
Abstract
Tafazzin, encoded by the TAZ gene, is a mitochondrial membrane-associated protein that remodels cardiolipin (CL), an important mitochondrial phospholipid. TAZ mutations are associated with Barth syndrome (BTHS). BTHS is an X-linked multisystemic disorder affecting usually male patients. Through sequence analysis of TAZ, we found one novel mutation c.39_60del p.(Pro14Alafs*19) by whole-exome sequencing and a reported missense mutation c.280C>T p.(Arg94Cys) by Sanger sequencing in two male patients (Pt1 and Pt2). Patient with c.280C>T mutation had dilated cardiomyopathy, while another patient with c.39_60del mutation had no feature of cardiomyopathy. A reported m.1555A>G homoplasmic variant was also identified in the patient having mutation c.39_60del by whole mitochondrial DNA sequencing method. This variant was not considered to be the main cause of mitochondrial dysfunction based on a cytoplasmic hybrid (cybrid) assay. Tafazzin expression was absent in both patient-derived fibroblast cells. Complementation of TAZ expression in fibroblasts from the patient with the novel mutation c.39_60del restored mitochondrial respiratory complex assembly. High-performance liquid chromatography–tandem mass spectrometry-based metabolic analysis revealed the decline of CL and the accumulation of monolysocardiolipin, indicating the loss of tafazzin activity. Owing to phenotypic variability, it is difficult to diagnose BTHS based on clinical features only. We conclude that genetic analysis should be performed to avoid underdiagnosis of this potentially life-threatening inborn error of metabolism.
Similar content being viewed by others
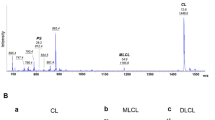
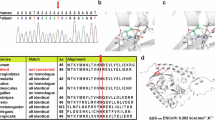
Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Vreken, P., Valianpour, F., Nijtmans, L. G., Grivell, L. a, Plecko, B., Wanders, R. J. et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382 (2000).
Johnston, J., Kelley, R. I., Feigenbaum, A, Cox, G. F., Iyer, G. S., Funanage, V. L. et al. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 61, 1053–1058 (1997).
Gonzalez, I. L. Barth syndrome:TAZ gene mutations, mRNAs, and evolution. Am. J. Med. Genet. Part A 134A, 409–414 (2005).
Aprikyan, A. A. & Khuchua, Z. Advances in the understanding of Barth syndrome. Br. J. Haematol. 161, 330–338 (2013).
Sakamoto, O., Ohura, T., Katsushima, Y., Fujiwara, I., Ogawa, E., Miyabayashi, S. et al. A novel intronic mutation of the TAZ (G4.5) gene in a patient with Barth syndrome: creation of a 5′ splice donor site with variant GC consensus and elongation of the upstream exon. Hum. Genet. 109, 559–563 (2001).
Ferri, L., Dionisi-Vici, C., Taurisano, R., Vaz, F. M., Guerrini, R. & Morrone, A. When silence is noise: infantile-onset Barth syndrome caused by a synonymous substitution affecting TAZ gene transcription. Clin. Genet. 90, 461–465 (2016).
Jefferies, J. L. Barth syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 163, 198–205 (2013).
Barth, P. G., Valianpour, F., Bowen, V. M., Lam, J., Duran, M., Vaz, F. M. et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am. J. Med. Genet. 126A, 349–354 (2004).
Roberts, A. E., Nixon, C., Steward, C. G., Gauvreau, K., Maisenbacher, M., Fletcher, M. et al. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. A 158A, 2726–2732 (2012).
Bowron, A., Honeychurch, J., Williams, M., Tsai-Goodman, B., Clayton, N., Jones, L. et al. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J. Inherit. Metab. Dis. 38, 279–286 (2015).
Ronvelia, D., Greenwood, J., Platt, J., Hakim, S. & Zaragoza, M. V. Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol. Genet. Metab. 107, 428–432 (2012).
Moric-Janiszewska, E. & Markiewicz-Łoskot, G. Genetic heterogeneity of left-ventricular noncompaction cardiomyopathy. Clin. Cardiol. 31, 201–204 (2008).
Chang, B., Momoi, N., Shan, L., Mitomo, M., Aoyagi, Y., Endo, K. et al. Gonadal mosaicism of a TAZ (G4.5) mutation in a Japanese family with Barth syndrome and left ventricular noncompaction. Mol. Genet. Metab. 100, 198–203 (2010).
Cosson, L., Toutain, A., Simard, G., Kulik, W., Matyas, G., Guichet, A. et al. Barth syndrome in a female patient. Mol. Genet. Metab. 106, 115–120 (2012).
Ferri, L., Donati, M. a, Funghini, S., Cavicchi, C., Pensato, V., Gellera, C. et al. Intra-individual plasticity of the TAZ gene leading to different heritable mutations in siblings with Barth syndrome. Eur. J. Hum. Genet. 23, 1708–1712 (2015).
Rigaud, C., Lebre, A.-S., Touraine, R., Beaupain, B., Ottolenghi, C., Chabli, A. et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J. Rare Dis. 8, 70 (2013).
Clarke, S. L. N., Bowron, A., Gonzalez, I. L., Groves, S. J., Newbury-Ecob, R., Clayton, N. et al. Barth syndrome. Orphanet J. Rare Dis. 8, 23 (2013).
Ferri, L., Donati, M. A., Funghini, S., Malvagia, S., Catarzi, S., Lugli, L. et al. New clinical and molecular insights on Barth syndrome. Orphanet J. Rare Dis. 8, 27 (2013).
Horvath, S. E. & Daum, G. Lipids of mitochondria. Prog. Lipid Res. 52, 590–614 (2013).
Houtkooper, R. H., Turkenburg, M., Poll-The, B. T., Karall, D., Pérez-Cerdá, C., Morrone, A. et al. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta Biomembr. 1788, 2003–2014 (2009).
Schlame, M., Ren, M., Xu, Y., Greenberg, M. L. & Haller, I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49 (2005).
Houtkooper, R. H. & Vaz, F. M. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol. Life Sci. 65, 2493–2506 (2008).
Pfeiffer, K., Gohil, V., Stuart, R. a., Hunte, C., Brandt, U., Greenberg, M. L. et al. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 (2003).
Vaz, F. M., Houtkooper, R. H., Valianpour, F., Barth, P. G. & Wanders, R. J. A. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J. Biol. Chem. 278, 43089–43094 (2003).
Kohda, M., Tokuzawa, Y., Kishita, Y., Nyuzuki, H., Moriyama, Y., Mizuno, Y. et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 12, e1005679 (2016).
Hazkani-Covo, E., Zeller, R. M. & Martin, W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6, e1000834 (2010).
Hayashi, J., Ohta, S., Kikuchi, A., Takemitsu, M., Goto, Y. & Nonaka, I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl Acad. Sci. USA 88, 10614–10618 (1991).
Kulik, W., Van Lenthe, H., Stet, F. S., Houtkooper, R. H., Kemp, H., Stone, J. E. et al. Bloodspot assay using HPLC-tandem mass spectrometry for detection of barth syndrome. Clin. Chem. 54, 371–378 (2008).
Mazurová, S., Tesařová, M., Magner, M., Houšťková, H., Hansíková, H., Augustínová, J. et al. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med. Rep. 114, 139–153 (2013).
Xu, Y., Malhotra, A., Claypool, S. M., Ren, M. & Schlame, M. Tafazzins from Drosophila and mammalian cells assemble in large protein complexes with a short half-life. Mitochondrion 21, 27–32 (2015).
Joshi, A. S., Thompson, M. N., Fei, N., Ttemann, M. H. & Greenberg, M. L. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287, 17589–17597 (2012).
Spencer, C. T., Bryant, R. M., Day, J., Gonzalez, I. L., Colan, S. D., Thompson, W. R. et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118, e337–e346 (2006).
Ren, M., Phoon, C. K. L. & Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 55, 1–16 (2014).
Schlame, M., Kelley, R. I., Feigenbaum, A., Towbin, J. a., Heerdt, P. M., Schieble, T. et al. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999 (2003).
Coman, D., Yaplito-Lee, J. & Boneh, A. New indications and controversies in arginine therapy. Clin. Nutr. 27, 489–496 (2008).
Kirwin, S. M., Manolakos, A., Barnett, S. S. & Gonzalez, I. L. Tafazzin splice variants and mutations in Barth syndrome. Mol. Genet. Metab. 111, 26–32 (2014).
Gonzalvez, F., D’Aurelio, M., Boutant, M., Moustapha, A., Puech, J. P., Landes, T. et al. Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta Mol. Basis Dis. 1832, 1194–1206 (2013).
Schlame, M., Rua, D. & Greenberg, M. L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 (2000).
Acehan, D., Xu, Y., Stokes, D. L. & Schlame, M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 87, 40–48 (2007).
Acehan, D., Vaz, F., Houtkooper, R. H., James, J., Moore, V., Tokunaga, C. et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 286, 899–908 (2011).
Prezant, T. R., Agapian, J. V., Bohlman, M. C., Bu, X., Oztas, S., Qiu, W. Q. et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 4, 289–294 (1993).
Acknowledgements
This work was supported by a grant of the Innovative Cell Biology by Innovative Technology (Cell Innovation Program) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan to YO; the Support Project and a grant of Strategic Research Center in Private Universities from the MEXT, Japan to Saitama Medical University Research Center for Genomic Medicine. This work was also supported by Grants-in-Aid of the Research on Intractable Diseases (Mitochondrial Disorder) from the Ministry of Health, Labor and Welfare of Japan to KM. YO received a Special research grant from Takeda Science Foundation. NNB is a recipient of the Saitama Medical University Research Fellowship. We acknowledge the Laboratory Genetic Metabolic Disease in the Academic Medical Center in Amsterdam for measuring the MLCL/CL ratio. We thank Atsuko Imai for providing valuable comments. We are grateful to Ryoko Nakamura, Department of Pediatric Neurology, Fukuoka Children’s Hospital, Fukuoka, Japan; who is an attending physician of one of our patients.
Author contributions
NNB, YK and YO designed the study. NNB, YK and KI acquired data. KN, J-IH, YT, MK, HN, YY-S, KM and AO gave critical comments. YO, NNB and YK wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Borna, N., Kishita, Y., Ishikawa, K. et al. A novel mutation in TAZ causes mitochondrial respiratory chain disorder without cardiomyopathy. J Hum Genet 62, 539–547 (2017). https://doi.org/10.1038/jhg.2016.165
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2016.165
This article is cited by
-
A high mutation load of m.14597A>G in MT-ND6 causes Leigh syndrome
Scientific Reports (2021)

{kind=link}