare diseases, it can be difficult to find an external validation cohort within a reasonable timeframe. In such situations, resampling methods, such as the two-step iterative resampling (TSIR) approach have been used to identify SNPs associated with the outcome of interest. However, the TSIR approach involves choosing several parameters in each step, which can influence the performance of the approach. In this paper, we undertook extensive simulation studies to assess the effect of choice of different parameters on the type I error and power for both binary and continuous phenotypes and also compared the TSIR approach with the traditional one-stage (OS) and two-stage (TS) GWAS analysis. We illustrate the usefulness of the TSIR approach by applying it to a GWAS of childhood cancer survivors. Our results indicate that the TSIR approach with an at least 70:30 split and a cutoff of discovering and replicating SNPs at least 20 times in 100 replications provides conservative type I error control and has near ‘optimal’ power for internally validated SNPs. Its performance is comparable with the TS GWAS for which an external validation cohort is available with only slight reduction in power in some situations. It has almost the same power as OS GWAS with conservative type I error which leads to fewer false positive findings. TSIR is a powerful and efficient method for identifying and internally validating SNPs for GWAS when independent cohorts for external validation may not be available.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Klein, R. J., Zeiss, C., Chew, E. Y., Tsai, J. Y., Sackler, R. S., Haynes, C. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Sladek, R., Rocheleau, G., Rung, J., Dina, C., Shen, L., Serre, D. et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885 (2007).
The Wellcome Trust Case Control Consortium (WTCCC) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Skol, A. D., Scott, L. J., Abecasis, G. R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 209–213 (2006).
Pahl, R., Schäfer, H. & Müller, H.-H. Optimal multistage designs—a general framework for efficient genome-wide association studies. Biostatistics 10, 297–309 (2008).
Rothman, N., Garcia-Closas, M., Chatterjee, N., Malats, N., Wu, X., Figueroa, J. D. et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet. 42, 978–984 (2010).
Gurney, J. G., Severson, R. K., Davis, S. & Robison, L. L. Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer 75, 2186–2195 (1995).
Wheeler, H. E., Maitland, M. L., Dolan, M. E., Cox, N. J. & Ratain, M. J. Cancer pharmacogenomics: strategies and challenges. Nat. Rev. Genet. 14, 23–34 (2013).
Hudson, M. M., Ness, K. K., Nolan, V. G., Armstrong, G. T., Green, D. M., Morris, E. B. et al. Prospective medical assessment of adults surviving childhood cancer: study design, cohort characteristics, and feasibility of the St. Jude Lifetime Cohort Study. Pediatr. Blood Cancer 56, 825–836 (2011).
Wilson, C. L., Liu, L., Yang, J. J., Kang, G., Ojha, R. P., Neale, G. et al. Genetic and clinical factors associated with obesity among adult survivors of childhood cancer: a report from the St. Jude Lifetime cohort. Cancer (e-pub ahead of print 11 May 2015).
Yang, J. J., Cheng, C., Devidas, M., Cao, X., Campana, D., Yang, W. et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood 120, 4197–4204 (2012).
Elliott, K. S., Chapman, K., Day-Williams, A., Panoutsopoulou, K., Southam, L., Lindgren, C. M. et alGIANT consortium Evaluation of the genetic overlap between osteoarthritis with body mass index and height using genome-wide association scan data. Ann. Rheum. Dis. 72, 935–941 (2013).
Hayes, M. G., Pluzhnikov, A., Miyake, K., Sun, Y., Ng, M. C., Roe, C. A. et al. Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes 56, 3033–3044 (2007).
Cheng, C. Internal validation inferences of significant genomic features in genome-wide screening. Comput. Stat. Data Anal. 53, 788–800 (2009).
Simón-Sánchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D. et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312 (2009).
Yue, W. H., Wang, H. F., Sun, L. D., Tang, F. L., Liu, Z. H., Zhang, H. X. et al. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11.2. Nat. Genet. 43, 1228–1231 (2011).
Kang, G., Bi, W., Zhao, Y., Zhang, J. F., Yang, J. J., Xu, H. et al. A new system identification approach to identify genetic variants in sequencing studies for a binary phenotype. Hum. Hered. 78, 104–116 (2014).
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M. & Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Igl, B. W., Konig, I. R. & Ziegler, A. What do we mean by 'replication' and 'validation' in genome-wide association studies? Hum. Hered. 67, 66–68 (2009).
Ioannidis, J. P. A., Gilles, T. & Daly, M. J. Validating, augmenting and refining genome-wide association signals. Nat. Rev. Genet. 10, 318–329 (2009).
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D. et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81, 559–575 (2007).
Song, K. & Elston, R. C. A powerful method of combining measures of association and Hardy-Weinberg disequilibrium for fine-mapping in case-control studies. Stat. Med. 25, 105–126 (2006).
Freidlin, B., Zheng, G., Li, Z. & Gastwirth, J. L. Trend tests for case-control studies of genetic markers: power, sample size and robustness. Hum. Hered. 53, 146–152 (2002) Erratum in Hum Hered 2009; 68: 220.
Kang, G., Lin, D., Hakonarson, H. & Chen, J. Two-stage extreme phenotype sequencing design for discovering and testing common and rare genetic variants: efficiency and power. Hum. Hered. 73, 139–147 (2012).
Lee, S., Emond, M. J., Bamshad, M. J., Barnes, K. C., Rieder, M. J., Nickerson, D. A. et alNHLBI GO Exome Sequencing Project—ESP Lung Project Team Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Kang, G., Gao, G., Shete, S., Redden, D. T., Chang, B.-L., Rebbeck, T. R. et al. Capitalizing on admixture in genome-wide association studies: A two-stage testing procedure and application to height in African-Americans. Front. Genet. 2, 11 (2011).
Chen, J., Kang, G., VanderWeele, T., Zhang, C. & Mukherjee, B. Efficient designs of gene-environment interaction studies: implications of Hardy-Weinberg equilibrium and gene-environment independence. Stat. Med. 31, 2516–2530 (2012).
Acknowledgements
We would like to thank two reviewers for their helpful comments which have significantly improved the paper. This research was supported by St. Jude Children’s Research Hospital Cancer Center Support (CORE) grant CA21765 from the National Cancer Institute and by the American Lebanese and Syrian Associated Charities (ALSAC). The research work of Jun J Yang was in part supported by the grant U01CA176063.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kang, G., Liu, W., Cheng, C. et al. Evaluation of a two-step iterative resampling procedure for internal validation of genome-wide association studies. J Hum Genet 60, 729–738 (2015). https://doi.org/10.1038/jhg.2015.110
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2015.110
This article is cited by
-
The association between prescription drugs and colorectal cancer prognosis: a nationwide cohort study using a medication-wide association study
BMC Cancer (2023)
-
Variable selection in social-environmental data: sparse regression and tree ensemble machine learning approaches
BMC Medical Research Methodology (2020)
-
A common polymorphism in the retinoic acid pathway modifies adrenocortical carcinoma age-dependent incidence
British Journal of Cancer (2020)
