
Abstract
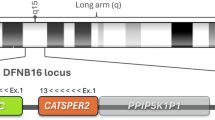
Approximately 50% of childhood deafness is caused by mutations in specific genes. Autosomal recessive loci account for approximately 80% of nonsyndromic genetic deafness1. Here we report the identification of a new transmembrane serine protease (TMPRSS3; also known as ECHOS1) expressed in many tissues, including fetal cochlea, which is mutated in the families used to describe both the DFNB10 and DFNB8 loci. An 8-bp deletion and insertion of 18 monomeric (∼68-bp) β-satellite repeat units, normally present in tandem arrays of up to several hundred kilobases on the short arms of acrocentric chromosomes, causes congenital deafness (DFNB10). A mutation in a splice-acceptor site, resulting in a 4-bp insertion in the mRNA and a frameshift, was detected in childhood onset deafness (DFNB8). This is the first description of β-satellite insertion into an active gene resulting in a pathogenic state, and the first description of a protease involved in hearing loss.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Kalatzis, V. & Petit, C. The fundamental and medical impacts of recent progress in research on hereditary hearing loss. Hum. Mol. Genet. 7, 1589–1597 (1998).
Veske, A. et al. Autosomal recessive non-syndromic deafness locus (DFNB8) maps on chromosome 21q22 in a large consanguineous kindred from Pakistan. Hum. Mol. Genet. 5, 165–168 (1996).
Scott, H.S. et al. Refined genetic mapping of the autosomal recessive non-syndromic deafness locus DFNB8 on human chromosome 21q22.3. Adv. Otorhinolaryngol. 56, 158–163 ( 2000).
Bonne-Tamir, B. et al. Linkage of congenital recessive deafness (gene DFNB10) to chromosome 21q22.3. Am. J. Hum. Genet. 58, 1254–1259 (1996).
Berry, A. et al. Refined localization of autosomal recessive nonsyndromic deafness DFNB10 locus using 34 novel microsatellite markers, genomic structure, and exclusion of six known genes in the region. Genomics 68, 22–29 (2000).
Michaud, J. et al. Isolation and characterization of a human chromosome 21q22.3 gene (WDR4) and its mouse homologue that code for a WD-repeat protein. Genomics 68, 71–79 ( 2000).
Bartoloni, L. et al. Cloning and characterization of a putative human glycerol 3-phosphate permease gene (SLC37A1 or G3PP) on 21q22.3: mutation analysis in 2 candidate phenotypes, DFNB10 and a glycerol kinase deficiency. Genomics 70, 190–200 ( 2000).
Wattenhofer, M. et al. Isolation and characterization of the UBASH3A gene on 21q22.3 encoding a potential nuclear protein with a novel combination of domains: mutation analysis in DFNB10. Hum. Genet. (in press).
The Chromosome 21 Mapping and Sequencing Consortium. The DNA sequence of human chromosome 21. Nature 405 , 311–319 (2000).
Paoloni-Giacobino, A., Chen, H., Peitsch, M.C., Rossier, C. & Antonarakis, S.E. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3 . Genomics 44, 309–320 (1997).
Rawlings, N.D. & Barrett, A.J. Families of serine peptidases. Methods Enzymol. 244, 19–61 (1994).
Tsuji, A. et al. Hepsin, a cell membrane-associated protease. Characterization, tissue distribution, and gene localization. J. Biol. Chem. 266, 16948–16953 (1991).
van Driel, I.R., Goldstein, J.L., Sudhof, T.C. & Brown, M.S. First cysteine-rich repeat in ligand-binding domain of low density lipoprotein receptor binds Ca2+ and monoclonal antibodies, but not lipoproteins . J. Biol. Chem. 262, 17443– 17449 (1987).
Resnick, D., Pearson, A. & Krieger, M. The SRCR superfamily: a family reminiscent of the Ig superfamily. Trends Biochem. Sci. 19, 5– 8 (1994).
Kawamura, S., Kurachi, S., Deyashiki, Y. & Kurachi, K. Complete nucleotide sequence, origin of isoform and functional characterization of the mouse hepsin gene. Eur. J. Biochem. 262, 755–764 (1999).
Handt, O., Sutherland, G.R. & Richards, R.I. Fragile sites and minisatellite repeat instability . Mol. Genet. Metab. 70, 99– 105 (2000).
Greig, G.M. & Willard, H.F. β satellite DNA: characterization and localization of two subfamilies from the distal and proximal short arms of the human acrocentric chromosomes. Genomics 12, 573–580 (1992).
Eichler, E.E. et al. Complex β-satellite repeat structures and the expansion of the zinc finger gene cluster in 19p12. Genome Res. 8, 791–808 (1998).
Shiels, C., Coutelle, C. & Huxley, C. Contiguous arrays of satellites 1, 3, and β form a 1.5-Mb domain on chromosome 22p. Genomics 44, 35–44 (1997).
Farrell, S.A., Winsor, E.J. & Markovic, V.D. Moving satellites and unstable chromosome translocations: clinical and cytogenetic implications. Am. J. Med. Genet. 46, 715–720 (1993).
Assum, G., Fink, T., Steinbeisser, T. & Fisel, K.J. Analysis of human extrachromosomal DNA elements originating from different β-satellite subfamilies. Hum. Genet. 91, 489– 495 (1993).
Gaubatz, J.W. Extrachromosomal circular DNAs and genomic sequence plasticity in eukaryotic cells. Mutat. Res. 237, 271– 292 (1990).
Wolff, D.J. & Schwartz, S. Characterization of Robertsonian translocations by using fluorescence in situ hybridization. Am. J. Hum. Genet. 50, 174–181 (1992).
Samonte, R.V., Conte, R.A., Ramesh, K.H. & Verma, R.S. Molecular cytogenetic characterization of breakpoints involving pericentric inversions of human chromosome 9. Hum. Genet. 98, 576–580 (1996).
Scott, H.S. et al. Identification of mutations in the α-L-iduronidase gene (IDUA) that cause Hurler and Scheie syndromes. Am. J. Hum. Genet. 53, 973–986 ( 1993).
Thalmann, R. & Thalmann, I. Source and role of endolymph macromolecules . Acta Otolaryngol. 119, 293– 296 (1999).
Chen, H. et al. Cloning of 559 potential exons of genes of human chromosome 21 by exon trapping. Genome Res. 6, 747– 760 (1996).
Dunnen, J.T. & Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12 ( 2000).
Acknowledgements
We thank S. Dahoun, C. Vieux and M.A. Morris for help with the FISH analyses; all members of the S.E.A. laboratory, past and present, for transcription mapping; and A. Shintani, T. Sasaki, K. Nagamine, M. Takahashi, M. Sasaki and all members of the genomic sequencing team in the Laboratory of Genomic Medicine, Keio University School of Medicine for their contribution to this work. The laboratory of S.E.A. is supported by grants from the Swiss FNRS, the OFES/EU, and funds from the University and Cantonal Hospital of Geneva. The laboratory of B.B.-T. is supported in part by grants from the Applebaum Foundation. The Laboratory of Genomic Medicine, Keio University School of Medicine was supported in part by a Fund for Human Genome Sequencing Project from the Japan Science and Technology Corporation; Grants in Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan; and Grants in Aid for Scientific Research and a Fund for “Research for the Future” Program from the Japan Society for the Promotion of Science. The laboratory of A.G. is supported in part by grants from the FAUN-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Figure A
Representative protein homologies with other transmembrane proteases are shown: human TMPRSS2 (O15393), mouse Tmprss2 (AAF64186), human TMPRSS4 (AAF74526), human TMPRSS1 (hepsin, P05981) and the human airway trypsin-like protease (ATRYPL, O60235). Domains, as detected in TMPRSS3, are boxed and labeled underneath with the active-site residues His257, Asp304 and Ser401 indicated (arrows). TMPRSS3 is predicted to cleave after the K or R residues, as it contains D395 at the base of the substrate-specificity-binding pocket (S1 subsite). Preceded by an R, the N terminus of the protease domain peptide sequence is IVGG. Proteolytic cleavage between R and I would result in protease activation similar to other serine protease zymogens11, converting TMPRSS3 to a non-catalytic and a catalytic subunit linked by a disulfide bond (probably C207 to C324). The TMPRSS3 serine protease domain contains six conserved cysteine residues which, by homology to other proteases and three-dimensional modeling, are likely to form the following intrasubunit disulfide bonds: C242-C258; C370-C386; C397-C425. The points of mutation in the DFNB8 and DFNB10 families are marked by 'Splice' and 'Ins', respectively. (GIF 59 kb)
Rights and permissions
About this article
Cite this article
Scott, H., Kudoh, J., Wattenhofer, M. et al. Insertion of β-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 27, 59–63 (2001). https://doi.org/10.1038/83768
Received:
Accepted:
Issue date:
DOI: https://doi.org/10.1038/83768
This article is cited by
-
Stable long-term outcomes after cochlear implantation in subjects with TMPRSS3 associated hearing loss: a retrospective multicentre study
Journal of Otolaryngology - Head & Neck Surgery (2023)
-
ARNSHL gene identification: past, present and future
Molecular Genetics and Genomics (2022)
-
Identification of autosomal recessive nonsyndromic hearing impairment genes through the study of consanguineous and non-consanguineous families: past, present, and future
Human Genetics (2022)
-
Molecular genetic landscape of hereditary hearing loss in Pakistan
Human Genetics (2022)
-
Evolutionary history of type II transmembrane serine proteases involved in viral priming
Human Genetics (2022)

{kind=link}