
Abstract
The past few decades are characterized by an explosive evolution of genetics and molecular cell biology. Advances in chemistry and engineering have enabled increased data throughput, permitting the study of complete sets of molecules with increasing speed and accuracy using techniques such as genomics, transcriptomics, proteomics, and metabolomics. Prediction of long-term outcomes in transplantation is hampered by the absence of sufficiently robust biomarkers and a lack of adequate insight into the mechanisms of acute and chronic alloimmune injury and the adaptive mechanisms of immunological quiescence that may support transplantation tolerance. Here, we discuss some of the great opportunities that molecular diagnostic tools have to offer both basic scientists and translational researchers for bench-to-bedside clinical application in transplantation medicine, with special focus on genomics and genome-wide association studies, epigenetics (DNA methylation and histone modifications), gene expression studies and transcriptomics (including microRNA and small interfering RNA studies), proteomics and peptidomics, antibodyomics, metabolomics, chemical genomics and functional imaging with nanoparticles. We address the challenges and opportunities associated with the newer high-throughput sequencing technologies, especially in the field of bioinformatics and biostatistics, and demonstrate the importance of integrative approaches. Although this Review focuses on transplantation research and clinical transplantation, the concepts addressed are valid for all translational research.
Key Points
-
The omics approach has accelerated biomarker discovery and improved understanding of multifactorial biological processes and diseases
-
The complex interplay between the genome, transcriptome, proteome and metabolome has mostly been overlooked in traditional translational research and in the field of transplantation
-
In transplantation research, data from high-throughput molecular diagnostic tools are shedding new light on many pathogenic processes (for example, acute cellular rejection, ischemia–reperfusion injury and chronic allograft damage)
-
Upcoming high-throughput techniques, such as massively parallel sequencing for genotyping and transcriptomics, are expected to further accelerate the pace at which translational research is performed
-
The challenge for the future is to translate the progress that is being made in molecular biology into health gains, an outcome that will only be possible with a holistic approach and integrative data analysis
Similar content being viewed by others

Introduction
The 50 years since the first successful solid organ transplantation in humans in 19531,2 have been marked by an exponential increase in our understanding of the basic immunological processes involved in organ transplantation, rejection and allograft tolerance.3 This knowledge has resulted in the identification of immunogenic drug targets and resultant rational drug design. Transplantation is now a well-established routine treatment strategy for end-stage kidney, heart, liver and lung diseases.4 Nevertheless, new diagnostic and predictive monitoring tools are desperately needed to customize the delivery of immunosuppressive drugs for optimal patient and organ care, such that graft life is extended and patient morbidity minimized.
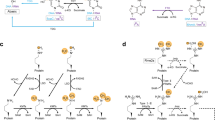
The past 50 years have also seen an explosive evolution in the fields of genetics and molecular biology: the discovery of the DNA double-stranded helix by Watson and Crick in 1953;5 the completion of the Human Genome Project in 2003;6 and the further refinement of the analytical tools that enable high-resolution sequence mapping of human structural variation.7 Using these insights and with the advent of high-throughput technical devices, molecular biology has undergone an impressive transformation, with the recent advent of informative fields of biology such as the elucidation of the following: the importance of epigenetics on the DNA transcription process (most importantly DNA methylation and histone modifications);8 alternative messenger (m)RNA splicing;9 RNA interference (RNAi; that is, RNA-mediated post-transcriptional regulation through noncoding microRNA [miRNA] or small interfering RNA [siRNA]);8,10,11 post-translational modifications;12,13 and regulation at the metabolic level14,15 (Figure 1).

DNA is packed in chromosomes in tertiary structures defined by the histones. Histone modifications and DNA methylation are epigenetic factors that can influence the transcription process. DNA is transcribed into pre-mRNA, which is spliced into different mRNA molecules by alternative splicing. Noncoding RNAs, such as siRNA and miRNA, are also transcribed from DNA, and these small RNA fragments affect mRNA stability and regulate the translation of mRNA into proteins. In addition, post-translational modifications of proteins (for example, folding, cleavage, and chemical modifications) contribute greatly to the diversity of the proteome. These proteins can function as enzymes, affecting DNA repair or replication, the transcription process or translation, or can act as a catalyst for metabolic reactions. Proteins are also involved in cell signaling and ligand binding, and as structural elements. Each level interacts closely with the other level: protein–DNA and RNA–DNA interactions and chemical modifications of DNA that do not affect the primary DNA sequence also affect the molecular biological processes in cell systems. Each of these molecular biological processes can be studied with specialized omics tools—genomics/epigenomics, transcriptomics, proteomics and metabolomics—which study the complete sets of genes, transcripts, proteins and metabolites. Abbreviations: dsRNA, double-stranded RNA; Me, methylation; miRNA, microRNA; mRNA, messenger RNA; pre-mRNA, precursor messenger RNA; siRNA, small interfering RNA.
“It is the responsibility of those of us involved in today's biomedical research enterprise to translate the remarkable scientific innovations we are witnessing into health gains”, stated Dr Elias Zerhouni, former Director of the NIH, in 2005.16 In this Review, we discuss some of the opportunities that molecular diagnostic tools have to offer both basic scientists and translational researchers for bench-to-bedside clinical applications in transplantation. Terms used in the text are defined in Box 1.
General concepts in molecular diagnostics
Single-molecule versus high-throughput strategies
The classic paradigm that the DNA of the cell will determine its fate is too simplistic. Variations in the genome sequences of humans or other species only partly explain the fundamental nature of the cells and organisms, and are often only marginally implicated in disease processes.17 The different levels of biomolecular organization and control, such as regulation of the transcriptome (the catalogue of RNA transcripts, including mRNA, siRNA and miRNA), the proteome (the complete set of proteins) and the metabolome (the complete set of small-molecule metabolites) (Figure 1) are interdependent and are greatly affected by environmental events and stresses throughout life. An analysis of the full spectrum of cellular organization is therefore necessary to gain insight into the mechanisms of disease.18
Each of these aspects can be studied at the level of individual molecules, but advances in engineering have allowed for increased data throughput, enabling the study of complete sets of molecules (omics) with improved speed, accuracy, and cost-effectiveness compared with previous experimental models of repeated single-molecule experiments, and with potential for the generation of new hypotheses. The omics approach has impressively accelerated biomarker discovery and our understanding of multifactorial biological processes and diseases. The major differences between omics strategies and single-molecule approaches obviously lies in throughput (hundreds of thousands versus one or a few), but also in experimental design. Classic single-molecule biomedical research is based on hypothesis testing, building new experiments based on prior observations and theory, such that classic scientific research tends towards a reductionist approach in understanding disease and disease processes, owing to the limitations of most technologies and the complex nature of pathological systems. This approach, which drills deeply into a focused area and eliminates confounding variables, had great importance in advancing our biological insights, and will continue to do so.
The application of omics technologies and the analysis of the genome, transcriptome, proteome or metabolome, however, creates unprecedented opportunities for improving understanding of complex systems and diseases at a global level, and enables hypothesis generation with fewer a priori assumptions. Therefore, the technological progress in genome-wide measurements has shifted the scientific discovery process from hypothesis-driven approaches towards more data-driven approaches. Nevertheless, omics approaches are also based on assumptions about the nature of the experiments (the theory underlying the central paradigms in molecular biology, the phenotypes of the studied samples and sometimes the pathways deduced from previous experiments), as well as assumptions about the functioning of the instruments used to perform the experiments. After a first omics hypothesis-generating approach, classic single-molecule experiments should ensue, in order to delve further into the mechanisms of the newly generated hypotheses.
Biomarker versus therapeutic target discovery
Omics technologies are currently used in two closely related areas: biomarker discovery and the elucidation of pathophysiologic processes to identify novel therapeutic targets. However, clear differences exist between these aspects of translational research in terms of the ultimate goals, optimal matrix, optimal platforms and preferred study design (Table 1).
Determining the immunologic threshold
The 1st International Conference on Transplantomics and Biomarkers in Organ Transplantation was held in San Francisco in 2010. Hosted by The Transplantation Society, the conference brought together myriad disciplines in transplantation research including genomics, transcriptomics, proteomics, metabolomics, informatics, next-generation sequencing technologies, imaging and clinical transplantation. Individualized therapy was the focus of the conference, and the prominent topic was the search for both diagnostic and predictive biomarkers for allograft dysfunction that could be used in personalizing treatment. Other themes highlighted throughout the conference included the use of protocol and for-cause biopsies for microarray and histological analysis, the use of noninvasive methods for biomarker discovery, the pitfalls of relying solely on gold standard criteria for biopsy classification, low-cost and time-efficient diagnostic and analytic tools, defining and identifying causative versus correlative biomarkers, and compiling and harnessing information in the public domain.
The term 'immunosuppression' derives from the general sense that the immune system is solely oriented in an offensive antiallograft response and thus needs continuous suppression. However, it has become increasingly apparent that alloimmunity is far more complex and diverse. For example, T regulatory (TREG) cells control the immune response in such a way that their suppression per se would actually augment rejection. Similarly, allospecific T cells are now recognized as having widely variable effector capabilities and activation requirements based on prior history: naive T cells seem to be exceptionally dependent on antigen presentation and co-stimulation, whereas antigen-experienced, or memory, T cells are triggered primarily through the T-cell receptor without substantial requirements for accessory or co-stimulatory molecule engagement.19 Additionally, nonalloantigen exposure is capable of influencing alloimmune reactivity through heterologous immunity, a process that has been postulated as an explanation for the difficulties in tolerance induction.20
The net result of these findings is that alloimmune competence is now recognized as being fluid, changing with ongoing immune experience, age, and disease state. Even donor characteristics, such as differing degrees of human leukocyte antigen (HLA) mismatch, donor age, cold ischemia time and organ type, change the degree to which antigens and accessory and co-stimulatory molecules are presented and thus alter the effective allospecific precursor frequency. Thus, each patient can be considered a mosaic of alloimmune potential that is specific for a particular donor–recipient pair at a particular time and situation. Simply declaring the immune system as requiring suppression defies the complexity inherent in the relevant biology; the concept of 'immunosuppression' should give way to deliberate and individualized 'immune modulation'.
The complexity of the immune responses after allotransplantation can initially seem daunting. Development of an immune repertoire is fundamentally a random process and responsiveness toward any particular complement of alloantigens can be considered equally stochastic. However, the aggregate threshold of alloimmune responsiveness changes in reasonably predictable ways. In general, alloimmune responsiveness is greatest at the time of transplantation, and wanes with time. As the need for more intense immunosuppression wanes, the overall need for maintenance immunosuppression varies with two predominant variables—precursor frequency and memory. Precursor frequency relates to T-cell repertoire compatibility and is influenced most notably by HLA mismatch.21 Memory is a function of prior exposures, either to alloantigens or to sufficiently cross-reactive environmental antigens. These standard variables help to establish rational choices for therapeutic agents based on increasingly well-defined susceptibilities that individual immune cell types have to commonly available immunosuppressive drugs.
Therapeutic decisions based on individualized immune modulation can be distilled down to a balanced appraisal of risk versus benefit. In transplantation, an unmet need exists for specific and sensitive, noninvasive biomarkers that can track graft injury and graft survival and replace the renal transplant biopsy as the gold standard. The application of molecular diagnostics may uncover novel biomarkers. Importantly, the discovery of such biomarkers would allow the dose (or load) of immunosuppression to be customized for individual patients, to identify the particular threshold between rejection and infection that avoids allorecognition yet retains normal immunity to infection and cancer.
Molecular diagnostics in transplantation
Long-term outcomes in transplantation are hampered by an incomplete understanding of the mechanisms driving acute and chronic alloimmune injury, immunological quiescence, operational tolerance and tissue injury. The absence of sufficiently robust biomarkers further complicates the clinical management of allograft recipients; better diagnostic biomarkers could potentially improve outcomes. Here, we provide an overview of transplantation research in humans that used different applied molecular cell biology tools for improving mechanistic insight and biomarker discovery (Figure 2).

The outcome of renal transplantation is determined by pretransplantation, peritransplantation and post-transplantation factors. Omics tools can be used either for biomarker discovery or for elucidation of the molecular mechanisms underlying pathophysiologic processes. Peripheral blood contains almost exclusively recipient-derived cells, and genetic profiles are recipient-driven. Transcriptomics, proteomics and metabolomics research in peripheral blood is promising for both biomarker and therapeutic target discovery. Genotype analysis in kidney biopsy samples gives information on the donor genotype, although extensive graft inflammation with recipient-derived cells could also lead to the presence of recipient's DNA in biopsy samples. Omics analyses in biopsy specimens can be used for both therapeutic target studies and for biomarker studies, although the invasive procedure represents a major drawback of potential tissue-derived biomarkers. Finally, urine analysis using omics tools offers a great window for biomarker discovery, but the low amount of DNA in urine samples makes it unsuitable for genomic studies. Abbreviations: BKV, BK virus; CMV, cytomegalovirus; HLA, human leukocyte antigen; UTI, urinary tract infection.
Genetics, genomics and epigenetics
Candidate gene approach for DNA polymorphisms
DNA polymorphisms are variations in the DNA sequence that are present in the population at a frequency >1%. These polymorphisms can be single nucleotide polymorphisms (SNPs), a variable number of tandem repeats (microsatellites and minisatellites), deletions or insertions. If such polymorphisms occur in the coding region of a gene, they may or may not alter the structure of the mature protein, depending on the impact of the gene sequence variation on the amino acid sequence. Polymorphisms in noncoding regions may also influence gene splicing, transcription and translation. Although the degree of health and disease determined by inherited DNA sequences outside of all the currently known genetic diseases is not known, the inherited genetic diseases do tell us that single gene molecular changes encoded for in DNA can have remarkable implications for health and can lead to multisystemic disease.
As in other medical disciplines, association studies between genetic variation and disease conditions have also been performed in the field of transplantation. Most of these studies have been performed using a candidate gene approach.
Over the past few decades, knowledge about the highly polymorphic HLA system, an important determinant of outcomes after transplantation, has increased greatly.22 This knowledge has been translated into routine clinical practice, with many laboratories routinely using DNA-based HLA typing (instead of serological typing),23 and has proven especially valuable in bone marrow transplantation.24,25 However, current HLA genotyping is limited to a small portion of the HLA region in the genome. Detailed, more comprehensive HLA genotyping might be important for immunology and transplantation, although this idea has not been studied to date.
The majority of the other studies addressing genetic variation in transplantation have been performed in the fields of rejection,26,27 ischemia–reperfusion injury,28,29,30,31 chronic allograft damage32,33 and pharmacogenetics.34 The well-known association between polymorphisms in the gene encoding thiopurine methyltransferase (TPMT) and azathioprine toxicity35 illustrates that genetic variation research using the candidate gene approach can be of high clinical importance; the FDA recommends that consideration is given to determining a patient's TPMT genotype or phenotype before azathioprine is used. Other pharmacogenetic markers (for example, ABCB1 and CYP3A5) also have potential value in the clinical setting.34,36,37,38 Validation of these early results is needed, however, and the long-term benefits of genotyping for these additional markers on patient outcome remains to be shown.
In contrast to the clinical success of HLA genotyping and pharmacogenetics, data linking clinical outcome parameters in transplantation with SNPs in genes encoding cytokines and cytokine receptors, chemokines and chemokine receptors, complement genes, co-stimulation molecules, growth factors and adhesion molecules, innate immune genes and genes implicated in ischemia–reperfusion injury and chronic allograft damage, have been inconsistent and remain without clinical application.26,27,33 No candidate genes are ready for their clinical value to be tested in prospective trials, mainly because studies to date have been relatively small, have exhibited selection bias, have insufficiently accounted for confounding factors and have had ill-defined and short-term outcome variables; such limitations have led to conflicting results and poor reproducibility. Better designed and adequately powered studies are urgently needed to overcome these issues and bring genotyping into the transplantation clinic.
Genome-wide association studies
Genetic analyses using the candidate gene approach have not been very successful in translating genotype analyses into the clinic. Apart from flaws with study design and underpowering in the majority of the studies, the manual selection of potentially interesting genes is not the most efficient way to link variation in genome sequence with outcome parameters.39,40 The number of SNPs recorded in the public HapMap database41,42 and the Single Nucleotide Polymorphism Database (dbSNP)43 has reached several millions, and upcoming massive parallel sequencing tools will enable the screening of these SNPs and many more, as will be discussed. Genome-wide association (GWA) studies, which use accessible tools for high-throughput genotyping, have now become a standard method for disease gene discovery,44 and the catalogue of published GWA studies is growing rapidly.45
One drawback with GWA studies is that a substantial number of recent GWA studies (albeit in fields other than transplantation) found that only a few common variants were implicated in human disorders, and that the associated SNPs explain only a small fraction of the genetic risk.46,47 The relevance of findings from standard GWA studies is indeed based on a 'common disease, common gene' hypothesis: the results are relevant when the polymorphisms associated with a disease are present sufficiently frequently in a population that is sufficiently large. In recent years, many GWA studies have successfully elucidated such polymorphisms,40 but many more GWA studies are necessary to elucidate the true influence of common DNA variants on disease susceptibility. However, some researchers argue that although many genes have been linked with disease processes, many have too low an impact on the risk of the disease to be biologically relevant.46,48 With this reasoning, the limited role of common variation in many heritable diseases would suggest that many rare or less frequent variants remain to be found. More in-depth study of these disease-associated rare variants might better reveal novel therapeutic targets or improve the tailoring of treatment regimens to an individual's risk profile, compared with the 'common disease, common gene' approach.39,48 In this light, the advent of newer tools such as exome arrays and whole-genome sequencing (through next-generation sequencing techniques, as will be discussed), and expanded reference panels such as the 1,000 Genomes project,49 will better determine the degree of complex diseases caused by alterations in DNA sequence. Surely GWA studies are a first but very important step in the process of identifying genes to improve patient management.
At least one multicenter GWA study is being conducted in clinical transplantation (a UK–Irish consortium project funded by the Wellcome Trust Case–Control Consortium),50 and is likely to help us to define the genetic variation in recipients' genomes that determines transplantation outcome. As each transplantation involves two genomes, donors' genomes are also likely to affect transplantation outcome, and interactions between donors' and recipients' genomes will need to be taken into account in GWA studies in transplantation.
Epigenetics and epigenomics
Epigenetic alterations encompass heritable regulation of gene expression that does not involve DNA nucleotide sequence variation. The epigenome constitutes an array of molecular modifications to both DNA and chromatin, the most extensively investigated of which are DNA methylation, and changes to the chromatin packaging of DNA by post-translational histone modifications.8,51 Interestingly, environmental exposure to nutritional, chemical and physical factors has the potential to affect gene expression and alter disease susceptibility through changes in the epigenome.
Epigenetics and epigenomics are exciting and critically important new areas of research. Recent years have seen development of novel techniques for studying chromatin state on a genome scale. Most of these techniques are based on chromatin immunoprecipitation (ChIP), with fragment identification by hybridization to a microarray (ChIP-chip). Nowadays, ChIP sequencing (ChIP-Seq) is becoming available, whereby ChIP DNA is directly sequenced using next-generation massively parallel sequencing tools.52 Aside from the use of ChIP-Seq in cancer research,53 one of the biological systems most studied using ChIP-Seq is T cells,54,55 which is of special interest in the field of transplantation. The first studies demonstrated the importance of histone modifications for T-cell responses54,56,57 and ischemia–reperfusion injury.58 T cells are not the only immune cells regulated by epigenetic phenomena: natural killer cell characteristics59 and B-cell development60 are also governed by epigenetic mechanisms.
Approaches for the analysis of genome-scale DNA methylation have also been developed, and the different approaches (for example, array-based versus massively parallel sequencing-based approaches) have competing strengths and weaknesses.61 DNA methylation analysis is also of interest in the field of immunology and transplantation. Methylation of CpG residues has been shown to repress expression of Foxp3, which is involved in TREG cell development and function, whereas complete demethylation is necessary for stable Foxp3 expression.62,63 Many more studies have investigated the influence of differential DNA methylation on T-cell responses, as is reviewed elsewhere.57 Another proof-of-concept study for the potential usefulness of DNA methylation for transplantation research demonstrated that modification of methylated cytosines at a regulatory site within the promoter of complement component C3 may occur as a result of ischemia–reperfusion injury.64
As the importance of the epigenome for gene expression, cell development and cell function is only beginning to be recognized, epigenetic and epigenomic research is expected to become important in the future; improved accessibility of newer tools for screening the epigenome will likely contribute to this success.52,61 However, epigenetics research in the field of transplantation and immunology has so far exclusively been in the form of basic research. Translational studies with more direct clinical relevance should be initiated to transfer the successes of epigenetics to the clinic.
Gene expression and transcriptomics
Candidate gene approach for gene expression
DNA is transcribed to RNA, and the complete set and quantity of RNA transcripts in a cell at a specific developmental stage or physiological condition is called the transcriptome. The transcriptome is dynamic and changes in different circumstances owing to different patterns of gene expression. Hypothesis-driven candidate mRNA real-time polymerase chain reaction (RT-PCR) studies have been performed in experimental and clinical transplantation and have contributed to current insight into the functioning of the immune system and rejection, the mechanisms of action of immunosuppressive drugs, the mechanisms involved in tissue injury and repair, cell senescence, epithelial–mesenchymal transition (EMT), and the study of chimerism. No field in transplantation research has been untouched by RNA studies, and quantitative (RT-PCR) or spatial (in situ hybridization) molecular analysis of transcripts in allograft biopsy samples provides a sensitive and powerful means of evaluating these specimens.
Gene expression studies are not only useful in the detection of underlying pathogenic processes. The concept that gene expression in transplanted tissue alters before histological damage is visible has been demonstrated for ischemia–reperfusion injury,65,66 acute rejection67 and chronic allograft damage,68,69 and could represent a means to foresee and perhaps prevent future injury by timely intervention.70 In addition, if validated in larger ongoing multicenter studies, analysis of mRNA in urine71 or peripheral blood72,73 could become a useful and, importantly, noninvasive biomarker for ongoing intragraft damage such as rejection or progressive chronic histological injury, as is reviewed elsewhere.74 The advent of high-throughput techniques does not mean that standard techniques are irrelevant. On the contrary, the targeted gene approach will probably be more cost-effective than the high-throughput microarray techniques for gene expression analysis in a clinical setting, as just a few well-selected gene targets should suffice as clinical biomarkers; the interpretation of microarray data is more complex.
Gene expression analysis with microarrays
Microarray technology has revolutionized gene expression research by allowing the screening of gene expression of thousands of genes simultaneously.75,76 Several commercial gene expression microarrays are currently available for researchers. Among these widely used platforms are Affymetrix's GeneChip®, Agilent's oligonucleotide microarrays, Illumina's microarrays, and Roche's Nimblegen® microarrays. Each of these platforms have strengths and weaknesses, and researchers must choose the appropriate platform based on their priorities.77
As the technical and biostatistical analytical tools are more complicated than candidate gene approaches, and because they must be controlled for multiple testing, gene expression microarrays have been used by only a few groups in the field of transplantation. Nevertheless, studies performed over the past decade have covered the full spectrum of transplantation biology—including acute rejection, operational tolerance, chronic allograft rejection, and the identification of biomarkers, in liver, heart, pancreas, kidney, and lung transplantation, as reviewed elsewhere.78,79,80 For example, the use of gene expression microarray technology on renal allograft biopsy samples has demonstrated the molecular heterogeneity of allografts at the time of transplantation,81,82 acute rejection83,84 and after chronic histological damage.85,86,87,88
Microarray expression analysis has also been used to study the periphery. This approach offers insight into the pathways of acute and chronic rejection, and of induced and operational tolerance.89,90,91 Additionally, the application of microarray technology on peripheral blood samples is a suitable platform for the efficient discovery of new noninvasive biomarkers for rejection or chronic allograft damage.92,93,94,95 However, gene expression microarray technology has its own disadvantages, such as a lower dynamic range than RT-PCR, a high cost, decreased specificity owing to cross-hybridization, errors in probe design, differences in concept and results between the different microarray platforms, insufficient coverage of splice variation and annotation difficulties, as is reviewed elsewhere.80 For implementation in clinical practice, a good compromise will likely be found in custom-made arrays or in commercial high-density oligoarrays with fewer but well-chosen probes.
The splice variant issues have been partly overcome by the development of exon expression or 'whole genome tiling' arrays. This approach has uncovered the immense complexity of the transcriptome: gene expression varies much more greatly than was previously believed.96 However, many of the limitations of standard DNA microarrays remain, including reliance upon existing knowledge about the genome sequence, high background levels owing to cross-hybridization, and a limited dynamic range.97 Moreover, akin to what is seen with standard DNA microarrays, comparing expression levels across different experiments is often difficult and can require complicated normalization methods.
Newer technologies using high-throughput (next-generation) sequencing tools, such as RNA sequencing (RNA-Seq), aim to overcome many of the above problems by revealing unprecedented detail while simultaneously improving quantification.97,98 The faster, cheaper and more reliable analysis of gene expression will potentially advance transcriptomics, and RNA-Seq is expected to replace microarrays for many applications that involve determining the structure and dynamics of the transcriptome. However, although these new technologies might improve the quality of transcriptome profiling, the greater challenge associated with omics research—how best to generate biologically meaningful interpretations of complex datasets that are sufficiently interesting to drive follow-up experimentation—will remain (Box 2).
Noncoding RNA (miRNA and siRNA)
The transcriptome contains many more RNA fragments than the mRNA that is translated into peptides and proteins. Among these noncoding RNAs, miRNAs are well known and form a populous class of evolutionarily conserved, naturally occurring RNA fragments (∼22 nucleotides long). Apart from miRNAs, other types of small RNAs have also been found, including endogenous siRNAs and Piwi-interacting RNAs (piRNAs). All of these small RNAs function as guide RNAs within the broad phenomenon known as RNA silencing (or RNAi), through their interaction with Argonaute proteins that regulate the translation process (translational repression) or guarantee mRNA cleavage (mRNA degradation).10,11,99,100,101 Since their discovery, hundreds of small noncoding RNAs have been identified, and it is predicted that the actual diversity is a plurality of this.
It is believed that miRNAs are involved in human disease;102 noncoding RNA fragments are also likely to be of importance in the field of transplantation and immunity. In addition, miRNAs are implicated in the regulation of immune cell development and in the modulation of innate and adaptive immune responses.103,104,105 Moreover, in clinical transplantation, an internally validated miRNA expression profiling study demonstrated that miRNA expression in renal allograft biopsy samples correlates with acute T-cell-mediated rejection and renal allograft function, with circumstantial evidence that the differentially expressed miRNAs are largely derived from infiltrating immune cells.106,107,108 Although this result confirms previous findings that different cell types and different cell activation states are associated with specific miRNAs,109 it remains to be elucidated whether these miRNA expression alterations are pathogenic or whether they represent only the infiltration of the transplanted organ with activated immune cells.
The ultimate goal of small noncoding RNA research is to discover disease-causing or disease-modifying small RNA fragments that could subsequently be used as biomarkers or for specific targeted therapy. Indeed, siRNAs have the potential to silence disease-relevant genes that cannot be shut down with available drugs. Moreover, specific miRNAs that promote disease processes could also themselves be targets for shut-down. Despite the relatively recent discovery of small noncoding RNA, RNAi is already being used therapeutically in human clinical trials, and biotechnology companies that focus on RNAi therapeutics are emerging.110 One of the most problematic issues is how to move small RNAs efficiently and specifically to their target sites in the human body; in addition, safety, effectiveness and reliability issues need to be addressed.110
Regardless of its great therapeutic potential, RNAi has already revolutionized basic biomedical research, allowing researchers to rapidly, efficiently and accurately repress the function of a gene of interest. The coming years promise to be exciting in the field of small noncoding RNA, both in basic and in translational research, and possibly even in clinical trials.
Proteins and antibodies
Gel-based and mass spectrometric approaches
The development of genomics and transcriptomics notwithstanding, gene polymorphisms and transcript levels correlate incompletely with the expression level of the functionally active proteins, which more accurately reflect actual cellular events than genomics or transcriptomics.111 This poor correlation between genotype, gene expression and the localization or activity of the ensuing proteins is caused by the complex regulation of the transcriptome and the broad range of post-translational modifications that change the properties of proteins (for example, activity, localization, turnover, kinetics, and interactions with other proteins).112,113 Proteins therefore provide a better picture of events that occur inside an organism and provide ideal biomarkers for disease conditions, as has been extensively demonstrated for proteins such as prostate-specific antigen, CA125, CA19-9 and carcinoembryonic antigen.114 Proteomics and peptidomics aim to identify, quantify, analyze and functionally define a large number of proteins and peptides in biological processes or disease states. However, as genomics and transcriptomics techniques and analysis tools are well established and have matured over a couple of decades, their strengths and weaknesses are relatively well addressed in study designs and analysis methods. By contrast, proteomics and peptidomics tools have only just begun to gain widespread use. These methods still suffer from issues such as a lack of widely accepted standards in quality control, normalization, data analysis, and protein identification criteria, which makes comparison of data from different groups difficult. More robust proteomics and peptidomics methods are evolving and the number of publications in this field is increasing exponentially.115,116,117
Over the past few decades, new tools have revolutionized proteomics and peptidomics. Proteomics tools include gel electrophoresis (for example, two-dimensional gel electrophoresis [2DE] and two-dimensional difference in gel electrophoresis [2D DIGE]) and gel-free methods using mass spectrometry (for example, surface-enhanced laser desorption/ionization with time-of-flight mass spectrometry [SELDI-TOF-MS], liquid chromatography mass spectrometry [LC-MS] and matrix-assisted laser desorption/ionization [MALDI]) are increasingly being used for proteomics and peptidomics.113,114,115,117 Mass spectrometry was initially used to identify and characterize isolated proteins and to profile the mass of components in biological or clinical samples, but has evolved into a technique for interrogating complex proteomes by matching mass spectra to sequence databases to identify proteins.118
Proteomics methods are also increasingly being used in the field of organ transplantation. Because urine is the ideal noninvasive specimen for renal diseases,119 the number of proteomic studies of urine has surged, and urine proteomics are a promising tool for the noninvasive diagnosis of acute rejection120,121,122,123,124 and chronic allograft histological damage.125,126,127 However, although sensitivity and accuracy of these tools have improved over the last decade, the complexities of tissues and biological fluid proteomes often exceed the capacity of proteomic analysis tools. Even the very best technologies available can interrogate only a small fraction of the total proteome in any sample and the more complex the sample, the more this limitation is a problem for discovery and interpretation of results. These detected proteins are also usually the most abundant, and efforts to resolve these issues and improve sensitivity and detection are underway. In proteomics and peptidomics, a compromise needs to be reached between in-depth analysis and identification of individual protein and peptide components on the one hand, and broad proteome coverage on the other hand. The most challenging step in proteomics and peptidomics research is the integration of seemingly unrelated findings of various protein fragments into a rational pathogenic pathway or into a reasonable biomarker. In addition, the value of urinary biomarkers is limited in anuric patients and in patients in whom urine may come from native rather than transplant kidneys.
Protein and antibody microarrays
Enzyme-linked immunosorbent assays (ELISAs) have long been the gold standard for quantifying protein expression in tissue or body fluids, and remain popular today. Multiplexed assays based on ELISA have become available and are especially useful when samples only need to be analyzed for a handful of molecules (for example, in biomarker qualification). Building on the same principle, high-density protein arrays have been introduced in recent years, and encompass any platform in which a large number of proteins are immobilized on a solid support in a spatially addressable manner (for example, planar arrays such as Protoarray® by Invitrogen or ProteoScan Cancer Lysate Arrays by Origene) or a spectrally addressable manner (for example, bead-based arrays such as Luminex® platform by Luminex Corporation).128 High-density microarrays enable the analysis of protein interaction networks, the generation of protein phosphorylation maps, and the identification of disease-specific autoantibodies. Protein arrays are therefore usually used for high-throughput profiling of secreted factors such as cytokines, chemokines and growth factors, as well as for screening for autoantibodies129 or cancer antibodies.130,131 As the diversity of proteins is greater than that of nucleic acids, true proteomic antibody arrays, targeting a significant portion of a tissue proteome, are not practical, unlike genomic or transcriptomic level arrays. Protein arrays will probably be developed for specific hypotheses in specific tissue types.
Antibody microarrays are also of special interest in transplantation research because pathogenic donor-directed alloantibodies can form. These antibodies can target HLA molecules and lead to antibody-mediated rejection, but non-HLA antibodies can also be implicated in allograft rejection. First screening experiments demonstrated the potential usefulness of the Protoarray® platform in human transplantation, and revealed increased levels of a large set of non-HLA antibodies in the post-transplantation serum of renal allograft recipients.132,133,134 In 2009, another study evaluated antibody responses using Protoarray® in tolerant recipients of combined kidney and bone marrow transplants from HLA haplo-identical donors, and showed the development of B-cell alloimmunity and autoimmunity in patients with T-cell tolerance to the donor graft.135 Although the data from these studies need confirmation and the pathogenic capacity of these antibodies needs to be examined, and although it is not known why so many autoreactive antibodies exist in the circulation, protein microarray technology offers an interesting approach for antibody research in transplantation.
Metabolomics
Mass spectrometry and NMR
Rather than characterizing and quantifying large macromolecules (DNA, RNA, proteins, and peptides), as is done in genomics, transcriptomics and proteomics, metabolomics focuses on small molecules (<1,000 Da), catabolic and metabolic products arising from the interactions of these large macromolecules.136 The correlation between transcriptomics or proteomics and metabolic changes is poorer than expected as the relationship between mRNA levels and protein expression is not simply linear, and even if the protein is expressed, its concentration may not correlate with its activity. The metabolome is a more direct reflection of cell functionality.
Researchers have already catalogued more than 7,900 metabolites in the human body; these molecules can be either endogenous or exogenous, and include minerals, elements, carbohydrates, hormones, and phospholipids, among others.137 Metabolites are not inert and are more than end products of metabolism. They have many biological functions and represent accurate reporters of an organism's physiology, being involved in processes such as glycolysis, gluconeogenesis and lipid metabolism. Metabolites such as citrate, lactate, and glucose can reflect situations such as apoptotic alterations, hypoxia and oxidative stress. Therefore, metabolite concentrations often track with specific disease conditions, turning metabolomics into an efficient tool for distinguishing health from disease.
The study of the whole metabolome (metabolomics, also known as metabonomics) was made possible by technological breakthroughs in small molecule separation and identification. Such breakthroughs include mass spectrometry, nuclear magnetic resonance (NMR), high-pressure liquid chromatography, capillary electrophoresis and ultrahigh pressure liquid chromatography. As outlined in a 2008 review,136 metabolomics can be approached in two very distinct ways: in a targeted profiling manner with identification of single metabolites, or in a pattern recognition fashion where spectral patterns and intensities are recorded rather than individual molecules. In order to promote and speed up targeted profiling studies, the Human Metabolome Database (HMDB) was created to provide reference mass spectrometry and NMR spectra, metabolite–disease associations, metabolic pathway data, and reference metabolite concentrations (for diagnostic comparisons) for hundreds of human metabolites.137,138
Metabolomics has been used by very few groups in transplantation, for the evaluation of metabolic processes involved in ischemia–reperfusion injury,139 acute rejection,140 drug toxicity,141 graft dysfunction and graft damage.141,142,143 Although these first studies showed some promising results, all were hampered by small sample sizes and insufficient validation. More systematic and collaborative research is necessary to bring metabolomics closer to the clinic.141
Chemical genetics and chemical genomics
Another area of critical future importance is chemical genetics and chemical genomics (also known as chemogenomics or chemical biology), the study of the influence of small molecules (rather than genetic intervention) on biological processes; that is, research on the cross-section between chemical space and biological activity. Chemical genomics can be seen as the omics counterpart of chemical genetics, being performed less on an ad hoc basis and more systematically than chemical genetics.144 In the past, the study of small molecules was hampered by the complexity and high cost of synthesizing and purifying these compounds. Nowadays, the advent of combinatorial chemistry has enabled the synthetic preparation of large collections of diverse compounds, using relatively few chemical steps. In addition, advances in robotic technology and informatics now allow hundreds of thousands of compounds to be screened in a single day, orders of magnitude greater than was possible a decade ago.
The integration of large collections of compounds with advanced high-throughput omics technologies (such as genomics, transcriptomics, proteomics, metabolomics) represents a powerful way to accelerate the discovery of potential drug targets.145 In this light, Chembank, the public, web-based informatics environment that contains freely available data derived from small-molecule screens performed by the National Cancer Institute's Initiative for Chemical Genetics,146 is likely to have a major role in future drug target development, both in academic and in pharmaceutical industry research. With respect to transplantation, it is interesting to note that many chemical genetics studies have been performed with sirolimus and tacrolimus, yielding novel insights into the downstream signaling pathways of the targeted proteins.144
Nanoparticles and functional imaging
Molecular imaging reporters of specific processes and activities are becoming available and offer exciting opportunities for the noninvasive tracking of disease processes. Fluorescent, nuclear or magnetic probes can be bound to metabolites involved in metabolic processes, and can act as in vivo markers of metabolic activity.147,148 Studies in cancer and atherosclerosis show that these techniques are easily translatable into the clinic.149,150 Targeted molecular imaging tools can also track immune cells, as was shown for lymphocytes with specific targeting of the deoxyribonucleotide salvage pathway151 and for monocytes,152 and this could be especially interesting in transplantation research. In 2009, molecular imaging reporters of either phagocytosis or protease activity detected cardiac allograft rejection noninvasively by providing a functional three-dimensional map of macrophage accumulation, at least in mice.153 Whether these targeted molecular imaging tools could be of interest for clinical transplantation has not yet been studied.
Future challenges and opportunities
Next-generation sequencing
The application of the newer genome-wide molecular diagnostics has led to a rapid increase in the complexity of translational biomedical research. Upcoming high-throughput techniques such as massively parallel sequencing for genotyping, epigenomics and transcriptomics are expected to further accelerate the pace of translational research.154,155,156,157,158,159 These high-throughput sequencing technologies ('next-generation sequencing' tools) include Illumina's Genome Analyzer systems, Applied Biosystems' SOLiD™ system, Roche's 454 sequencing system, Helicos Biosciences' tSMS™ system156 and Pacific Biosciences' SMRT® system.160 With these new technologies, the cost of omics technologies is fast approaching a threshold at which omics will become a routine part of the diagnostic armamentarium, and with a modest investment, data acquisition for any genomic project can become comprehensive, integrating multiple genome-wide data sets with great depth and resolution.
Clinical application of omics data
The flood of research data concerning the genetic basis of health and disease, and the analytical and interpretative problems that come with this wealth of data, risk the further alienation of clinicians from basic sciences. True translational research should be multidimensional and collaborative, to ensure adequate data retrieval, correct data flow analysis and relevant clinical interpretation. The complexity of the clinical parameters, the complex physiology and immunology of many disease processes, the multitude of data per sample and the complex data analyses can only lead to biologically and clinically meaningful results with a multidisciplinary, holistic approach involving clinicians, molecular biologists, bioinformaticists and biostatisticians. Truly showing a 'return on investment' from functional genomics will require findings to be taken beyond the hypothesis-generating stage and integrated in downstream research applications, in discovery pipelines, or as biomarkers in clinical practice (Figure 3).

A growing gap exists between the generation of high-throughput molecular data and the translation of omics features into clinically meaningful concepts. Standard clinical research is performed by associating clinical features with pathophysiologic processes, histological alterations and identification of risk factors or comorbidities. The integration of these clinical data leads to the identification of disease networks of sometimes related phenomena such as acute and chronic ABMR or chronic ischemia with calcineurin-inhibitor nephrotoxicity. Omics research aims to integrate molecular diagnostic data into molecular networks. The great challenge in translational research today is to integrate the complex clinical networks with the even more complex molecular networks, in order to identify clinically relevant molecular networks and reclassify diseases on the basis of the underlying molecular alterations. Abbreviation: ABMR, antibody-mediated rejection.
A first step to bridge clinical (evidence-based) medicine and genomics-based translational science would be to collect and store DNA from patients enrolled in clinical trials, which would provide a resource for interrogating the role of genomic variation in treatment response and disease evolution. Trials that use genomic data for guiding clinical decision making would also be needed. As is outlined by the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group,161 the analytical validity, clinical validity and clinical utility of genomic tests will need assessing extensively before these tests can be adopted into clinical practice.
As outlined above, the currently accepted paradigms in molecular biology offer an integrated view on health and disease processes, with sophisticated regulation at each molecular level and indispensable feedback regulation between the different molecular levels. This complex interplay between the genome, transcriptome, proteome and metabolome18 has mostly been overlooked in traditional translational research and in the field of transplantation. Research groups often have access to few molecular diagnostic tools and collect information from single types of genomics data. The integration of different omics tools (integrative genomics), however, offers great advances in understanding human disease: each technology interrogates different aspects of gene function and each technology tends to produce noisy data and can be associated with its own inherent experimental limitations.114,162 Previous studies have demonstrated that data integration can improve the sensitivity and specificity for detecting true functional relationships among genes,163,164 and the use of integrated approaches is increasing rapidly, also in transplantation research.88,95,132
Bioinformatics and biostatistics
Multiple testing corrections
The important discrepancy between the vast amount of data obtained per sample and the often limited number of samples included in genomics studies is, however, a major problem for the translation of omics technologies to clinical practice (Box 2). This problem is known as the 'curse of dimensionality' (too many features in one sample), combined with the 'curse of dataset sparsity' (too few clinical samples).165 Bioinformatics and biostatistics have provided new tools to partly overcome this problem; false discovery rates are now recognized as marker of significance in omics studies, while standard P values should be reserved for testing individual hypotheses in classic experiments.166,167 Proper and sufficiently large validation sets will, however, still be necessary to obtain reliable and interpretable data. Many easy-to-use analysis packages have now been developed to avoid the need for tiresome programming that is out of the reach of many laboratories. Important tools for bioinformatics, such as Significance Analysis of Microarrays (SAM),168 GenePattern,169 GenMAPP,170 R software facilities and Bioconductor,171 are often free to download and are widely used in translational science.
Confounder analysis
Omics studies in transplantation typically include only a small number of transplant recipients. If the complexity of the patient population and the number of potential confounding factors is taken into account, the statistical power of many of these studies must be considered insufficient.141 This problem could be partly overcome by compiling and analyzing publicly available data in a sort of meta-analysis. Indeed, most omics data are included in publicly available databases such as the Gene Expression Omnibus (GEO),172 which offers unique opportunities for a more systematic meta-study of combined smaller studies, with unprecedented power for revealing commonalities and differences across different organ systems and different diseases.173,174 However, although expression and sequencing data are often publicly available, clinical data are often not, which obviates the integration of the molecular measurements with complex clinical features and phenotypes.
Molecule annotation and pathway analysis
The evolving annotation of probesets (that is, the mapping of probes in an omics platform to established gene identifiers), is a second major issue in bioinformatics for omics data. Probeset annotation often changes as the annotation for the underlying sequences changes over time, and care should be taken to use updated annotation files for analysis.175,176,177 It is not only the annotation of probes that constitute an omics platform that change over time. In pathway analysis, results depend on the validity of the pathways under consideration, and inconsistencies or evolution of the annotations of different molecules and their biological processes, location or function, will erratically be adopted in the final conclusions of pathway analysis.178,179
In addition, the integration of single-molecule data into biologically relevant networks is needed (Figure 3). Molecular networks and genesets can be manually curated, based on prior literature findings, or can be newly assembled using data obtained from omics techniques. The gene sets used for network analysis can be divided into five broad categories: curated gene sets (canonical pathways defined by domain experts); locational gene sets (based on chromosomal location); motif gene sets (based on the sharing of regulatory motifs); computational gene sets (based on prior experimental data); and Gene Ontology gene sets (based on Gene Ontology annotations for cellular components, biological processes or molecular functions). Many tools have been developed to map these functional networks, to assess the importance of candidate networks in complex disease processes and to evaluate single molecules as potential therapeutic targets within these networks. For this purpose, pathway analysis tools have been developed, enabling easy data mining without labor-intensive customized programming: GenePattern and the included Gene Set Enrichment Analysis tool (GSEA); the Database for Annotation, Visualization and Integrated Discovery (DAVID); Onto-Tools' PathwayExpress; GeneGo Metacore; Ingenuity Pathway Analysis; and BIOBASE ExPlain Analysis.
Absence of anatomical detail and cell specificity
Finally, typical genomics experiments collapse the cells in a given tissue, be it peripheral blood, biopsy tissue, urine or other samples. Although the tools seem to be sensitive for differential molecular regulation, they are not specific for localization within tissues. This lack of specificity is a major drawback of many omics studies, and spatial/anatomical information will only be reached with additional sample visualization techniques, microdissection and cell sorting.180,181 Obviously, these strategies are time-consuming and expensive. Recently, a novel method—cell-specific significance analysis of microarrays (csSAM)—was used to show that statistical deconvolution approaches can estimate cell-type-specific gene expression from heterogeneous samples, by weighted computational analysis of specific white cell compartment compositions available from an individual's differential white cell count.182 This approach helps in the virtual dissection of gene expression localization, and illustrates how the integration of clinical and laboratory values with molecular diagnostics can enhance understanding without additional effort.
Conclusions
Improved cross-linking and public repository of available data sets, together with in-depth, integrative data mining and smart biological interpretation, will ultimately lead to the more rapid development of translational research, easier validation of small studies in larger cohorts and a more efficient pipeline from hypothesis generation to validation and application. In 2009, Hirschhorn wrote: “Each discovery of a biologically relevant locus is a potential first step in a translational journey, and some journeys will be shorter than others. With a more complete collection of relevant genes and pathways, we can hope to shorten the interval between biologic knowledge and improved patient care.”40 We fully agree with this statement.
Review criteria
We searched PubMed and Google Scholar using the following terms: “transplantation”, “immune system”, “genome” and “genomics”, “genome-wide association”, “DNA polymorphisms”, “epigenetics” and “epigenomics”, “DNA methylation”, “gene expression” and “transcriptomics”, “mRNA degradation”, “translational repression”, “RNA interference”, “micro-array”, “next-generation sequencing”, “proteomics” and “peptidomics”, “metabolomics” and “chemical genomics”. Full-text publications relevant to the field were selected and relevant articles from reference lists and relevant review articles were also included. The search was limited to papers published in the English language.
References
Merrill, J., Murray, J., Harrison, J. & Guild, W. R. Successful homotransplantation of the human kidney between identical twins. J. Am. Med. Assoc. 160, 277–282 (1956).
Morris, P. J. Transplantation—a medical miracle of the 20th century. N. Engl. J. Med. 351, 2678–2680 (2004).
Starzl, T. E. & Zinkernagel, R. M. Transplantation tolerance from a historical perspective. Nat. Rev. Immunol. 1, 233–239 (2001).
Kahan, B. D. Individuality: the barrier to optimal immunosuppression. Nat. Rev. Immunol. 3, 831–838 (2003).
Watson, J. D. & Crick, F. H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953).
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Kidd, J. M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008).
Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
Sultan, M. et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321, 956–960 (2008).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Kim, D. H. & Rossi, J. J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 8, 173–184 (2007).
Cohen, P. The regulation of protein function by multisite phosphorylation—-a 25 year update. Trends Biochem. Sci. 25, 596–601 (2000).
Jensen, O. N. Interpreting the protein language using proteomics. Nat. Rev. Mol. Cell Biol. 7, 391–403 (2006).
Fiehn, O. Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171 (2002).
Nicholson, J. K., Connelly, J., Lindon, J. C. & Holmes, E. Metabonomics: a platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 1, 153–161 (2002).
Zerhouni, E. A. Translational and clinical science—time for a new vision. N. Engl. J. Med. 353, 1621–1623 (2005).
Hemminki, K., Lorenzo, B. J. & Forsti, A. The balance between heritable and environmental aetiology of human disease. Nat. Rev. Genet. 7, 958–965 (2006).
Nicholson, J. K., Holmes, E., Lindon, J. C. & Wilson, I. D. The challenges of modeling mammalian biocomplexity. Nat. Biotechnol. 22, 1268–1274 (2004).
Farber, D. L. Biochemical signaling pathways for memory T cell recall. Semin. Immunol. 21, 84–91 (2009).
Adams, A. B. et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Invest. 111, 1887–1895 (2003).
Smith, J. B. Frequency in human peripheral blood of T cells which respond to self, modified self and alloantigens. Immunology 50, 181–187 (1983).
Erlich, H. A., Opelz, G. & Hansen, J. HLA DNA typing and transplantation. Immunity 14, 347–356 (2001).
Hurley, C. K., Maiers, M., Marsh, S. G. & Oudshoorn, M. Overview of registries, HLA typing and diversity, and search algorithms. Tissue Antigens 69 (Suppl. 1), 3–5 (2007).
Sasazuki, T. et al. Effect of matching of class I HLA alleles on clinical outcome after transplantation of hematopoietic stem cells from an unrelated donor. Japan Marrow Donor Program. N. Engl. J. Med. 339, 1177–1185 (1998).
Rubinstein, P. HLA matching for bone marrow transplantation—how much is enough? N. Engl. J. Med. 345, 1842–1844 (2001).
Nickerson, P. The impact of immune gene polymorphisms in kidney and liver transplantation. Clin. Lab. Med. 28, 455–468, vii (2008).
Girnita, D. M., Webber, S. A. & Zeevi, A. Clinical impact of cytokine and growth factor genetic polymorphisms in thoracic organ transplantation. Clin. Lab. Med. 28, 423–440 (2008).
St Peter, S. D. et al. Genetic determinants of delayed graft function after kidney transplantation. Transplantation 74, 809–813 (2002).
Ullrich, R. et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter and cardiac allograft vasculopathy. J. Heart Lung Transplant. 24, 1600–1605 (2005).
Haimila, K. et al. Association of genetic variation in inducible costimulator gene with outcome of kidney transplantation. Transplantation 87, 393–396 (2009).
Kruger, B. et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl Acad. Sci. USA 106, 3390–3395 (2009).
Brown, K. M. et al. Influence of donor C3 allotype on late renal-transplantation outcome. N. Engl. J. Med. 354, 2014–2023 (2006).
Kruger, B., Schroppel, B. & Murphy, B. T. Genetic polymorphisms and the fate of the transplanted organ. Transplant. Rev. (Orlando) 22, 131–140 (2008).
de Jonge, H. & Kuypers, D. R. Pharmacogenetics in solid organ transplantation: current status and future directions. Transplant. Rev. (Orlando) 22, 6–20 (2008).
Lennard, L., Van Loon, J. A. & Weinshilboum, R. M. Pharmacogenetics of acute azathioprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin. Pharmacol. Ther. 46, 149–154 (1989).
Naesens, M. et al. Donor age and renal P-glycoprotein expression associate with chronic histological damage in renal allografts. J. Am. Soc. Nephrol. 20, 2468–2480 (2009).
Burckart, G. J. & Amur, S. Update on the clinical pharmacogenomics of organ transplantation. Pharmacogenomics 11, 227–236 (2010).
Thervet, E. et al. Optimization of initial tacrolimus dose using pharmacogenetic testing. Clin. Pharmacol. Ther. 87, 721–726 (2010).
Kruglyak, L. The road to genome-wide association studies. Nat. Rev. Genet. 9, 314–318 (2008).
Hirschhorn, J. N. Genomewide association studies—illuminating biologic pathways. N. Engl. J. Med. 360, 1699–1701 (2009).
Frazer, K. A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
International HapMap Project [online], (2010).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Manolio, T. A., Brooks, L. D. & Collins, F. S. A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 118, 1590–1605 (2008).
Hindorff, L. A., Junkins, H. A., Hall, P. N., Mehta, J. P. & Manolio, T. A. A Catalog of Published Genome-Wide Association Studies [online], (2010).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Cantor, R. M., Lange, K. & Sinsheimer, J. S. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am. J. Hum. Genet. 86, 6–22 (2010).
Goldstein, D. B. Common genetic variation and human traits. N. Engl. J. Med. 360, 1696–1698 (2009).
Genomes: a Deep Catalog of Human Genetic Variation [online], (2010).
The United Kingdom and Ireland Renal Transplant Consortium. WTCCC3—Wellcome Trust Case-Control Consortium 3: Defining the genetic basis of interactions between donor and recipient DNA that determine early and late renal transplant dysfunction [online], (2010).
Jirtle, R. L. & Skinner, M. K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 8, 253–262 (2007).
Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 (2009).
Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 358, 1148–1159 (2008).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Cuddapah, S., Barski, A. & Zhao, K. Epigenomics of T cell activation, differentiation, and memory. Curr. Opin. Immunol. 22, 341–347 (2010).
Araki, Y. et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 30, 912–925 (2009).
Wilson, C. B., Rowell, E. & Sekimata, M. Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 9, 91–105 (2009).
Granger, A. et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 22, 3549–3560 (2008).
Uhrberg, M. Shaping the human NK cell repertoire: an epigenetic glance at KIR gene regulation. Mol. Immunol. 42, 471–475 (2005).
Parra, M. Epigenetic events during B lymphocyte development. Epigenetics 4, 462–468 (2009).
Laird, P. W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 11, 191–203 (2010).
Floess, S. et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5, e38 (2007).
Lal, G. & Bromberg, J. S. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 114, 3727–3735 (2009).
Parker, M. D., Chambers, P. A., Lodge, J. P. & Pratt, J. R. Ischemia–reperfusion injury and its influence on the epigenetic modification of the donor kidney genome. Transplantation 86, 1818–1823 (2008).
Hoffmann, S. C. et al. Molecular and immunohistochemical characterization of the onset and resolution of human renal allograft ischemia-reperfusion injury. Transplantation 74, 916–923 (2002).
Avihingsanon, Y. et al. On the intraoperative molecular status of renal allografts after vascular reperfusion and clinical outcomes. J. Am. Soc. Nephrol. 16, 1542–1548 (2005).
Strehlau, J. et al. Quantitative detection of immune activation transcripts as a diagnostic tool in kidney transplantation. Proc. Natl Acad. Sci. USA 94, 695–700 (1997).
Shihab, F. et al. Transforming growth factor-beta and matrix protein expression in acute and chronic rejection of human renal allografts. J. Am. Soc. Nephrol. 6, 286–294 (1995).
Sharma, V. K. et al. Intragraft TGF-beta 1 mRNA: a correlate of interstitial fibrosis and chronic allograft nephropathy. Kidney Int. 49, 1297–1303 (1996).
Strom, T. B. & Suthanthiran, M. Transcriptional profiling to assess the clinical status of kidney transplants. Nat. Clin. Pract. Nephrol. 2, 116–117 (2006).
Li, B. et al. Noninvasive diagnosis of renal-allograft rejection by measurement of messenger RNA for perforin and granzyme B in urine. N. Engl. J. Med. 344, 947–954 (2001).
Simon, T., Opelz, G., Wiesel, M., Ott, R. C. & Susal, C. Serial peripheral blood perforin and granzyme B gene expression measurements for prediction of acute rejection in kidney graft recipients. Am. J. Transplant. 3, 1121–1127 (2003).
Aquino-Dias, E. C. et al. Non-invasive diagnosis of acute rejection in kidney transplants with delayed graft function. Kidney Int. 73, 877–884 (2008).
Anglicheau, D. & Suthanthiran, M. Noninvasive prediction of organ graft rejection and outcome using gene expression patterns. Transplantation 86, 192–199 (2008).
Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470 (1995).
Derisi, J. et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 14, 457–460 (1996).
Shi, L. et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161 (2006).
Weintraub, L. A. & Sarwal, M. M. Microarrays: a monitoring tool for transplant patients? Transpl. Int. 19, 775–788 (2006).
Ying, L. & Sarwal, M. In praise of arrays. Pediatr. Nephrol. 24, 1643–1659 (2009).
Khatri, P. & Sarwal, M. M. Using gene arrays in diagnosis of rejection. Curr. Opin. Organ Transplant. 14, 34–39 (2009).
Mueller, T. F. et al. The transcriptome of the implant biopsy identifies donor kidneys at increased risk of delayed graft function. Am. J. Transplant. 8, 78–85 (2008).
Naesens, M. et al. Expression of complement components differs between kidney allografts from living and deceased donors. J. Am. Soc. Nephrol. 20, 1839–1851 (2009).
Sarwal, M. et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N. Engl. J. Med. 349, 125–138 (2003).
Mueller, T. F. et al. Microarray analysis of rejection in human kidney transplants using pathogenesis-based transcript sets. Am. J. Transplant. 7, 2712–2722 (2007).
Mas, V. et al. Establishing the molecular pathways involved in chronic allograft nephropathy for testing new noninvasive diagnostic markers. Transplantation 83, 448–457 (2007).
Park, W., Griffin, M., Grande, J. P., Cosio, F. & Stegall, M. D. Molecular evidence of injury and inflammation in normal and fibrotic renal allografts one year posttransplant. Transplantation 83, 1466–1476 (2007).
Mengel, M. et al. Molecular correlates of scarring in kidney transplants: the emergence of mast cell transcripts. Am. J. Transplant. 9, 169–178 (2009).
Nakorchevsky, A. et al. Molecular mechanisms of chronic kidney transplant rejection via large-scale proteogenomic analysis of tissue biopsies. J. Am. Soc. Nephrol. 21, 362–373 (2010).
Brouard, S. et al. Identification of a peripheral blood transcriptional biomarker panel associated with operational renal allograft tolerance. Proc. Natl Acad. Sci. USA 104, 15448–15453 (2007).
Martinez-Llordella, M. et al. Using transcriptional profiling to develop a diagnostic test of operational tolerance in liver transplant recipients. J. Clin. Invest. 118, 2845–2857 (2008).
Zarkhin, V. & Sarwal, M. M. Microarrays: monitoring for transplant tolerance and mechanistic insights. Clin. Lab. Med. 28, 385–410 (2008).
Li, L. et al. Interference of globin genes with biomarker discovery for allograft rejection in peripheral blood samples. Physiol. Genomics 32, 190–197 (2008).
Gunther, O. P. et al. Functional genomic analysis of peripheral blood during early acute renal allograft rejection. Transplantation 88, 942–951 (2009).
Lin, D. et al. Whole blood genomic biomarkers of acute cardiac allograft rejection. J. Heart Lung Transplant. 28, 927–935 (2009).
Kurian, S. M. et al. Biomarkers for early and late stage chronic allograft nephropathy by proteogenomic profiling of peripheral blood. PLoS ONE 4, e6212 (2009).
Kwan, T. et al. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 40, 225–231 (2008).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Richard, H. et al. Prediction of alternative isoforms from exon expression levels in RNA-Seq experiments. Nucleic Acids Res. 38, e112 (2010).
Grosshans, H. & Filipowicz, W. Molecular biology: the expanding world of small RNAs. Nature 451, 414–416 (2008).
Heo, I. & Kim, V. N. Regulating the regulators: posttranslational modifications of RNA silencing factors. Cell 139, 28–31 (2009).
Ghildiyal, M. & Zamore, P. D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108 (2009).
McManus, M. T. MicroRNAs and cancer. Semin. Cancer Biol. 13, 253–258 (2003).
McManus, M. T. Small RNAs and immunity. Immunity 21, 747–756 (2004).
Gantier, M. P., Sadler, A. J. & Williams, B. R. Fine-tuning of the innate immune response by microRNAs. Immunol. Cell Biol. 85, 458–462 (2007).
Lodish, H. F., Zhou, B., Liu, G. & Chen, C. Z. Micromanagement of the immune system by microRNAs. Nat. Rev. Immunol. 8, 120–130 (2008).
Anglicheau, D. et al. MicroRNA expression profiles predictive of human renal allograft status. Proc. Natl Acad. Sci. USA 106, 5330–5335 (2009).
Cohen, C. D. Will non-coding RNAs help to decipher renal allograft failure? Nephrol. Dial. Transplant. 24, 2325–2327 (2009).
Harris, A., Krams, S. M. & Martinez, O. M. MicroRNAs as immune regulators: implications for transplantation. Am. J. Transplant. 10, 713–719 (2010).
Rodriguez, A. et al. Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611 (2007).
Castanotto, D. & Rossi, J. J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 457, 426–433 (2009).
Abbott, A. A post-genomic challenge: learning to read patterns of protein synthesis. Nature 402, 715–720 (1999).
Mann, M. & Jensen, O. N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 (2003).
Cristea, I. M., Gaskell, S. J. & Whetton, A. D. Proteomics techniques and their application to hematology. Blood 103, 3624–3634 (2004).
Ludwig, J. A. & Weinstein, J. N. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 5, 845–856 (2005).
Graham, D. R., Elliott, S. T. & Van Eyk, J. E. Broad-based proteomic strategies: a practical guide to proteomics and functional screening. J. Physiol. 563, 1–9 (2005).
Hanash, S. M., Pitteri, S. J. & Faca, V. M. Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 (2008).
Sigdel, T. K., Klassen, R. B. & Sarwal, M. M. Interpreting the proteome and peptidome in transplantation. Adv. Clin. Chem. 47, 139–169 (2009).
Jenkins, J. K., Huang, H., Ndebele, K. & Salahudeen, A. K. Vitamin E inhibits renal mRNA expression of COX II, HO I, TGFbeta, and osteopontin in the rat model of cyclosporine nephrotoxicity. Transplantation 71, 331–334 (2001).
O'Riordan, E., Gross, S. S. & Goligorsky, M. S. Technology Insight: renal proteomics—at the crossroads between promise and problems. Nat. Clin. Pract. Nephrol. 2, 445–458 (2006).
Clarke, W. et al. Characterization of renal allograft rejection by urinary proteomic analysis. Ann. Surg. 237, 660–664 (2003).
Schaub, S. et al. Proteomic-based detection of urine proteins associated with acute renal allograft rejection. J. Am. Soc. Nephrol. 15, 219–227 (2004).
O'Riordan, E. et al. Bioinformatic analysis of the urine proteome of acute allograft rejection. J. Am. Soc. Nephrol. 15, 3240–3248 (2004).
Borozdenkova, S. et al. Use of proteomics to discover novel markers of cardiac allograft rejection. J. Proteome Res. 3, 282–288 (2004).
Ling, X. B. et al. Integrative urinary peptidomics in renal transplantation identifies biomarkers for acute rejection. J. Am. Soc. Nephrol. 21, 646–653 (2010).
Quintana, L. F. et al. Urine proteomics to detect biomarkers for chronic allograft dysfunction. J. Am. Soc. Nephrol. 20, 428–435 (2009).
Banon-Maneus, E. et al. Two-dimensional difference gel electrophoresis urinary proteomic profile in the search of nonimmune chronic allograft dysfunction biomarkers. Transplantation 89, 548–558 (2010).
Quintana, L. F., Banon-Maneus, E., Sole-Gonzalez, A. & Campistol, J. M. Urine proteomics biomarkers in renal transplantation: an overview. Transplantation 88, S45–S49 (2009).
Yu, X., Schneiderhan-Marra, N., Hsu, H. Y., Bachmann, J. & Joos, T. O. Protein microarrays: effective tools for the study of inflammatory diseases. Methods Mol. Biol. 577, 199–214 (2009).
Robinson, W. H. et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 8, 295–301 (2002).
Hudson, M. E., Pozdnyakova, I., Haines, K., Mor, G. & Snyder, M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc. Natl Acad. Sci. USA 104, 17494–17499 (2007).
Wang, X. et al. Autoantibody signatures in prostate cancer. N. Engl. J. Med. 353, 1224–1235 (2005).
Li, L. et al. Identifying compartment-specific non-HLA targets after renal transplantation by integrating transcriptome and “antibodyome” measures. Proc. Natl Acad. Sci. USA 106, 4148–4153 (2009).
Sutherland, S. M. et al. Protein microarrays identify antibodies to protein kinase Czeta that are associated with a greater risk of allograft loss in pediatric renal transplant recipients. Kidney Int. 76, 1277–1283 (2009).
Li, L. et al. Compartmental localization and clinical relevance of MICA antibodies after renal transplantation. Transplantation 89, 312–319 (2010).
Porcheray, F. et al. B-cell immunity in the context of T-cell tolerance after combined kidney and bone marrow transplantation in humans. Am. J. Transplant. 9, 2126–2135 (2009).
Wishart, D. S. Metabolomics: a complementary tool in renal transplantation. Contrib. Nephrol. 160, 76–87 (2008).
Wishart, D. S. et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 37, D603–D610 (2009).
Frolkis, A. et al. SMPDB: The small molecule pathway database. Nucleic Acids Res. 38, D480–D487 (2010).
Serkova, N., Fuller, T. F., Klawitter, J., Freise, C. E. & Niemann, C. U. H-NMR-based metabolic signatures of mild and severe ischemia/reperfusion injury in rat kidney transplants. Kidney Int. 67, 1142–1151 (2005).
Wang, J. N., Zhou, Y., Zhu, T. Y., Wang, X. & Guo, Y. L. Prediction of acute cellular renal allograft rejection by urinary metabolomics using MALDI-FTMS. J. Proteome Res. 7, 3597–3601 (2008).
Christians, U. et al. Toxicodynamic therapeutic drug monitoring of immunosuppressants: promises, reality, and challenges. Ther. Drug Monit. 30, 151–158 (2008).
Foxall, P. J., Mellotte, G. J., Bending, M. R., Lindon, J. C. & Nicholson, J. K. NMR spectroscopy as a novel approach to the monitoring of renal transplant function. Kidney Int. 43, 234–245 (1993).
Serkova, N. J. et al. Early detection of graft failure using the blood metabolic profile of a liver recipient. Transplantation 83, 517–521 (2007).
Butcher, R. A. & Schreiber, S. L. Using genome-wide transcriptional profiling to elucidate small-molecule mechanism. Curr. Opin. Chem. Biol. 9, 25–30 (2005).
Strausberg, R. L. & Schreiber, S. L. From knowing to controlling: a path from genomics to drugs using small molecule probes. Science 300, 294–295 (2003).
Seiler, K. P. et al. ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 36, D351–D359 (2008).
Weissleder, R., Tung, C. H., Mahmood, U. & Bogdanov, A. Jr. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat. Biotechnol. 17, 375–378 (1999).
Sosnovik, D. E., Nahrendorf, M. & Weissleder, R. Targeted imaging of myocardial damage. Nat. Clin. Pract. Cardiovasc. Med. 5 (Suppl. 2), S63–S70 (2008).
Harisinghani, M. G. et al. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N. Engl. J. Med. 348, 2491–2499 (2003).
Kooi, M. E. et al. Accumulation of ultrasmall superparamagnetic particles of iron oxide in human atherosclerotic plaques can be detected by in vivo magnetic resonance imaging. Circulation 107, 2453–2458 (2003).
Radu, C. G. et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2′-deoxycytidine analog. Nat. Med. 14, 783–788 (2008).
Wildgruber, M. et al. Monocyte subset dynamics in human atherosclerosis can be profiled with magnetic nano-sensors. PLoS ONE 4, e5663 (2009).
Christen, T. et al. Molecular imaging of innate immune cell function in transplant rejection. Circulation 119, 1925–1932 (2009).
Rogers, Y. H. & Venter, J. C. Genomics: massively parallel sequencing. Nature 437, 326–327 (2005).
Rothberg, J. M. & Leamon, J. H. The development and impact of 454 sequencing. Nat. Biotechnol. 26, 1117–1124 (2008).
Tucker, T., Marra, M. & Friedman, J. M. Massively parallel sequencing: the next big thing in genetic medicine. Am. J. Hum. Genet. 85, 142–154 (2009).
Aparicio, S. A. & Huntsman, D. G. Does massively parallel DNA resequencing signify the end of histopathology as we know it? J. Pathol. 220, 307–315 (2010).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Shendure, J. The beginning of the end for microarrays? Nat. Methods 5, 585–587 (2008).
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009).
Teutsch, S. M. et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet. Med. 11, 3–14 (2009).
Giallourakis, C., Henson, C., Reich, M., Xie, X. & Mootha, V. K. Disease gene discovery through integrative genomics. Annu. Rev. Genomics Hum. Genet. 6, 381–406 (2005).
Mootha, V. K. et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl Acad. Sci. USA 100, 605–610 (2003).
Lee, I., Date, S. V., Adai, A. T. & Marcotte, E. M. A probabilistic functional network of yeast genes. Science 306, 1555–1558 (2004).
Somorjai, R. L., Dolenko, B. & Baumgartner, R. Class prediction and discovery using gene microarray and proteomics mass spectroscopy data: curses, caveats, cautions. Bioinformatics 19, 1484–1491 (2003).
Pawitan, Y., Michiels, S., Koscielny, S., Gusnanto, A. & Ploner, A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics 21, 3017–3024 (2005).
Butte, A. The use and analysis of microarray data. Nat. Rev. Drug Discov. 1, 951–960 (2002).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001).
Reich, M. et al. GenePattern 2.0. Nat. Genet. 38, 500–501 (2006).
Dahlquist, K. D., Salomonis, N., Vranizan, K., Lawlor, S. C. & Conklin, B. R. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat. Genet. 31, 19–20 (2002).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
National Center for Biotechnology Information. Gene Expression Omnibus [online], (2010).
Butte, A. J. & Kohane, I. S. Creation and implications of a phenome-genome network. Nat. Biotechnol. 24, 55–62 (2006).
Thorisson, G. A., Muilu, J. & Brookes, A. J. Genotype-phenotype databases: challenges and solutions for the post-genomic era. Nat. Rev. Genet. 10, 9–18 (2009).
Perez-Iratxeta, C. & Andrade, M. A. Inconsistencies over time in 5% of NetAffx probe-to-gene annotations. BMC Bioinformatics 6, 183 (2005).
Noth, S. & Benecke, A. Avoiding inconsistencies over time and tracking difficulties in Applied Biosystems AB1700/Panther probe-to-gene annotations. BMC Bioinformatics 6, 307 (2005).
Chen, R., Li, L. & Butte, A. J. AILUN: reannotating gene expression data automatically. Nat. Methods 4, 879 (2007).
Rhee, S. Y., Wood, V., Dolinski, K. & Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 9, 509–515 (2008).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Schulze, A. & Downward, J. Navigating gene expression using microarrays—a technology review. Nat. Cell Biol. 3, E190–E195 (2001).
Lieberfarb, M. E. et al. Genome-wide loss of heterozygosity analysis from laser capture microdissected prostate cancer using single nucleotide polymorphic allele (SNP) arrays and a novel bioinformatics platform dChipSNP. Cancer Res. 63, 4781–4785 (2003).
Shen-Orr, S. S. et al. Cell type-specific gene expression differences in complex tissues. Nat. Methods 7, 287–289 (2010).
Acknowledgements
We apologize to colleagues whose work we were unable to cite due to space constraints.
Author information
Authors and Affiliations
Contributions
Both authors contributed to researching data for the article, discussing the content, writing the article, and reviewing and editing the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Naesens, M., Sarwal, M. Molecular diagnostics in transplantation. Nat Rev Nephrol 6, 614–628 (2010). https://doi.org/10.1038/nrneph.2010.113
Published:
Issue date:
DOI: https://doi.org/10.1038/nrneph.2010.113
This article is cited by
-
Advances in Detection of Kidney Transplant Injury
Molecular Diagnosis & Therapy (2019)
-
Identification of Candidate Biomarkers for Transplant Rejection from Transcriptome Data: A Systematic Review
Molecular Diagnosis & Therapy (2019)
-
Integrated and global pseudotargeted metabolomics strategy applied to screening for quality control markers of Citrus TCMs
Analytical and Bioanalytical Chemistry (2017)
-
Lipidomics comparing DCD and DBD liver allografts uncovers lysophospholipids elevated in recipients undergoing early allograft dysfunction
Scientific Reports (2015)
-
Managing diabetes with nanomedicine: challenges and opportunities
Nature Reviews Drug Discovery (2015)
