
Abstract
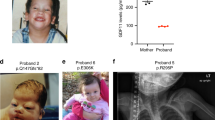
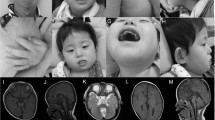
We report on a Brachydactyly Type C (BDC) patient with clinically inconspicuous parents. Molecular genetic analyses revealed compound heterozygosity for two GDF5 variants. The variant c.956G>T (p.Gly319Val) was inherited from her mother and has been reported in exome sequencing projects, whereas c.1073T>C (p.Ile358Thr) has never been reported so far. In silico, both variants were predicted to be ‘disease-causing’, but the fact that p.Ile358Thr was predicted by SIFT to be ‘tolerated’ raised our suspicion. Therefore, we performed in vitro assays. To our surprise, GDF5G319V showed pronounced loss of function in luciferase reporter assays and in vitro chondrogenesis, whereas GDF5I358T and GDF5WT had comparable biological activities. Western blot analyses revealed decreased protein levels after overexpression of GDF5G319V. In absence of linkage or de novo mutation, several scenarios could explain the underlying mechanism of the patient’s phenotype. Owing to reduced activity of GDF5G319V in our functional assays, p.Gly319Val might be causative for BDC, but typically evoke an unrecognizably mild phenotype or even nonpenetrance. Another possibility is that our assays failed to pinpoint the disease-causing mechanism of the p.Ile358Thr allele. A final possibility is that compound heterozygosity for p.Ile358Thr and p.Gly319Val is more deleterious to GDF5 activity than either variant alone. Until all possible explanations can be rigorously tested experimentally, a precise recurrence risk counseling for the parents and the affected child is not possible.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Bell, J. in Treasury of Human Inheritance (ed Penros, L .) 1–31 (Cambridge Univ. Press, London, UK, 1951).
Everman, D. B., Bartels, C. F., Yang, Y., Yanamandra, N., Goodman, F. R., Mendoza-Londono, J. R. et al. The mutational spectrum of brachydactyly type C. Am. J. Med. Genet. 112, 291–296 (2002).
Luyten, F. P. Cartilage-derived morphogenetic protein-1. Int. J. Biochem. Cell Biol. 29, 1241–1244 (1997).
Ploger, F., Seemann, P., Schmidt-von Kegler, M., Lehmann, K., Seidel, J., Kjaer, K. W. et al. Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum. Mol. Genet. 17, 1222–1233 (2008).
Nickel, J., Sebald, W., Groppe, J. C. & Mueller, T. D. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev. 20, 367–377 (2009).
Schwabe, G. C., Turkmen, S., Leschik, G., Palanduz, S., Stover, B. & Goecke, T. O. et al. Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1. Am. J. Med. Genet. A 124A, 356–363 (2004).
Camera, G., Camera, A., Costa, M. & Mantero, R. Pitfalls of genetic counselling in brachydactyly type C. Am. J. Med. Genet. 53, 199–201 (1994).
Savarirayan, R., White, S. M., Goodman, F. R., Graham, J. M. Jr ., Delatycki, M. B., Lachman, R. S. et al. Broad phenotypic spectrum caused by an identical heterozygous CDMP-1 mutation in three unrelated families. Am. J. Med. Genet. A 117A, 136–142 (2003).
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP), Seattle, WAURL http://evs.gs.washington.edu/EVS/).
Exome Aggregation Consortium (ExAC), Cambridge, MAURL http://exac.broadinstitute.org.
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Degenkolbe, E., Konig, J., Zimmer, J., Walther, M., Reissner, C., Nickel, J. et al. A GDF5 point mutation strikes twice—causing BDA1 and SYNS2. PLoS Genet. 9, e1003846 (2013).
Stange, K., Thieme, T., Hertel, K., Kuhfahl, S., Janecke, A. R., Piza-Katzer, H. et al. Molecular analysis of two novel missense mutations in the GDF5 proregion that reduce protein activity and are associated with brachydactyly type C. J. Mol. Biol. 426, 3221–3231 (2014).
Kuhfahl, S., Hauburger, A., Thieme, T., Groppe, J., Ihling, C., Tomic, S. et al. Identification of a core domain within the proregion of bone morphogenetic proteins that interacts with the dimeric, mature domain. Biochem. Biophys. Res. Commun. 408, 300–305 (2011).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Jonk, L. J., Itoh, S., Heldin, C. H., ten Dijke, P. & Kruijer, W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 273, 21145–21152 (1998).
Heinecke, K., Seher, A., Schmitz, W., Mueller, T. D., Sebald, W. & Nickel, J. Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC Biol. 7, 59 (2009).
Kotzsch, A., Nickel, J., Seher, A., Sebald, W. & Muller, T. D. Crystal structure analysis reveals a spring-loaded latch as molecular mechanism for GDF-5-type I receptor specificity. EMBO J. 28, 937–947 (2009).
Seemann, P., Schwappacher, R., Kjaer, K. W., Krakow, D., Lehmann, K., Dawson, K. et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 115, 2373–2381 (2005).
Ibrahim, D. M., Hansen, P., Rodelsperger, C., Stiege, A. C., Doelken, S. C., Horn, D. et al. Distinct global shifts in genomic binding profiles of limb malformation-associated HOXD13 mutations. Genome Res. 23, 2091–2102 (2013).
Graul-Neumann, L. M., Deichsel, A., Wille, U., Kakar, N., Koll, R., Bassir, C. et al. Homozygous missense and nonsense mutations in BMPR1B cause acromesomelic chondrodysplasia-type Grebe. Eur. J. Hum. Genet. 22, 726–733 (2014).
Dathe, K., Kjaer, K. W., Brehm, A., Meinecke, P., Nurnberg, P., Neto, J. C. et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 84, 483–492 (2009).
Haws, D. V. Inherited brachydactyly and hypoplasia of the bones of the extremities. Ann. Hum. Genet. 26, 201–212 (1963).
Sanz, J. G. S. Type C brachydactyly transmitted through four generations. Ann. Genet. 31, 43–46 (1988).
Galjaard, R. J., van der Ham, L. I., Posch, N. A., Dijkstra, P. F., Oostra, B. A., Hovius, S. E. et al. Differences in complexity of isolated brachydactyly type C cannot be attributed to locus heterogeneity alone. Am. J. Med. Genet. 98, 256–262 (2001).
Robin, N. H., Gunay-Aygun, M., Polinkovsky, A., Warman, M. L. & Morrison, S. Clinical and locus heterogeneity in brachydactyly type C. Am. J. Med. Genet. 68, 369–377 (1997).
Thieme, T., Patzschke, R., Job, F., Liebold, J., Seemann, P., Lilie, H. et al. Biophysical and structural characterization of a folded core domain within the proregion of growth and differentiation factor-5. FEBS J. 281, 4866–4877 (2014).
Stenson, P. D., Mort, M., Ball, E. V., Shaw, K., Phillips, A. & Cooper, D. N. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9 (2014).
Acknowledgements
We thank Johannes Grünhagen and Gundula Leschik for technical assistance and Lutz Schomburg for critical remarks on the manuscript. This work was funded by a doctoral fellowship of the Sonnenfeld-Stiftung to KS. Contributions were made possible by DFG funding through the Berlin-Brandenburg School for Regenerative Therapies GSC 203 (KS) and by DFG funding MU880/11-01 (CEO). All funding sources had no influence on any aspect of the presented study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Stange, K., Ott, CE., Schmidt-von Kegler, M. et al. Brachydactyly Type C patient with compound heterozygosity for p.Gly319Val and p.Ile358Thr variants in the GDF5 proregion: benign variants or mutations?. J Hum Genet 60, 419–425 (2015). https://doi.org/10.1038/jhg.2015.48
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2015.48
