
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is a rare genetic disease with an estimated prevalence of no more than 1 in 8000; however, it is among the most common myopathies affecting global populations. This condition is classically categorised into two genetic types, FSHD1 (MIM: 158900) and FSHD2 (MIM: 158901), which, although have different genetic causes, are phenotypically indistinguishable, manifesting as progressive muscle weakness primarily affecting the face and periscapular muscles, as well as other muscle groups in later stages. The intense efforts of clinical and basic studies to understand this disease have revealed the critical necessity for disease manifestation: ectopic activation of the embryogenic and germline gene DUX4 (double homeobox 4, MIM: 606009) in skeletal muscles and the genetic and epigenetic backgrounds allowing DUX4 expression. Thus, the potential target therapies of FSHD include silencing DUX4 transcription or blocking its translation. Although the central role of DUX4 in FSHD pathology has almost reached a consensus, the mechanism of its activation remains largely unclear. Notably, the clinical dissection of genotype–epigenotype-phenotype observations, including non-penetrant and asymptomatic carriers of permissive genetic backgrounds, highlights the yet unsolved clinical diversity with potential additional layers of DUX4 regulation or other disease-modifying factors. This review provides an overview of essential findings with potential implications for further understanding the mechanisms underlying diverse clinical cases of FSHD and endogenous DUX4 activation in FSHD pathology.
Similar content being viewed by others

Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal inherited genetic disorder characterised by initial symptoms and signs of skeletal muscle atrophy and weakness in the face, shoulders, and upper arms [1]. Muscle weakness slowly progresses throughout the lifespan, sometimes in an asymmetric manner [2], often affecting other parts, including the lower limbs, pulmonary function-related muscles, and trunk muscles, leading to disabilities such as the loss of independent ambulation, wheelchair use, chronic respiratory dysfunction necessitating an assistance device [3, 4], sleep disorders, chronic pain and fatigue as well [5]. Facial weakness typically serves as an early indicator of disease onset, whereas lower-extremity involvement and wheelchair dependency indicate more advanced disease severity, although these are not observed in all cases as progression patterns vary among individuals. The disease often manifests in the second or third decade of life; however, the age at onset, levels of local penetrance, and total disease severity largely vary among individuals with FSHD. Early onset FSHD is defined by the clinical onset of facial involvement before 5 years of age and manifestations of shoulder girdle weakness before 10 years of age, often showing faster disease progression and more frequent dependence on wheelchair use [6, 7]. Extramuscular and systemic symptoms, such as high-frequency hearing loss [8, 9], retinal abnormalities resembling Coat’s disease [10,11,12], spinal deformities [13], epilepsy/mental retardation [14], and mild cardiac involvement [15] have rarely been observed. Early-onset or infantile FSHD averagely accounts for 10% of the total FSHD population [16]. These cases often present with more severe muscle phenotypes and more frequent systemic symptoms [13, 17], representing the severe end of the FSHD disease spectrum [18].
The prevalence of FSHD is estimated at 15:10,000 in the Dutch population, the highest to date [19], whereas it is lower in other populations partially due to fewer opportunities for access to genetic diagnosis, especially for asymptomatic or paucisymptomatic cases [20], possibly depending on the active research environment, or otherwise due to potential race differences [21, 22]. The sex difference in disease severity is implied where FSHD affects males more severely and more frequently than females [23,24,25], although other studies reported that females were more likely to progress to wheelchair use and predominantly occupied a category with severe phenotype [26,27,28], requiring further study with attention to differences in the detailed classification of disease severity, age related to hormonal activity and race. Furthermore, 10–30% cases are considered de novo, including those derived from parental germline mutations and other cases of gonadosomatic mosaicism occurring during early embryogenesis [29], whereas a recent family-based study in a cohort of Italian families estimated a frequency of as low as 8% for truly de novo cases [30].
Basic genetic classification and common local epigenetic de-repression by complex genetic backgrounds cause FSHD onset
Numerous investigations in clinical genetics and epigenetics have reported the central role of ectopic activation of embryogenic and germline gene DUX4 (double homeobox 4) in muscle-specific pathology and the genetic necessity of the ‘permissive allele’ to allow ectopic DUX4 expression in skeletal muscles [31]. Earlier efforts to understand the genetic cause of this disease using linkage analysis of FSHD families identified an FSHD-associated region in 4q35, a subtelomeric region on chromosome 4 [32]. The 4q35 region contains a type of large tandem repeat unit named the D4Z4 macrosatellite or simply a D4Z4 repeat, each unit of which consists of a 3.3 kb CpG-rich sequence. A wide range of copy number variations, up to 150 units, are found in D4Z4 repeats in the human population. Several early studies have demonstrated that the level of DNA methylation on D4Z4 repeats in at least one allele is significantly low, that is, hypomethylated, in cells derived from patients with FSHD, including muscle cells and other cell types from different tissue origins, such as blood, skin, and saliva [33,34,35,36,37,38]. Several studies have reported that the level of DNA hypomethylation in the D4Z4 region is correlated with disease severity (details are described later).
Two major factors are linked to differences in DNA methylation levels of D4Z4 repeats in the 4q35 region: copy number of D4Z4 repeat units (RUs) and mutations in chromatin regulators of this repeat. Most of FSHD cases (~95%) are found with the lower D4Z4 RU within the ‘contraction’ range of 1–10 RU, where the significant DNA hypomethylation of D4Z4 repeat on the contraction-associated allele coincides in clinical FSHD cases [37]. FSHD cases associated with D4Z4 repeat contractions were classified as FSHD type 1 (FSHD1). The other cases without contracted D4Z4 repeat observed are classified into FSHD type 2 (FSHD2), where the additional mutations can be found most frequently (80%) in SMCHD1 (the Structural Maintenance of Chromosomes flexible Hinge Domain-containing protein 1, OMIM: 614982) on chromosome 18p11 [39], or less frequently in DNMT3B (DNA methyltransferase 3B, OMIM: 602900) on chromosome 20q11 [40] and LRIF1 (the ligand-dependent nuclear receptor-interacting factor 1, OMIM: 615354) on chromosome 1p13 [41]. The D4Z4 repeat copy numbers in FSHD2 cases have been previously recognised as 10 RU to 20 RU. However, cases involving > 20 RUs have been reported, albeit less frequently [42]. The clinical features of patients with FSHD1 and FSHD2 were indistinguishable, suggesting that FSHD1 and FSHD2 result from the same pathophysiologic process [43]. SMCHD1 mutations are found in more severe symptomatic cases with 9–10 RU of D4Z4, a range conventionally categorised as FSHD1, than in FSHD1 cases with larger FSHD1 alleles (8–10 RU) [44,45,46]. This combined case was referred to as FSHD1 + 2. Symptomatic cases with relatively milder severity, asymptomatic and non-penetrant cases are often found among the individuals with 8–10 RU plus without SMCHD1 mutations [20, 47, 48], highlighting 8–10 RU as the ‘borderline’ or ‘grey zone’ of FSHD1 penetrance [49]. Interestingly, FSHD1 patients have been rarely found in the ‘grey zone’ in a Japanese and a Korean cohort [22]. This suggests potential differences in disease susceptibility among ethnic populations, though further research is needed to clarify whether these differences reflect ascertainment bias and diagnostic practices, genuine genetic background effects, or cultural factors such as diet. In addition, mutations in DNMT3B have been attributed to the worsening of disease progression as disease modifiers in FSHD1 [40]. Based on these genotype-phenotype observations, it is proposed that FSHD should be considered a disease continuum or spectrum depending on multiple factors, such as D4Z4 repeat copy numbers, DNA methylation levels in the D4Z4 region, and mutations in chromatin regulator genes, rather than undergoing a traditional simple classification into two discrete categories, FSHD1 and FSHD2 [46]. Therefore, the current common genetic diagnosis protocol allows for the overlapping category of FSHD1 and FSHD2 with 8–10 RU [49].
This concept of FSHD as a disease continuum also fits the clinical variability, in which a certain population of patients manifests earlier and/or more severe disease progression. A rough and inverse correlation between disease severity and the number of D4Z4 RUs in FSHD1 among 1–10 RU is observed [48, 50], although intrafamilial clinical variability and asymptomatic/non-penetrant cases are often observed over 4 RU [25]. Early-onset FSHD cases tend to be found within a shorter range of D4Z4 repeat (1–3 RU), although this shorter range of contraction does not always lead to a severe disease outcome or, rather, even an asymptomatic case in certain relatives carrying the same short allele as the proband [51]. In addition, even monozygotic twins with FSHD show extreme variability in the phenotype [52]. In contrast, affected carriers of a disease array of 7–10 D4Z4 RU, but not familial non-penetrant mutation carriers, have a greater reduction in D4Z4 DNA methylation [38]. Another study reported that DNA methylation in FSHD1-affected subjects was lower, with a higher response to epigenetically derepressing chemicals, than in non-manifesting familial subjects in the context of encoded gene activation [53]. These studies indicate that epigenetic derepression, represented by DNA hypomethylation, can serve as a significant hallmark and reliable predictor of clinical FSHD progression.
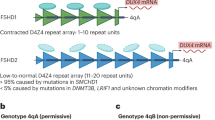
Each unit of the D4Z4 repeat contains a single open reading frame of a retrogene named DUX4 their oriented forward [54]. Each ORF in the repeat lacks a polyadenylation signal (PAS) sequence to stabilise its transcripts, except for the most distal unit, which allows for stable transcription and subsequent translation, depending on the sequence patterns with potential PAS-containing elements outside of the D4Z4 repeat. Human DUX4, and its murine orthologous gene Dux as well, is transiently activated during a limited window of early embryonic stages [55,56,57] and is expressed in tissues such as the testis and thymus (possibly reflecting on expression in the lymphocytes) in humans [58, 59], suggesting potential unknown functions, but is normally tightly silenced in most later developmental stages and somatic tissues. In an evolutionarily conserved context, DUX4 can utilise a PAS-containing exon located 10 kbp downstream of the D4Z4 repeat (in the T2T-CHM13 reference genome, but varies among haplotypes), and this splicing pattern is considered irrelevant to FSHD [58]. Intense genetic investigations revealed that although the 4q35 region has a variety of haplotypes in the human population [60], only a class of haplotypes called 4qA is relevant to FSHD manifestation, and other haplotypes, such as 4qB, are not [47, 61]. The 4qA haplotypes exclusively contain a segment of DNA called pLAM and a 6.2 kb β-satellite repeat [62]. The SNP present in the pLAM region creates a functional but non-canonical PAS (AUUAAA) sequence that is proximally located after the last D4Z4 unit, stabilises DUX4 transcripts to produce mature mRNAs, and allows DUX4 protein production [31]. Thus, the requirements of genetically typical cases of FSHD are conceptually summarised as the combination of DNA hypomethylation of the D4Z4 repeat and its cis-connected 4qA haplotype, which forms a permissive 4qA allele that potentially allows DUX4 activation and FSHD onset (Fig. 1).

Schematic representation of the D4Z4 repeat array on chromosome 4q35.2, the genetic locus associated with FSHD. Each D4Z4 unit is 3.3 kb in length. FSHD is caused by contraction of these repeats [1,2,3,4,5,6,7,8,9,10] or mutations in chromatin regulators such as SMCHD1, DNMT3B and LRIF1, leading to hypomethylation of the region and aberrant expression of the DUX4 gene in the most distal repeat. DUX4 expression additionally requires a permissive 4qA allele containing a polyadenylation signal to stabilize its transcript. Cases caused by D4Z4 contraction per se are categorized as FSHD1, while those associated with variants in other genes are categorized as FSHD2. These standard criteria accommodate cases that overlap both categories (such as FSHD1 with modifier effects from SMCHD1 variants). In FSHD1, disease severity roughly correlates inversely with the number of D4Z4 repeats: cases with 1–3 repeats tend to be most severe including more early onset cases, those with 4–7 repeats moderately severe, and those with 8–10 repeats relatively mild. Asymptomatic cases are more frequently observed among individuals with longer repeat arrays
DUX4 may have evolutionarily conserved biological functions that are essential during the early embryonic stages and in specific tissues that express its protein; however, when activated in skeletal muscle cells, DUX4 exerts its phenotypic effects through its transcriptional activity followed by multiple molecular pathways, leading to tissue-level degradation [63,64,65,66,67,68,69,70]. The currently available cell and animal models for investigating DUX4 function and FSHD have been summarised in other review articles [71, 72]. The molecular and cellular consequences of DUX4 activation in FSHD have been summarised in these review articles [73, 74]. Importantly, recent studies support the association between inflammatory magnetic resonance imaging (MRI) characteristics and the expression of genes regulated by DUX4 and other gene categories associated with FSHD disease activity at the local level within skeletal muscle tissues, indicating detailed disease progression, from inflammation or oedema to fat infiltration [75]. In contrast, a meta-analysis of published FSHD muscle biopsy gene expression studies revealed that PAX7 target gene repression in FSHD correlated with disease severity, independent of onsite DUX4 target gene expression [68, 76]. This apparent discrepant signature may occur via the epigenetic footprint of historical DUX4 activation during development and growth before the onset, when DUX4 is already on [77] and later accumulatively affects stem cell function after growth and during regeneration [78]. Thus, based on this supporting knowledge of the central role of DUX4 in pathology, methodologies to silence DUX4 activation, degrade DUX4 mRNA, and prevent or halt DUX4 protein transcriptional activity and downstream adverse toxic effects are being developed as promising target therapeutic approaches for FSHD [79]. In addition, supplementation strategy for muscle cell resources may be necessary for functional improvement of damaged muscles in severely degraded stages.
Learning from genetically atypical cases to understand essential genetic factors for FSHD onset
As described above, the genetic background of FSHD consists of large genomic loci and mutations at different loci. Although several clinical cases demonstrating FSHD symptoms can be genetically confirmed based on the typical requirements explained above, certain cases fall outside these criteria and yet may still arise from the same mechanism, in which a hypomethylated D4Z4 repeat with a cis-linked PAS-containing sequence forms a permissive allele that enables stabilised DUX4 expression. In addition, certain asymptomatic and non-penetrating cases, which are often found among family cohorts of patients with FSHD, can guide a more precise dissection of essential genetic/epigenetic elements and disease modifiers of FSHD onset. Intense investigations of FSHD cases by molecular combing revealed complex patterns of rearrangements other than the typical sequence of the reference genome, which can be overlooked with standard genetic diagnosis by Southern blot [80] (Fig. 2).

Schematic representation of the D4Z4 region in atypical FSHD and non-FSHD cases. In the duplication type, although one D4Z4 repeat array is contracted, traditional Southern blotting may misinterpret the duplicated units as a single long repeat array, masking the contraction. While D4Z4 repeats exist throughout the genome, a similar repeat array occurs on chromosome 10q. Contractions in 10qD4Z4 alone do not cause FSHD; however, translocation of a permissive 4qA allele to the 10q locus can result in 10q-linked FSHD. In the D4Z4 proximally extended deletion (DPED) cases, a large proximal deletion removes the Southern blot probe region, making detection of the pathogenic allele difficult. In mosaicism, only a fraction of somatic cells harbour D4Z4-contracted alleles, leading to DUX4 expression and FSHD symptoms that are typically milder than in contraction-matched cases without mosaicism
Subgroups of haplotypes
Although the haplotypes of 4q35 and 10q26 are often categorised into groups such as 4qA, 4qB, and 10qB, according to the necessity of PAS immediately distal to the D4Z4 repeat for FSHD manifestation, more small subtypes and subgroups with sequence variations inside and outside the D4Z4 repeats exist [47, 60]. A detailed analysis of these haplotypes revealed the necessity of PAS for DUX4 activation and FSHD onset [31]. Although at least 17 unique 4q haplotypes have been identified, only 4A161S, 4A161L, 4A159, and 4A168 have been reported to be associated with FSHD [73], and most of the other haplotypes, including 4qB and 10q, lack functional PAS and are not permissive. Interestingly, 4A166, among PAS-containing 4qA haplotypes, is not considered to be associated with FSHD, as no clinical FSHD cases have been found even when it had repeat contraction [47], indicating that certain critical genetic elements required for enhancing DUX4 activation may be altered or that certain de novo elements may be formed to affect DUX4 activation in this haplotype allele. However, another report claimed that 4A166 haplotypes with clinical phenotypes were observed [25, 81], thus requiring further clarification among different studies. A molecular genetic study reported a significant role of other nearby sequences in the 4qA β-satellite as a cis-acting element for DUX4 mRNA production [62], in addition to the PAS in 4qA pLAM. Further comparative investigations among subtypes may help identify the essential functional elements for DUX4 activation in FSHD muscle cells. Gene editing strategies for PAS in the permissive allele to achieve DUX4 reduction have yielded inconsistent results [82,83,84], possibly because distinct approaches might affect the molecular behaviours of regulators by modifying or provoking surrounding genetic elements associated with polyadenylation and transcription.
D4Z4 proximally extended deletion (DPED)
Genetic analysis in certain clinical cases demonstrated longer genomic deletions extending from contracted D4Z4 repeats to include sequences immediately upstream of the repeat, called D4Z4 proximally extended deletions (DPED) [80, 85,86,87]. These DPED cases manifested typical FSHD phenotypes, indicating that the deleted genomic region most likely does not play an essential role in FSHD pathology. An in vitro study suggested that myogenic regulatory elements that can activate DUX4 lie within the deleted region, although their significance is controversial based on this clinical genetic observation [88]. In addition, this deleted region contains DUX4c, FRG2, and long non-coding DBE-T [87], genes previously suggested as contributing factors to FSHD. However, their central significance remains uncertain, although FRG2B, another FRG2 family member, is located near 10q D4Z4 with potential compensation by their functional redundancy. In contrast, as several studies have reported long-range three-dimensional conformational changes between non-FSHD and FSHD chromatin [88,89,90,91,92,93], the DPED cases do not exclude the possibility of any regulatory elements that can activate DUX4 beyond the deleted region [94].
Translocation of 4qA to 10q
The D4Z4 repeat and its partial fragment are scattered throughout the whole human genome [95]. 10q26, the subtelomeric region of chromosome 10, contains the D4Z4 repeat, with each unit having a sequence difference that is technically recognisable by restriction enzyme sensitivity in genetic tests [60]. 10q D4Z4, similar to 4q D4Z4, also has variability in copy number, although contraction of 10q D4Z4 was not pathogenic when it was cis-linked to the prevalent typical 10q haplotypes [47]. Globally, population genetic studies have revealed that translocation events occur between 4q and 10q in non-FSHD and FSHD populations [80, 96,97,98,99]. The translocation of 4qA to 10q allows for a permissive allele on chromosome 10 and FSHD manifestation when the allele has a repeat contraction [30, 100]. The 10q-linked FSHD cases displaying a classical FSHD phenotype raise a question about the central pathological contribution of other 4 qter genes, such as WWC2, SORBS2, FAT1, and FRG1, as dysregulation of these genes did not appear to be consistently affected in those cells; however, upregulation of DUX4 and its direct target FRG2 was conserved. Although these cases may justify DUX4 and D4Z4 repeats as the only critical coding elements explaining FSHD manifestation, DUX4 could be activated by three-dimensional access of cis-active regulatory elements near D4Z4 on chromosome 10, which may again be muscle-specifically active, considering that enhancer-promoter communication can be triggered without proper chromatin regulation [101].
Duplication
In certain cases with clinical FSHD manifestations, cis D4Z4 array duplication or even triplication alleles with repeat contraction in either array and 4qA haplotype were found [80, 102,103,104]. Those alleles can cause FSHD onset both with or without variants of the FSHD2 genes. A typical Southern blot test may overlook those permissive alleles as the spacer sequence flanked by two D4Z4 arrays and repeat contraction on either array is not recognised, whereas the molecular combing assay and long-read sequencing assay can resolve this genetic rearrangement [80, 104]. These duplication cases also fit the model of the necessity of the permissive allele for FSHD onset, and DNA hypomethylation in the contracted array of the duplication alleles was also detected by nanopore analysis [104].
Although duplication cases can occur de novo in any of the permissive 4qA haplotypes, most cases were linked to 4qA-L, a European-specific 4qA class haplotype, indicating that these cases were derived from a limited number of ancestral origins in local regions. Interestingly, in one family, the proximal 17U array of the 17U + 2U duplication allele showed a methylation profile comparable to that of the distal 2U allele and lower than that of the 17U array of the 17U + 9U duplication allele [104], implying that DUX4 is transcribed from both the proximal and distal arrays or may even be predominantly from either array, which could be transcriptionally more actively regulated by distal elements as it is closer than the other. A detailed analysis of the duplicated alleles may be informative for understanding the mechanism of DUX4 activation, as chromatin conformation may also be affected, although hardly predicted, due to the potential insulator function of the D4Z4 sequence.
Gonosomal mosaicism
Mosaicism is often observed in FSHD, in which more than two chromosome 4 alleles and at least one permissive allele with a reduced proportion are found in a single proband [29, 30, 80, 105]. Rearrangements that produce repeat contractions causing FSHD1 can occur during early cell division, leading to gonosomal mosaicism, explaining approximately half of the de novo cases [30, 106]. In theory, although the rate of transmission of the FSHD1 allele from a parent with gonosomal mosaicism to the offspring is lower than that of a non-mosaic patient, the offspring will be more severely affected than the mosaic parent because all cells in the offspring contain the permissive allele. These mosaicism cases emphasise that permissive alleles are critical to FSHD manifestation; however, it is unclear whether the muscle cells with the permissive allele directly or indirectly affect the others without the permissive allele in the tissue environment, or whether the clinical impact of the permissive allele and DUX4 activation are limited locally within the positive cells and their niches. Mosaicism often leads to disease manifestation in males, but not so in females [29, 30], indicating sex-oriented regulatory mechanisms or disease susceptibility.
Given the standard classifications of FSHD1 and FSHD2, these complex genetically atypical cases can compromise result interpretation using the common diagnostic method of pulsed-field gel electrophoresis (PFGE) with Southern blotting. A recent consensus publication outlines the minimal requirements for genetic confirmation of both FSHD1 and FSHD2 [49]. The atypical rearrangements described above can be pathogenic per se (similar to FSHD1) or in combination with variants of genes causing FSHD2, which will be discussed in details next.
FSHD2 genes
Currently, variants that cause FSHD2 have been identified in SMCHD1, DNMT3B, and LRIF1. These factors are thought to regulate chromatin by promoting a closed state in the D4Z4 region. The pathogenic variants of SMCHD1 in FSHD2 are found over the whole coding region, the 3′UTR region and the introns of the gene, leading to haploinsufficiency or dominant negative effect [38, 39, 107, 108]. In addition, mutations in SMCHD1 cause the unrelated disorder Bosma arhinia microphthalmia syndrome (BAMS) [109], in which mutations in SMCHD1 are enriched exclusively in the extended ATPase domain, and DNA hypomethylation is often observed in D4Z4 [110]. Certain identical pathogenic variants of SMCHD1 are associated with these two seemingly unrelated disorders, suggesting that BAMS is likely caused by complex oligogenic or multifactorial mechanisms that only partially overlap at the SMCHD1 level, similar to FSHD2 [111]. Heterozygous mutations in DNMT3B (DNA methyltransferase 3 beta, MIM:602900) have been found in FSHD2 cases, depending on the PAS containing the 4qA allele and repeat number [40]. Biallelic DNMT3B mutations have been reported in autosomal recessive immunodeficiency, centromeric instability, and facial anomaly syndrome type 1 (ICF1 [OMIM: 242860]), in which DNMT3B activity is reduced, leading to DNA hypomethylation in a variety of repeat structures, including D4Z4 [40, 112,113,114]. The homozygous mutation in ligand-dependent nuclear receptor interacting factor 1 (LRIF1, also referred to as HBiX1, MIM:615354) was found in a Japanese FSHD2 case with 13 RUs in the permissive allele affecting only the longer isoform LRIF1L expression [41], whereas the heterozygous mutation may function as a disease modifier in the FSHD1 family. FSHD2 appears to be involved in the establishment of high DNA methylation in D4Z4 repeat. Interestingly, SMCHD1 knockout in FSHD myoblasts allowed DUX4 activation but did not recapitulate the DNA hypomethylation observed in the cells derived from patients with FSHD2, suggesting that SMCHD1 is not actively involved in the maintenance of DNA methylation in somatic cells [115]. This indicates that SMCHD1 plays a critical role in gene regulation and establishment of DNA methylation.
There still exist genetically undiagnosed cases of typical FSHD clinical manifestations without repeat contractions or mutations in known FSHD2 genes. Considering that all these known FSHD2 genes are related to X chromosome inactivation (Xi) in females [116, 117], the mechanism of chromatin regulation in both biological contexts likely partially overlaps, and other unknown FSHD2 genes might be found in Xi-associated factors because certain FSHD2 cases remain unsolved for conclusive causative variants in any gene. Clinical and molecular investigations of binding proteins on the D4Z4 repeat have provided a list of potential FSHD2 candidates. Of the variants in these genes, CTCF, DNMT1, DNMT3A, EZH2, and SUV39H1 have been reported to potentially contribute to FSHD pathology in clinical FSHD cohorts with permissive alleles [118], whereas further investigation is needed with functional analysis for confirmation as FSHD2 genes and/or disease modifiers of FSHD1. To date, the prevalence of FSHD2 variants in candidate genes, except SMCHD1 and DNMT3B, is extremely rare. This might be because the other candidate genes likely have more general functions in gene regulation broadly on the genome compared to SMCHD1 and DNMT3B, which have limited targets on the genome and/or in certain biological contexts, and may have minimal tissue-specific impacts on developmental progress when mutated.
DNA hypomethylation: a potential predictor of symptom outcome
Despite their distinct genetic backgrounds described above, significant DNA hypomethylation in the D4Z4 repeat of the permissive allele is common among FSHD1, FSHD2, and all atypical FSHD cases, indicating its potentially critical contribution to pathology. In FSHD1 cases, DNA hypomethylation occurs in the permissive allele but not in other alleles without repeat contraction, as the reduction in methylation is associated with repeat contraction. In contrast, in FSHD2, hypomethylation can occur in all D4Z4 repeats in both permissive and non-permissive alleles of chromosomes 4 and 10, as variants of chromatin regulators such as SMCHD1 potentially affect all arrays [37, 38]. Although numerous studies are available on the analysis of D4Z4 DNA methylation comparing FSHD cases, non-FSHD controls, and non-penetrant carriers with distinct methods, such as methylation-sensitive restriction enzyme-based, bisulphite conversion-based, and long-read sequencing, certain studies might apply protocols that may produce potentially significant bias in PCR amplicons targeting D4Z4 sites after bisulfite conversion, observing caution when interpreting the published results and selecting a proper method for analysis [119].
Jones et al. carefully compared two of the published protocols with identical genetically confirmed samples and concluded that their protocol successfully reproduced the expected detection of differences in D4Z4 methylation, where FSHD1, FSHD2, and non-FSHD cases could be distinguished, potentially offering a widely accessible diagnostic for FSHD using saliva DNA. This method consists of two distinct assays: the ‘BSX’ assay analyses the DR1 regions to represent the average level of DNA methylation in the whole D4Z4 units with certain preference to 4q D4Z4 due to the primers with mismatches to typical 10q D4Z4 and the ‘BSSA’ and ‘BSSL’ assays the distal most D4Z4 unit of 4qA and 4qA-L haplotype, respectively, to represent the methylation level of the distal unit of the permissive allele [119]. This method is compatible with high-throughput sequencing, as well as Sanger sequencing [120]. The combined outcomes of these assays distinguished between non-FSHD, FSHD1 and FSHD2, as only the BSSA/L assay revealed reduced methylation in FSHD1 samples, whereas both BSSA/L and BSSX assays demonstrated reduced methylation in FSHD2.
Recently, several independent studies have demonstrated that the methylation level of the most distal D4Z4 unit of the permissive allele is correlated with disease severity and progression, similar to or rather than the number of D4Z4 RUs of the permissive allele, motivating the use of this regional DNA methylation level as a predictor of disease progression [120, 121]. Another study reported that distal hypomethylation of the permissive allele was detected in a case of DPED, supporting its diagnostic application for differential diagnosis in patients with suspected FSHD, including complex structural variants [122].
Long-read sequencing methods have emerged as promising diagnostic tools as alternatives to bisulfite PCR-based methods. Several studies have shown that long-read sequencing can successfully identify the number of D4Z4 repeats, haplotypes and DNA methylation levels in each D4Z4 unit along the sequential order of all alleles without PCR amplification bias and DNA fragmentation [95, 123,124,125,126,127]. In this methodology, all alleles, even if one donor sample contains the same haplotypes, such as 4qA/4qA, potentially mixing contracted and non-contracted alleles in FSHD1, can be separately assembled and analysed for DNA methylation if reads that are long enough to cover the regions proximal and distal to the D4Z4 repeat in one read are obtained. In theory, this methodology appears tolerant to the atypical FSHD cases described above, which would otherwise require other types of more specialised methods, such as optical genome mapping and molecular combing for genotyping. However, using those methods, it might be difficult to separately analyse DNA methylation levels of more than one distal region of each D4Z4 array. Therefore, long-read sequencing can potentially offer an efficient, regular diagnostic method in the future if the run cost becomes more feasible to obtain statistically sufficient numbers of high-quality long reads for assembling each allele with a quantitative methylation level. Several published cases now apply Cas9-mediated enrichment of D4Z4-associated fragments, requiring additional procedures compared to the protocol for whole-genome sequencing [95, 123, 126], whereas whole-genome sequencing or other types of enrichment protocols may allow the detection of variants in FSHD2 genes and the feasible application of this technique with a few days of simple workflow.
Interestingly, long-read sequencing studies demonstrated that DNA methylation increased from the proximal to the distal unit within the D4Z4 repeat, suggesting a mechanistic dependency of distal D4Z4 repeat methylation on the number of D4Z4 RU [95, 123]. The age of onset correlated with both the D4Z4 RU number and methylation in FSHD1 [128]. Statistical modelling studies have demonstrated that shorter D4Z4 repeats are predictive of an earlier age at onset, although longer disease duration may better explain more severe disease progression than repeat number [28]. Recent clinical studies, some of which used the FSHD Comprehensive Clinical Evaluation Form (CCEF) for the systematic dissection of disease phenotype [129], still suggest significant clinical variation in FSHD1, especially in a longer range of contracted D4Z4 repeat [130,131,132,133]. This variation implies an unknown disease modifier of FSHD. On the other hand, medical comorbidities and medication use appear to be highly linked to potentially severe outcomes such as wheelchair dependence [28]. Double trouble, referring to a condition with other coexisting neuromuscular genetic diseases, may explain certain atypical cases [133]. These observations call for careful specification of the intrinsic phenotype of FSHD [133] and its secondary risk factors that accelerate disability. Evidence of anticipation, an intergenerational worsening of the disease, has been observed [134,135,136], which may be explained by parental mosaicism and/or intergenerational changes in distal D4Z4 methylation [127, 137].
DUX4 activation in muscles: leakage rather than active mode
Although D4Z4 hypomethylation has been observed in a variety of cells, such as blood cells, saliva, skin fibroblasts, pluripotent cells and myoblasts derived from patients with FSHD [37, 138], the robust DUX4 activation is typically only observed in skeletal muscle biopsies, cultured differentiated myoblasts and cultured lymphocyte lineages of patients with FSHD [58, 64, 139, 140], demonstrating tissue/cell type specificity in the activation mode. Moreover, DUX4 is not equally activated in a muscle cell population; however, it seems that DUX4 is expressed during a certain stage of muscle differentiation sporadically with a very low frequency (highly variable, more or less than ~1 in 1000), as revealed by immunostaining [58, 141,142,143] and the single cell/nucleus transcriptome [144,145,146,147]. Although DUX4 activation in somatic muscle cells is not necessarily limited to patients with FSHD, it sometimes occurs at a significantly lower frequency [141]. The correlation between local inflammatory MRI scores and DUX4 expression in FSHD muscle tissues [75, 148, 149] may indicate that disease progression depends on the sporadic probability of DUX4 activation per cell. The mathematical simulations demonstrate DUX4 drives significant cell death despite expression in only 0.8% of live cells [150]. Interestingly, the frequency of DUX4-positive cells in cell culture appears to be conserved per clone, independent of cell passage [53], indicating a deterministic regulatory mechanism underlying sporadicity. A reporter assay monitoring the DUX4 promoter showed sporadic activation, particularly for sense orientation [151]. Importantly, in this study, the lentivirus-mediated promoter fragment (several fragments appeared to be randomly integrated into the genome per cell) showed obvious clonal and cell-type differences in burst rate, emphasising the importance of cellular context and surrounding genomic landscape which can mediate enhancer-promoter communication, rather than direct activation of the promoter, and eventually determining transcriptional burst frequency. However, in the promoter, given the correlation between the degree of epigenetic D4Z4 de-repression and DUX4 expression in an in vitro test among familial cohorts [53] and between disease severity and distal D4Z4 hypomethylation [120, 122], the level of hypomethylation likely regulates disease progression by affecting DUX4 frequency, as the promoter of translatable DUX4 lies in the most distal D4Z4.
However, because methylation scores in bulk BSS analysis roughly and inversely represent the ratio of cells with hypomethylated permissive alleles, and therefore majority of FSHD muscle cells likely show significant DNA hypomethylation, there exists a clear discrepancy between the DUX4 frequency (less than 1%) and the ratio of hypomethylated alleles/cells (more than 50%). This indicates that D4Z4 DNA hypomethylation is not sufficient for myogenic activation of DUX4, which requires more factors specific to the cell type and stage in limited contexts of DUX4 activation. Several studies are available on potential context-dependent factors and pathways that directly activate DUX4 such as telomere shortening [152], PARP1 [153], herpesviruses infection [154, 155], oxidative stress [138], DNA damage [138, 156, 157], unstable G-quadruplexes [158], active chromatin regulators [159,160,161,162] and SIX transcription family [163]; and also suppress DUX4 such as Wnt/β-catenin signalling [164], β2-adrenergic receptor (β2AR) pathway via cyclic adenosine monophosphate (cAMP) dependent on p38 mitogen-activated protein kinases (MAPKs) and/or protein kinase A (PKA) [161, 165,166,167,168] and repressive chromatin regulators [159, 169,170,171,172]. Most studies have not evaluated whether the observed changes in DUX4 expression due to these stresses and by targeting the signals reflected the frequency of DUX4-positive cells in a pool or expression per cell. Nevertheless, for instance, the physiological level of oxidative stress increased DUX4 expression in FSHD muscle cells, which was correlated with frequency [138]. DUX4 activators can be theoretically categorised into two groups: the first is essential for the basal expression, and the second has only an additional impact above the basal level. The SIX transcription family is involved in skeletal muscle development, although it is not exclusively expressed in the muscle lineage [173]. The latter group is unlikely to be a major driver of myogenic DUX4 activation in FSHD but may explain variations in disease progression, such as left-right asymmetricity. If a strong transcriptional activator binds directly to the DUX4 promoter, it produces a positive state more frequently than the widely observed rare ratios. There may be distal regulatory machinery that is less efficient and allows the leakage of DUX4 activation. Thus, the cell-type specificity of DUX4 activation in FSHD is not well understood, which is highly linked to the question of why FSHD is a ‘muscle-restricted’ disease.
FSHD as a muscle disease
Based on clinical observations, FSHD affects a limited number of tissues, primarily the skeletal muscles. The concept of DUX4 in pathology theoretically suggests that disease conditions and tissue specificity can be determined by DUX4 activation, represented by the level/frequency of DUX4 expression and DUX4 biological effects, including toxicity through transcriptional activity and myogenesis perturbation by DUX4 binding [174] (Fig. 3). Based on this, the question of why the FSHD genetic background ‘does not’ affect most other organs can be answered by the absence or negligible levels of DUX4 activation and/or DUX4 biological effects.

Model of DUX4 expression and downstream responses leading to FSHD onset and progression. The expression of DUX4 requires contraction of the D4Z4 repeat array, subsequent DNA hypomethylation and the presence of a permissive 4qA haplotype. Additionally, mutations in chromatin modifiers known to cause FSHD2, along with other intracellular signalling pathways or external stress factors, may contribute to a permissive epigenetic environment for DUX4 expression in the pre-DUX4 phase. Sporadic DUX4 expression in muscle tissue induces FSHD-specific pathological phenotypes downstream. On top of DUX4 intrinsic cytotoxicity, the cascade of various molecular events it triggers contributes to irreversible pathological changes in muscle tissues in the post-DUX4 phase. Moreover, the progression of pathology may be influenced not only by intracellular responses, but also by environmental factors through daily behaviours, which may either exacerbate or mitigate disease severity. Intrinsic genetic factors that influence disease severity at various points along these pathways act as disease modifiers in FSHD, potentially including genes and regulatory elements
The whole atlas of somatic DUX4 expression patterns in FSHD is currently not available; however, most cells in most organs in FSHD appear negative; however, certain skeletal muscle lineages and peripheral blood mononuclear cells, especially lymphocytes are positive [175], suggesting cell type-specific regulation. Extramuscular symptoms provoke questions regarding the possibility of any other somatic cell type expressing DUX4. Interestingly, a recent mouse study with muscle-specific DUX4 overexpression demonstrated a retinal abnormality resembling that of FSHD, implying a muscular phenotype as the origin and interorgan mechanism [70]. Hearing loss is bilateral [9], in contrast to the widely observed asymmetry in the muscle phenotype. The cell types directly affected by these extramuscular symptoms require further investigation to determine the possibility of DUX4 expression in these cells or the effects of secreted/systemic factors derived from the affected muscles. Moreover, skeletal muscle fibres are among the few cell types with multinucleate potential. This multinucleate state, resulting from the fusion of mononucleate myocytes that share the cytoplasm, allows DUX4 to diffuse into other nuclei, causing the entire cell to undergo deterioration [176]. This characteristic may explain why skeletal muscles are uniquely vulnerable to DUX4 toxicity compared with other organs composed of mononuclear cells.
Heterogeneity in intra-individual muscles
Certain muscles are affected earlier and others are affected later in FSHD, which was recently elegantly captured using whole-body MRI scans for regional fat infiltration [177]. However, mechanisms underlying this heterogeneity remain largely unknown. The facial muscles are among the earliest regions affected by typical clinical FSHD. Facial and trunk muscles differ in their developmental origin during mesodermal specification [178]. Differences in the characteristics of muscle stem cells with different developmental origins [179] may explain the regional preference for active disease. DUX4 leaves muscle cells in a biologically vulnerable state to metabolic stress, such as oxidative stress [180,181,182,183] and abnormal iron accumulation [70]. Hyaluronic acid pathway and autophagy pathways are involved in DUX4 toxicity and prevention, respectively [184, 185]. The intrinsic susceptibility to such stresses and differences in the metabolic capacity of each pathway may explain the regional preference of affected muscles and phenotypic differences in individuals, implying potential protective disease modifiers and lifestyle-linked environmental factors behind these pathways (Fig. 3). Regarding sex bias, controversial insights into sex hormones as protective factors were made from experimental and clinical studies [186,187,188,189,190], necessitating further clinical observations with consideration of the relevant temporal functions of sex hormones on skeletal muscles, including the establishment of a stem cell pool [191]. This potential protective effect of female hormones and melatonin may be mediated by an increase in miR-675, followed by both the inhibition of DUX4 toxicity and reduction in DUX4 expression [192]. Sexual dimorphisms in mitochondrial protein adaptation to exercise may also contribute to potential sex differences as well [193]. Muscle fibre types may contribute to regional heterogeneity because a reduction in sarcomeric force in type II but not type I muscle fibres was observed in FSHD [194], with potentially higher susceptibility to DUX4 stress or frequency of DUX4 activation in type II fibres. The topography of muscle abnormalities caused by Fat1 loss-of-function in mice resembled that of patients with FSHD, and thus, lower FAT1 expression in regional muscles that are affected at early stages of FSHD progression was proposed to be relevant to heterogeneity [195, 196].
The involvement of the perturbed immune system and inflammation in the pathology of the FSHD muscle has been pronounced. Immune cell infiltration [149, 197, 198] and elevated complement levels in the plasma and biopsies of FSHD subjects [149, 199]. Ectopic DUX4 expression in muscle cells activates immune mediators [63] and suppresses MHC Class I expression [200]. Muscle-specific chronic DUX4 expression in mice resembles the pathological features of FSHD muscles, including inflammation and fibrosis [201]. Bilateral comparison revealed that, in contrast to the often-observed intra-individual left–right asymmetric symptomatic manifestations captured by histopathological features in muscle biopsy, whole-muscle fat infiltration, proportion of immune cell populations, and D4Z4 DNA methylation were likely symmetric [149], suggesting a systemic mechanism of disease progression that might be relevant to extra-muscular symptoms through circulation.
Therapeutic development
Based on the current understanding of DUX4 central role, various therapeutic approaches have been demonstrated in preclinical studies, with some progressing to clinical trials. In alignment with the central dogma, these include: small molecules to suppress DUX4 transcription such as a p38 inhibitor, which was tested in recent clinical trials but showed insufficient efficacy [166,167,168] (refer to NCT05397470 at ClinicalTrials.gov); epigenetic editing strategies to silence transcription by modifying the gene to a repressive state using inactive CRISPR (clustered regularly interspaced short palindromic repeat)-based targeting approaches [202,203,204], with one approach planned for clinical trials (refer to NCT06907875 at ClinicalTrials.gov); genetic editing strategies targeting the DUX4 locus, including removal of the PAS sequence to reduce DUX4 mRNA; antisense oligonucleotides to degrade DUX4 mRNA, for which several clinical trials are ongoing or planned [79] (refer to NCT07038200 and NCT06131983 at ClinicalTrials.gov). Apart from DUX4 targeting strategies, clinical trials are ongoing for an antibody against myostatin, an Interleukin-6 receptor antagonist, and an adrenergic β2 receptor agonist, which aim to achieve efficacy by blocking the suppressive effect of myostatin on muscle volume and by mitigating inflammation and potentially curtailing fibrofatty degeneration in FSHD, respectively (refer to NCT05548556, NCT06222827, and NCT06721299 at ClinicalTrials.gov). In summary, while no established treatment currently exists, numerous therapeutic approaches are in various stages of development for FSHD.
Conclusion
Research has revealed the essential mechanisms underlying FSHD onset, thereby accelerating the development of targeted treatments focused on DUX4 pathology. However, the diverse clinical presentations among individuals with FSHD, stemming from specific genetic conditions that lead to different outcomes, call for deeper investigations of this disease from a broader perspective. In this sense, it is essential to uncover the mechanism of sporadic DUX4 activation and the epigenetic regulation of the D4Z4 repeat, providing insights into potential factors that influence disease outcome through gene regulation. In addition, understanding the extent to which genetic factors can or cannot explain the clinical diversity is critical. This is particularly important when considering intrafamilial clinical differences and the observed variations across sex and race. This knowledge will support not only the development of better drug targets and therapeutic options but also improve individual care management on a daily basis. It may reveal strategies to delay disease progression without costly medical interventions and help foster an inclusive society for all individuals with genetically confirmed FSHD. Although beyond the scope of this review, the effects of daily behaviours, such as exercise, nutrition, and mental state on disease progression, are also important areas for exploration. The combination of cutting-edge scientific approaches and insights from the lived experiences of individuals with FSHD such as patient-reported outcomes with large cohorts will promote a harmonised understanding and comprehensive solutions spanning genetics and epigenetics to patient experience.
References
Wang LH, Tawil R. Facioscapulohumeral dystrophy. Curr Neurol Neurosci Rep. 2016;16:66.
Mul K, Lassche S, Voermans NC, Padberg GW, Horlings CG, van Engelen BG. What’s in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pr Neurol. 2016;16:201–7.
Santos DB, Boussaid G, Stojkovic T, Orlikowski D, Letilly N, Behin A, et al. Respiratory muscle dysfunction in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2015;25:632–9.
Henke C, Spiesshoefer J, Kabitz H-J, Herkenrath S, Randerath W, Brix T, et al. Respiratory muscle weakness in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2019;60:679–86.
Morís G, Wood L, FernáNdez-Torrón R, González Coraspe JA, Turner C, Hilton-Jones D, et al. Chronic pain has a strong impact on quality of life in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2018;57:380–7.
Brouwer OF, Padberg GW, Wijmenga C, Frants RR. Facioscapulohumeral muscular dystrophy in early childhood. Arch Neurol. 1994;51:387–94.
Klinge L, Eagle M, Haggerty ID, Roberts CE, Straub V, Bushby KM. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2006;16:553–8.
Brouwer OF, Padberg GW, Ruys CJ, Brand R, de LaatJA, Grote JJ. Hearing loss in facioscapulohumeral muscular dystrophy. Neurology. 1991;41:1878–81.
Lutz KL, Holte L, Kliethermes SA, Stephan C, Mathews KD. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology. 2013;81:1374–7.
Fitzsimons RB, Gurwin EB, Bird AC. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain. 1987;110:631–48.
Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology. 2013;80:1247–50.
Goselink RJM, Schreur V, van Kernebeek CR, Padberg GW, van der Maarel SM, van Engelen BGM, et al. Ophthalmological findings in facioscapulohumeral dystrophy. Brain Commun. 2019;1:fcz023.
Goselink RJM, Mul K, van Kernebeek CR, Lemmers RJLF, van der Maarel SM, Schreuder THA, et al. Early onset as a marker for disease severity in facioscapulohumeral muscular dystrophy. Neurology. 2019;92:e378–85.
Funakoshi M, Goto K, Arahata K. Epilepsy and mental retardation in a subset of early onset 4q35-facioscapulohumeral muscular dystrophy. Neurology. 1998;50:1791–4.
Laforêt P, de Toma C, Eymard B, Becane HM, Jeanpierre M, Fardeau M, et al. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology. 1998;51:1454–6.
Chen T-H, Wu Y-Z, Tseng Y-H. Early-onset infantile facioscapulohumeral muscular dystrophy: A timely review. Int J Mol Sci. 2020;21:7783.
Chen T-H, Lai Y-H, Lee P-L, Hsu J-H, Goto K, Hayashi YK, et al. Infantile facioscapulohumeral muscular dystrophy revisited: expansion of clinical phenotypes in patients with a very short EcoRI fragment. Neuromuscul Disord. 2013;23:298–305.
Goselink RJM, Voermans NC, Okkersen K, Brouwer OF, Padberg GW, Nikolic A, et al. Early onset facioscapulohumeral dystrophy - a systematic review using individual patient data. Neuromuscul Disord. 2017;27:1077–83.
Deenen JCW, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJGM, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–9.
Wohlgemuth M, Lemmers RJ, Jonker M, van der Kooi E, Horlings CG, van Engelen BG, et al. A family-based study into penetrance in facioscapulohumeral muscular dystrophy type 1. Neurology. 2018;91:e444–54.
Wang Z, Qiu L, Lin M, Chen L, Zheng F, Lin L, et al. Prevalence and disease progression of genetically-confirmed facioscapulohumeral muscular dystrophy type 1 (FSHD1) in China between 2001 and 2020: a nationwide population-based study. Lancet Reg Health West Pac. 2022;18:100323.
Matsumura T, Hashimoto H, Takizawa H, Yoshioka W, Mori-Yoshimura M, Saito Y, et al. Clinical and genetic characteristics based on the Japanese patient registry for facioscapulohumeral muscular dystrophy: a nationwide analysis. Neuromuscul Disord. 2025;50:105346.
Zatz M, Marie SK, Cerqueira A, Vainzof M, Pavanello RC, Passos-Bueno MR. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am J Med Genet. 1998;77:155–61.
Tonini MMO, Passos-Bueno MR, Cerqueira A, Matioli SR, Pavanello R, Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. 2004;14:33–8.
Ricci G, Scionti I, Sera F, Govi M, D’Amico R, Frambolli I, et al. Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain. 2013;136:3408–17.
Lin F, Wang Z-Q, Lin M-T, Murong S-X, Wang N. New insights into genotype-phenotype correlations in Chinese facioscapulohumeral muscular dystrophy: A retrospective analysis of 178 patients: A retrospective analysis of 178 patients. Chin Med J (Engl). 2015;128:1707–13.
Park HJ, Hong J-M, Lee JH, Lee HS, Shin HY, Kim SM, et al. Low D4Z4 copy number and gender difference in Korean patients with facioscapulohumeral muscular dystrophy type 1. Neuromuscul Disord. 2015;25:859–64.
Katz NK, Hogan J, Delbango R, Cernik C, Tawil R, Statland JM. Predictors of functional outcomes in patients with facioscapulohumeral muscular dystrophy. Brain. 2021;144:3451–60.
van der Maarel SM, Deidda G, Lemmers RJ, van Overveld PG, van der Wielen M, Hewitt JE, et al. De novo facioscapulohumeral muscular dystrophy: frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am J Hum Genet. 2000;66:26–35.
Strafella C, Colantoni L, Megalizzi D, Trastulli G, Piorgo EP, Primiano G, et al. Characterization of D4Z4 alleles and assessment of de novo cases in Facioscapulohumeral dystrophy (FSHD) in a cohort of Italian families. Clin Genet. 2024;105:335–9.
Lemmers RJLF, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3.
van Deutekom JC, Wijmenga C, van Tienhoven EA, Gruter AM, Hewitt JE, Padberg GW, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2:2037–42.
van Overveld PGM, Lemmers RJFL, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–7.
de Greef JC, Lemmers RJLF, van Engelen BGM, Sacconi S, Venance SL, Frants RR, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–59.
Hartweck LM, Anderson LJ, Lemmers RJ, Dandapat A, Toso EA, Dalton JC, et al. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology. 2013;80:392–9.
Gaillard M-C, Roche S, Dion C, Tasmadjian A, Bouget G, Salort-Campana E, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology. 2014;83:733–42.
Jones TI, Yan C, Sapp PC, McKenna-Yasek D, Kang PB, Quinn C, et al. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin Epigenetics. 2014;6:23.
Lemmers RJLF, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24:659–69.
Lemmers RJLF, Tawil R, Petek LM, Balog J, Block GJ, Santen GWE, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4.
van den Boogaard ML, Lemmers RJLF, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet. 2016;98:1020–9.
Hamanaka K, Šikrová D, Mitsuhashi S, Masuda H, Sekiguchi Y, Sugiyama A, et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology. 2020;94:e2441–7.
Stence AA, Thomason JG, Pruessner JA, Sompallae RR, Snow AN, Ma D, et al. Validation of optical genome mapping for the molecular diagnosis of facioscapulohumeral muscular dystrophy. J Mol Diagn. 2021;23:1506–14.
de Greef JC, Lemmers RJLF, Camaño P, Day JW, Sacconi S, Dunand M, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–54.
Sacconi S, Lemmers RJLF, Balog J, van der Vliet PJ, Lahaut P, van Nieuwenhuizen MP, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93:744–51.
Larsen M, Rost S, El Hajj N, Ferbert A, Deschauer M, Walter MC, et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet. 2015;23:808–16.
Sacconi S, Briand-Suleau A, Gros M, Baudoin C, Lemmers RJLF, Rondeau S, et al. FSHD1 and FSHD2 form a disease continuum. Neurology. 2019;92:e2273–85.
Lemmers RJLF, Wohlgemuth M, van der Gaag KJ, van der Vliet PJ, van Teijlingen CMM, de Knijff P, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–94.
Statland JM, Donlin-Smith CM, Tapscott SJ, Lemmers RJLF, van der Maarel SM, Tawil R. Milder phenotype in facioscapulohumeral dystrophy with 7-10 residual D4Z4 repeats. Neurology. 2015;85:2147–50.
Giardina E, Camaño P, Burton-Jones S, Ravenscroft G, Henning F, Magdinier F, et al. Best practice guidelines on genetic diagnostics of facioscapulohumeral muscular dystrophy: Update of the 2012 guidelines. Clin Genet. 2024;106:13–26.
Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet. 1995;4:951–8.
Nikolic A, Ricci G, Sera F, Bucci E, Govi M, Mele F, et al. Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1-3 D4Z4 reduced alleles: experience of the FSHD Italian National Registry. BMJ Open. 2016;6:e007798.
Tawil R, Storvick D, Feasby TE, Weiffenbach B, Griggs RC. Extreme variability of expression in monozygotic twins with FSH muscular dystrophy. Neurology. 1993;43:345–8.
Jones TI, King OD, Himeda CL, Homma S, Chen JCJ, Beermann ML, et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin Epigenetics. 2015;7:37.
Gabriëls J, Beckers MC, Ding H, De Vriese A, Plaisance S, van der Maarel SM, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32.
Hendrickson PG, Doráis JA, Grow EJ, Whiddon JL, Lim J-W, Wike CL, et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet. 2017;49:925–34.
De Iaco A, Planet E, Coluccio A, Verp S, Duc J, Trono D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet. 2017;49:941–5.
Whiddon JL, Langford AT, Wong C-J, Zhong JW, Tapscott SJ. Conservation and innovation in the DUX4-family gene network. Nat Genet. 2017;49:935–40.
Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181.
Das S, Chadwick BP. Influence of repressive histone and DNA methylation upon D4Z4 transcription in non-myogenic cells. PLoS One. 2016;11:e0160022.
Lemmers RJLF, van der Vliet PJ, van der Gaag KJ, Zuniga S, Frants RR, de Knijff P, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet. 2010;86:364–77.
Lemmers RJFL, Wohlgemuth M, Frants RR, Padberg GW, Morava E, van der Maarel SM. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2004;75:1124–30.
Peart N, Wagner EJ. A distal auxiliary element facilitates cleavage and polyadenylation of Dux4 mRNA in the pathogenic haplotype of FSHD. Hum Genet. 2017;136:1291–301.
Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38–51.
Yao Z, Snider L, Balog J, Lemmers RJLF, Van Der Maarel SM, Tawil R, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. 2014;23:5342–52.
Bosnakovski D, Choi SH, Strasser JM, Toso EA, Walters MA, Kyba M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet Muscle. 2014;4:4.
Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, et al. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife [Internet]. 2015 Jan;4. Available from: https://doi.org/10.7554/eLife.04996.
Rickard AM, Petek LM, Miller DG. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet. 2015;24:5901–14.
Banerji CRS, Panamarova M, Hebaishi H, White RB, Relaix F, Severini S, et al. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat Commun. 2017;8:2152.
Karpukhina A, Galkin I, Ma Y, Dib C, Zinovkin R, Pletjushkina O, et al. Analysis of genes regulated by DUX4 via oxidative stress reveals potential therapeutic targets for treatment of facioscapulohumeral dystrophy. Redox Biol. 2021;43:102008.
Nakamura K, Ortuste-Quiroga H-P, Horii N, Fujimaki S, Moroishi T, Nakayama KI, et al. Iron supplementation alleviates pathologies in a mouse model of facioscapulohumeral muscular dystrophy. J Clin Invest [Internet]. 2025 July [cited 2025 July 2]; Available from: https://doi.org/10.1172/JCI181881.
DeSimone AM, Cohen J, Lek M, Lek A. Cellular and animal models for facioscapulohumeral muscular dystrophy. Dis Model Mech. 2020;13:dmm046904.
Yoshizawa T, Sasaki-Honda M, Sakurai H, Kosho T. Model animals and attempts to develop therapeutic drugs for facioscapulohumeral muscular dystrophy. Transl Regulatory Sci. 2025;7:15–25.
Greco A, Goossens R, van Engelen B, van der Maarel SM. Consequences of epigenetic derepression in facioscapulohumeral muscular dystrophy. Clin Genet. 2020;97:799–814.
Schätzl T, Kaiser L, Deigner H-P. Facioscapulohumeral muscular dystrophy: genetics, gene activation and downstream signalling with regard to recent therapeutic approaches: an update. Orphanet J Rare Dis. 2021;16:129.
Wang LH, Friedman SD, Shaw D, Snider L, Wong C-J, Budech CB, et al. MRI-informed muscle biopsies correlate MRI with pathology and DUX4 target gene expression in FSHD. Hum Mol Genet. 2019;28:476–86.
Banerji CRS, Zammit PS. PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single-cell level. Hum Mol Genet. 2019;28:2224–36.
Ferreboeuf M, Mariot V, Bessières B, Vasiljevic A, Attié-Bitach T, Collardeau S, et al. DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum Mol Genet. 2014;23:171–81.
Banerji CRS, Zammit PS. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: roles of DUX4 and PAX7. EMBO Mol Med. 2021;13:e13695.
Beck SL, Yokota T. Oligonucleotide therapies for facioscapulohumeral muscular dystrophy: Current preclinical landscape. Int J Mol Sci. 2024;25:9065.
Delourme M, Charlene C, Gerard L, Ganne B, Perrin P, Vovan C, et al. Complex 4q35 and 10q26 rearrangements: A challenge for molecular diagnosis of patients with facioscapulohumeral dystrophy. Neurol Genet. 2023;9:e200076.
Scionti I, Greco F, Ricci G, Govi M, Arashiro P, Vercelli L, et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am J Hum Genet. 2012;90:628–35.
Joubert R, Mariot V, Charpentier M, Concordet JP, Dumonceaux J. Gene editing targeting the DUX4 polyadenylation signal: a therapy for FSHD?. J Pers Med. 2020;11:7.
Šikrová D, Cadar VA, Ariyurek Y, Laros JFJ, Balog J, van der Maarel SM. Adenine base editing of the DUX4 polyadenylation signal for targeted genetic therapy in facioscapulohumeral muscular dystrophy. Mol Ther Nucleic Acids. 2021;25:342–54.
Mariot V, Dumonceaux J. Gene editing to tackle facioscapulohumeral muscular dystrophy. Front Genome Ed. 2022;4:937879.
Deak KL, Lemmers RJLF, Stajich JM, Klooster R, Tawil R, Frants RR, et al. Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E-11 and D4Z4. Neurology. 2007;68:578–82.
Nguyen K, Broucqsault N, Chaix C, Roche S, Robin JD, Vovan C, et al. Deciphering the complexity of the 4q and 10q subtelomeres by molecular combing in healthy individuals and patients with facioscapulohumeral dystrophy. J Med Genet. 2019;56:590–601.
Lemmers RJLF, van der Vliet PJ, Granado DSL, van der Stoep N, Buermans H, van Schendel R, et al. High-resolution breakpoint junction mapping of proximally extended D4Z4 deletions in FSHD1 reveals evidence for a founder effect. Hum Mol Genet. 2022;31:748–60.
Himeda CL, Debarnot C, Homma S, Beermann ML, Miller JB, Jones PL, et al. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol Cell Biol. 2014;34:1942–55.
Pirozhkova I, Petrov A, Dmitriev P, Laoudj D, Lipinski M, Vassetzky Y. A functional role for 4qA/B in the structural rearrangement of the 4q35 region and in the regulation of FRG1 and ANT1 in facioscapulohumeral dystrophy. PLoS One. 2008;3:e3389.
Bodega B, Ramirez GDC, Grasser F, Cheli S, Brunelli S, Mora M, et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009;7:41.
Robin JD, Ludlow AT, Batten K, Gaillard M-C, Stadler G, Magdinier F, et al. SORBS2 transcription is activated by telomere position effect-over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy. Genome Res. 2015;25:1781–90.
Cortesi A, Pesant M, Sinha S, Marasca F, Sala E, Gregoretti F, et al. 4q-D4Z4 chromatin architecture regulates the transcription of muscle atrophic genes in facioscapulohumeral muscular dystrophy. Genome Res. 2019;29:883–95.
Gaillard M-C, Broucqsault N, Morere J, Laberthonnière C, Dion C, Badja C, et al. Analysis of the 4q35 chromatin organization reveals distinct long-range interactions in patients affected with Facio-Scapulo-Humeral Dystrophy. Sci Rep. 2019;9:10327.
Xu X, Tsumagari K, Sowden J, Tawil R, Boyle AP, Song L, et al. DNaseI hypersensitivity at gene-poor, FSH dystrophy-linked 4q35.2. Nucleic Acids Res. 2009;37:7381–93.
Butterfield RJ, Dunn DM, Duval B, Moldt S, Weiss RB. Deciphering D4Z4 CpG methylation gradients in fascioscapulohumeral muscular dystrophy using nanopore sequencing [Internet]. bioRxivorg. 2023. Available from: http://biorxiv.org/lookup/doi/10.1101/2023.02.17.528868.
van Deutekom JC, Bakker E, Lemmers RJ, van der Wielen MJ, Bik E, Hofker MH, et al. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum Mol Genet. 1996;5:1997–2003.
van Overveld PG, Lemmers RJ, Deidda G, Sandkuijl L, Padberg GW, Frants RR, et al. Interchromosomal repeat array interactions between chromosomes 4 and 10: a model for subtelomeric plasticity. Hum Mol Genet. 2000;9:2879–84.
Matsumura T, Goto K, Yamanaka G, Lee JH, Zhang C, Hayashi YK, et al. Chromosome 4q;10q translocations; comparison with different ethnic populations and FSHD patients. BMC Neurol. 2002;2:7.
Wu Z-Y, Wang Z-Q, Murong S-X, Wang N. FSHD in Chinese population: characteristics of translocation and genotype-phenotype correlation. Neurology. 2004;63:581–3.
Lemmers RJLF, van der Vliet PJ, Blatnik A, Balog J, Zidar J, Henderson D, et al. Chromosome 10q-linked FSHD identifies DUX4 as principal disease gene. J Med Genet. 2022;59:180–8.
Ealo T, Sanchez-Gaya V, Respuela P, Muñoz-San Martín M, Martin-Batista E, Haro E, et al. Cooperative insulation of regulatory domains by CTCF-dependent physical insulation and promoter competition. Nat Commun. 2024;15:7258.
Nguyen K, Puppo F, Roche S, Gaillard M-C, Chaix C, Lagarde A, et al. Molecular combing reveals complex 4q35 rearrangements in Facioscapulohumeral dystrophy. Hum Mutat. 2017;38:1432–41.
Lemmers RJLF, van der Vliet PJ, Vreijling JP, Henderson D, van der Stoep N, Voermans N, et al. Cis D4Z4 repeat duplications associated with facioscapulohumeral muscular dystrophy type 2. Hum Mol Genet. 2018;27:3488–97.
Lemmers RJLF, Butterfield R, van der Vliet PJ, de Bleecker JL, van der Pol L, Dunn DM, et al. Autosomal dominant in cis D4Z4 repeat array duplication alleles in facioscapulohumeral dystrophy. Brain. 2024;147:414–26.
Qiu L, Ye Z, Lin L, Wang L, Lin X, He J, et al. Clinical and genetic features of somatic mosaicism in facioscapulohumeral dystrophy. J Med Genet. 2020;57:777–85.
Lemmers RJLF, Van Overveld PGM, Sandkuijl LA, Vrieling H, Padberg GW, Frants RR, et al. Mechanism and timing of mitotic rearrangements in the subtelomeric D4Z4 repeat involved in facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2004;75:44–53.
Goossens R, van den Boogaard ML, Lemmers RJLF, Balog J, van der Vliet PJ, Willemsen IM, et al. Intronic SMCHD1 variants in FSHD: testing the potential for CRISPR-Cas9 genome editing. J Med Genet. 2019;56:828–37.
Strafella C, Caputo V, Galota RM, Campoli G, Bax C, Colantoni L, et al. The variability of SMCHD1 gene in FSHD patients: evidence of new mutations. Hum Mol Genet. 2019;28:3912–20.
Gordon CT, Xue S, Yigit G, Filali H, Chen K, Rosin N, et al. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat Genet. 2017;49:249–55.
Lemmers, van der Stoep RJLF, Vliet N, van der PJ, Moore SA, San Leon Granado D, et al. SMCHD1 mutation spectrum for facioscapulohumeral muscular dystrophy type 2 (FSHD2) and Bosma arhinia microphthalmia syndrome (BAMS) reveals disease-specific localisation of variants in the ATPase domain. J Med Genet. 2019;56:693–700.
Mul K, Lemmers RJLF, Kriek M, van der Vliet PJ, van den Boogaard ML, Badrising UA, et al. FSHD type 2 and Bosma arhinia microphthalmia syndrome: Two faces of the same mutation. Neurology. 2018;91:e562–70.
Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–91.
Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14412–7.
Kondo T, Bobek MP, Kuick R, Lamb B, Zhu X, Narayan A, et al. Whole-genome methylation scan in ICF syndrome: hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum Mol Genet. 2000;9:597–604.
Kong X, Nguyen NV, Li Y, Sakr JS, Williams K, Sharifi S, et al. Engineered FSHD mutations results in D4Z4 heterochromatin disruption and feedforward DUX4 network activation. iScience. 2024;27:109357.
Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607.
Nozawa R-S, Nagao K, Igami K-T, Shibata S, Shirai N, Nozaki N, et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat Struct Mol Biol. 2013;20:566–73.
Strafella C, Caputo V, Bortolani S, Torchia E, Megalizzi D, Trastulli G, et al. Whole exome sequencing highlights rare variants in CTCF, DNMT1, DNMT3A, EZH2 and SUV39H1 as associated with FSHD. Front Genet. 2023;14:1235589.
Gould T, Jones TI, Jones PL. Precise epigenetic analysis using targeted bisulfite genomic sequencing distinguishes FSHD1, FSHD2, and healthy subjects. Diagnostics (Basel). 2021;11:1469.
Erdmann H, Scharf F, Gehling S, Benet-Pagès A, Jakubiczka S, Becker K, et al. Methylation of the 4q35 D4Z4 repeat defines disease status in facioscapulohumeral muscular dystrophy. Brain. 2023;146:1388–402.
Zheng F, Qiu L, Chen L, Zheng Y, Lin X, He J, et al. Association of 4qA-specific distal D4Z4 hypomethylation with disease severity and progression in facioscapulohumeral muscular dystrophy. Neurology. 2023;101:e225–37.
Xia X, Cheng N, Liu Y, Yue D, Gao M, Hu C, et al. 4qA D4Z4 methylation test as a valuable complement for differential diagnosis in patients with a facioscapulohumeral muscular dystrophy-like phenotype. J Mol Diagn. 2025;27:405–18.
Hiramuki Y, Kure Y, Saito Y, Ogawa M, Ishikawa K, Mori-Yoshimura M, et al. Simultaneous measurement of the size and methylation of chromosome 4qA-D4Z4 repeats in facioscapulohumeral muscular dystrophy by long-read sequencing. J Transl Med. 2022;20:517.
Yeetong P, Kulsirichawaroj P, Kumutpongpanich T, Srichomthong C, Od-Ek P, Rakwongkhachon S, et al. Long-read Nanopore sequencing identified D4Z4 contractions in patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2023;33:551–6.
Huang M, Zhang Q, Jiao J, Shi J, Xu Y, Zhang C, et al. Comprehensive genetic analysis of facioscapulohumeral muscular dystrophy by Nanopore long-read whole-genome sequencing. J Transl Med. 2024;22:451.
Xiao LC, Semwal A, St John B, Zeglinski K, Su S, Lancaster J, et al. D4Z4End2End: complete genetic and epigenetic architecture of D4Z4 macrosatellites in FSHD, BAMS and reference cohorts [Internet]. medRxiv. 2025. p. 2025.04.24.25326320. Available from: https://doi.org/10.1101/2025.04.24.25326320v1.
Erdmann H, Scharf F, Hallermayr A, Barseghyan H, Walter MC, Holinski-Feder E, et al. Reply: An epigenetic basis for genetic anticipation in facioscapulohumeral muscular dystrophy type 1. Brain. 2023;146:e111–4.
Zheng F, Lin Y, Qiu L, Zheng Y, Zeng M, Lin X, et al. Age at onset mediates genetic impact on disease severity in facioscapulohumeral muscular dystrophy. Brain. 2025;148:613–25.
Ricci G, Ruggiero L, Vercelli L, Sera F, Nikolic A, Govi M, et al. A novel clinical tool to classify facioscapulohumeral muscular dystrophy phenotypes. J Neurol. 2016;263:1204–14.
Ricci G, Cammish P, Siciliano G, Tupler R, Lochmuller H, Evangelista T. Phenotype may predict the clinical course of facioscapolohumeral muscular dystrophy. Muscle Nerve. 2019;59:711–3.
Banerji CRS, Cammish P, Evangelista T, Zammit PS, Straub V, Marini-Bettolo C. Facioscapulohumeral muscular dystrophy 1 patients participating in the UK FSHD registry can be subdivided into 4 patterns of self-reported symptoms. Neuromuscul Disord. 2020;30:315–28.
Ruggiero L, Mele F, Manganelli F, Bruzzese D, Ricci G, Vercelli L, et al. Phenotypic variability among patients with D4Z4 reduced allele facioscapulohumeral muscular dystrophy. JAMA Netw Open. 2020;3:e204040.
Puma A, Tammam G, Ezaru A, Slioui A, Torchia E, Tasca G, et al. Double trouble: a comprehensive study into unrelated genetic comorbidities in adult patients with Facioscapulohumeral Muscular Dystrophy Type I. Eur J Hum Genet. 2025; 1–9.
Goto K, Lee JH, Matsuda C, Hirabayashi K, Kojo T, Nakamura A, et al. DNA rearrangements in Japanese facioscapulohumeral muscular dystrophy patients: clinical correlations. Neuromuscul Disord. 1995;5:201–8.
Tawil R, Forrester J, Griggs RC, Mendell J, Kissel J, McDermott M, et al. Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. FSH-DY Group Ann Neurol. 1996;39:744–8.
Alavi A, Esmaeili S, Nafissi S, Kahrizi K, Najmabadi H. Genotype and phenotype analysis of 43 Iranian facioscapulohumeral muscular dystrophy patients; Evidence for anticipation. Neuromuscul Disord. 2018;28:303–14.
Zheng F, Qiu L, Chen L, Zheng Y, He Q, Lin X, et al. An epigenetic basis for genetic anticipation in facioscapulohumeral muscular dystrophy type 1. Brain. 2023;146:e107–10.
Sasaki-Honda M, Jonouchi T, Arai M, Hotta A, Mitsuhashi S, Nishino I, et al. A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum Mol Genet. 2018;27:4024–35.
Jones TI, Himeda CL, Perez DP, Jones PL. Large family cohorts of lymphoblastoid cells provide a new cellular model for investigating facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2017;27:221–38.
Banerji CRS, Panamarova M, Zammit PS. DUX4 expressing immortalized FSHD lymphoblastoid cells express genes elevated in FSHD muscle biopsies, correlating with the early stages of inflammation. Hum Mol Genet. 2020;29:2285–99.
Jones TI, Chen JCJ, Rahimov F, Homma S, Arashiro P, Beermann ML, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2012;21:4419–30.
Tassin A, Laoudj-Chenivesse D, Vanderplanck C, Barro M, Charron S, Ansseau E, et al. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy?. J Cell Mol Med. 2013;17:76–89.
Beermann ML, Homma S, Miller JB. Proximity ligation assay to detect DUX4 protein in FSHD1 muscle: a pilot study. BMC Res Notes. 2022;15:163.
van den Heuvel A, Mahfouz A, Kloet SL, Balog J, van Engelen BGM, Tawil R, et al. Single-cell RNA sequencing in facioscapulohumeral muscular dystrophy disease etiology and development. Hum Mol Genet. 2019;28:1064–75.
Jiang S, Williams K, Kong X, Zeng W, Nguyen NV, Ma X, et al. Single-nucleus RNA-seq identifies divergent populations of FSHD2 myotube nuclei. PLoS Genet. 2020;16:e1008754.
Zheng D, Wondergem A, Kloet S, Willemsen I, Balog J, Tapscott SJ, et al. snRNA-seq analysis in multinucleated myogenic FSHD cells identifies heterogeneous FSHD transcriptome signatures associated with embryonic-like program activation and oxidative stress-induced apoptosis. Hum Mol Genet. 2024;33:284–98.
Chen L, Kong X, Johnston KG, Mortazavi A, Holmes TC, Tan Z, et al. Single-cell spatial transcriptomics reveals a dystrophic trajectory following a developmental bifurcation of myoblast cell fates in facioscapulohumeral muscular dystrophy. Genome Res. 2024;34:665–79.
van den Heuvel A, Lassche S, Mul K, Greco A, San León Granado D, Heerschap A, et al. Facioscapulohumeral dystrophy transcriptome signatures correlate with different stages of disease and are marked by different MRI biomarkers. Sci Rep. 2022;12:1426.
Wong C-J, Friedman SD, Snider L, Bennett SR, Jones TI, Jones PL, et al. Regional and bilateral MRI and gene signatures in facioscapulohumeral dystrophy: implications for clinical trial design and mechanisms of disease progression. Hum Mol Genet. 2024;33:698–708.
Cowley MV, Pruller J, Ganassi M, Zammit PS, Banerji CRS. An in silico FSHD muscle fiber for modeling DUX4 dynamics and predicting the impact of therapy. Elife [Internet]. 2023;12:e88345. https://doi.org/10.7554/eLife.88345.
Block GJ, Petek LM, Narayanan D, Amell AM, Moore JM, Rabaia NA, et al. Asymmetric bidirectional transcription from the FSHD-causing D4Z4 array modulates DUX4 production. PLoS One. 2012;7:e35532.
Stadler G, Rahimov F, King OD, Chen JCJ, Robin JD, Wagner KR, et al. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol. 2013;20:671–8.
Sharma V, Pandey SN, Khawaja H, Brown KJ, Hathout Y, Chen Y-W. PARP1 differentially interacts with promoter region of DUX4 gene in FSHD myoblasts. J Genet Syndr Gene Ther [Internet]. Aug;7) (2016). Available from: https://doi.org/10.4172/2157-7412.1000303.
Neugebauer E, Walter S, Tan J, Drayman N, Franke V, van Gent M, et al. Herpesviruses mimic zygotic genome activation to promote viral replication. Nat Commun. 2025;16:710.
Inoue K, Bostan H, Browne MR, Bevis OF, Bortner CD, Moore SA, et al. DUX4 double whammy: The transcription factor that causes a rare muscular dystrophy also kills the precursors of the human nose. Sci Adv. 2023;9:eabq7744.
Grow EJ, Weaver BD, Smith CM, Guo J, Stein P, Shadle SC, et al. p53 convergently activates Dux/DUX4 in embryonic stem cells and in facioscapulohumeral muscular dystrophy cell models. Nat Genet. 2021;53:1207–20.
Salsi V, Losi F, Salani M, Kaufman PD, Tupler R. Posttranscriptional RNA stabilization of telomeric RNAs FRG2, DBE-T, D4Z4 at human 4q35 in response to genotoxic stress and D4Z4 macrosatellite repeat length. Clin Epigenetics. 2025;17:73.
Ciszewski L, Lu-Nguyen N, Slater A, Brennan A, Williams HEL, Dickson G, et al. G-quadruplex ligands mediate downregulation of DUX4 expression. Nucleic Acids Res. 2020;48:4179–94.
Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–31.
Himeda CL, Jones TI, Virbasius C-M, Zhu LJ, Green MR, Jones PL. Identification of epigenetic regulators of DUX4-fl for targeted therapy of facioscapulohumeral muscular dystrophy. Mol Ther. 2018;26:1797–807.
Campbell AE, Oliva J, Yates MP, Zhong JW, Shadle SC, Snider L, et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet Muscle. 2017;7:16.
Mocciaro E, Giambruno R, Micheloni S, Cernilogar FM, Andolfo A, Consonni C, et al. WDR5 is required for DUX4 expression and its pathological effects in FSHD muscular dystrophy. Nucleic Acids Res. 2023;51:5144–61.
Fox A, Oliva J, Vangipurapu R, Sverdrup FM. SIX transcription factors are necessary for the activation of DUX4 expression in facioscapulohumeral muscular dystrophy. Skelet Muscle. 2024;14:30.
Block GJ, Narayanan D, Amell AM, Petek LM, Davidson KC, Bird TD, et al. Wnt/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum Mol Genet. 2013;22:4661–72.
Cruz JM, Hupper N, Wilson LS, Concannon JB, Wang Y, Oberhauser B, et al. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J Biol Chem. 2018;293:11837–49.
Oliva J, Galasinski S, Richey A, Campbell AE, Meyers MJ, Modi N, et al. Clinically advanced p38 inhibitors suppress DUX4 expression in cellular and animal models of facioscapulohumeral muscular dystrophy. J Pharm Exp Ther. 2019;370:219–30.
Rojas LA, Valentine E, Accorsi A, Maglio J, Shen N, Robertson A, et al. P38α regulates expression of DUX4 in a model of facioscapulohumeral muscular dystrophy. J Pharm Exp Ther. 2020;374:489–98.
Vangipurapu R, Oliva J, Fox A, Sverdrup FM. Temporal variation in p38-mediated regulation of DUX4 in facioscapulohumeral muscular dystrophy. Sci Rep. 2024;14:26437.
Zeng W, de Greef JC, Chen Y-Y, Chien R, Kong X, Gregson HC, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009;5:e1000559.
Campbell AE, Shadle SC, Jagannathan S, Lim J-W, Resnick R, Tawil R, et al. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife [Internet]. 2018;7:e31023. https://doi.org/10.7554/eLife.31023.
Casa V, Runfola V, Micheloni S, Aziz A, Dilworth FJ, Gabellini D. Polycomb repressive complex 1 provides a molecular explanation for repeat copy number dependency in FSHD muscular dystrophy. Hum Mol Genet. 2017;26:753–67.
Paatela EM, St Amant FG, Hamm DC, Bennett SR, Gujral TS, van der Maarel SM, et al. A discrete region of the D4Z4 is sufficient to initiate epigenetic silencing. Hum Mol Genet [Internet]. 2025 July [cited 2025 July 15]; Available from: https://doi.org/10.1093/hmg/ddaf114.
Meurer L, Ferdman L, Belcher B, Camarata T. The SIX family of transcription factors: Common themes integrating developmental and cancer biology. Front Cell Dev Biol. 2021;9:707854.
Bosnakovski D, Gearhart MD, Toso EA, Ener ET, Choi SH, Kyba M. Low level DUX4 expression disrupts myogenesis through deregulation of myogenic gene expression. Sci Rep. 2018;8:16957.
Engquist EN, Greco A, Joosten LAB, van Engelen BGM, Zammit PS, Banerji CRS. FSHD muscle shows perturbation in fibroadipogenic progenitor cells, mitochondrial function and alternative splicing independently of inflammation. Hum Mol Genet. 2024;33:182–97.
Ferreboeuf M, Mariot V, Furling D, Butler-Browne G, Mouly V, Dumonceaux J. Nuclear protein spreading: implication for pathophysiology of neuromuscular diseases. Hum Mol Genet. 2014;23:4125–33.
Riem L, DuCharme O, Cousins M, Feng X, Kenney A, Morris J, et al. AI driven analysis of MRI to measure health and disease progression in FSHD. Sci Rep. 2024;14:15462.
Sambasivan R, Kuratani S, Tajbakhsh S. An eye on the head: the development and evolution of craniofacial muscles. Development. 2011;138:2401–15.
Ono Y, Boldrin L, Knopp P, Morgan JE, Zammit PS. Muscle satellite cells are a functionally heterogeneous population in both somite-derived and branchiomeric muscles. Dev Biol. 2010;337:29–41.
Turki A, Hayot M, Carnac G, Pillard F, Passerieux E, Bommart S, et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic Biol Med. 2012;53:1068–79.
Dmitriev P, Bou Saada Y, Dib C, Ansseau E, Barat A, Hamade A, et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic Biol Med. 2016;99:244–58.
Lek A, Zhang Y, Woodman KG, Huang S, DeSimone AM, Cohen J, et al. Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci Transl Med. 2020;12:eaay0271.
Heher P, Ganassi M, Weidinger A, Engquist EN, Pruller J, Nguyen TH, et al. Interplay between mitochondrial reactive oxygen species, oxidative stress and hypoxic adaptation in facioscapulohumeral muscular dystrophy: Metabolic stress as potential therapeutic target. Redox Biol. 2022;51:102251.
DeSimone AM, Leszyk J, Wagner K, Emerson CP Jr. Identification of the hyaluronic acid pathway as a therapeutic target for facioscapulohumeral muscular dystrophy. Sci Adv. 2019;5:eaaw7099.
Cohen J, Huang S, Koczwara K, Ho V, Woodman K, Lek A, et al. Flavones provide resistance to DUX4-induced toxicity via an mTor-independent mechanism. Res Sq [Internet]. 2023 Feb [cited 2025 July 13]; Available from: https://doi.org/10.21203/rs.3.rs-2452222/v1.
Teveroni E, Pellegrino M, Sacconi S, Calandra P, Cascino I, Farioli-Vecchioli S, et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J Clin Invest. 2017;127:1531–45.
Mul K, Horlings CGC, Voermans NC, Schreuder THA, van Engelen BGM. Lifetime endogenous estrogen exposure and disease severity in female patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2018;28:508–11.
Hangül C, Bozkurt S, Bilge U, Özdem S, Altunbaş H, Uysal H, et al. The Ratios of Estradiol and Progesterone to Testosterone Influence the Severity of Facioscapulohumeral Muscular Dystrophy. Neurol Sci Neurophysiol. 2020;37:190–6.
Maiullari S, Mele G, Calandra P, di Blasio G, Valentini S, Torcinaro A, et al. Estrogen rescues muscle regeneration impaired by DUX4 in a humanized xenograft mouse model. Cell Death Dis. 2025;16:1–13.
Hangul C, Ozcan F, Darbas S, Uysal H, Koc AF, Berker Karauzum S. Progesterone may be a regulator and B12 could be an indicator of the proximal D4Z4 repeat methylation status on 4q35ter. J Neurochem. 2024;168:3209–20.
Kim J-H, Han G-C, Seo J-Y, Park I, Park W, Jeong H-W, et al. Sex hormones establish a reserve pool of adult muscle stem cells. Nat Cell Biol. 2016;18:930–40.
Saad NY, Al-Kharsan M, Garwick-Coppens SE, Chermahini GA, Harper MA, Palo A, et al. Human miRNA miR-675 inhibits DUX4 expression and may be exploited as a potential treatment for Facioscapulohumeral muscular dystrophy. Nat Commun. 2021;12:7128.
Nishimura Y, Bittel A, Jagan A, Chen Y-W, Burniston J. Proteomic profiling uncovers sexual dimorphism in the muscle response to wheel running exercise in the FLExDUX4 Murine model of facioscapulohumeral muscular dystrophy. Mol Cell Proteom. 2025;24:101013.
Lassche S, Stienen GJM, Irving TC, van der Maarel SM, Voermans NC, Padberg GW, et al. Sarcomeric dysfunction contributes to muscle weakness in facioscapulohumeral muscular dystrophy. Neurology. 2013;80:733–7.
Caruso N, Herberth B, Bartoli M, Puppo F, Dumonceaux J, Zimmermann A, et al. Deregulation of the protocadherin gene FAT1 alters muscle shapes: implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet. 2013;9:e1003550.
Mariot V, Roche S, Hourdé C, Portilho D, Sacconi S, Puppo F, et al. Correlation between low FAT1 expression and early affected muscle in facioscapulohumeral muscular dystrophy: Involvement of FAT1 in FSHD. Ann Neurol. 2015;78:387–400.
Arahata K, Ishihara T, Fukunaga H, Orimo S, Lee JH, Goto K, et al. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): immunocytochemical and genetic analyses. Muscle Nerve Suppl. S56-66 1995.
Frisullo G, Frusciante R, Nociti V, Tasca G, Renna R, Iorio R, et al. CD8(+) T cells in facioscapulohumeral muscular dystrophy patients with inflammatory features at muscle MRI. J Clin Immunol. 2011;31:155–66.
Wong C-J, Wang L, Holers VM, Frazer-Abel A, van der Maarel SM, Tawil R, et al. Elevated plasma complement components in facioscapulohumeral dystrophy. Hum Mol Genet. 2022;31:1821–9.
Chew G-L, Campbell AE, De Neef E, Sutliff NA, Shadle SC, Tapscott SJ, et al. DUX4 suppresses MHC class I to promote cancer immune evasion and resistance to checkpoint blockade. Dev Cell. 2019;50:658–671.e7.
Bosnakovski D, Shams AS, Yuan C, da Silva MT, Ener ET, Baumann CW, et al. Transcriptional and cytopathological hallmarks of FSHD in chronic DUX4-expressing mice. J Clin Invest. 2020;130:2465–77.
Himeda CL, Jones TI, Jones PL. CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol Ther. 2016;24:527–35.
Himeda CL, Jones TI, Jones PL. Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic DUX4-fl expression in FSHD. Mol Ther Methods Clin Dev. 2021;20:298–311.
Sasaki-Honda M, Jonouchi T, Arai M, He J, Okita K, Sakurai S, et al. Hit-and-run silencing of endogenous DUX4 by targeting DNA hypomethylation on D4Z4 repeats in facioscapulohumeral muscular dystrophy [Internet]. bioRxiv. 2022. Available from: https://doi.org/10.1101/2022.04.12.487997.
Acknowledgements
This work was partially supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant 22KJ1636 to MSH). This work was partly supported by a grant from the Pathological Elucidation and Drug Discovery Utilizing Disease-specific iPS cells (JP23bm1423001 to HS), which was part of the Acceleration Programme of R&D and Implementation for Regenerative Medicine and Cell and Gene Therapy by the Japan Agency for Medical Research and Development (AMED). This work was supported by JST SPRING (JPMJSP2110 to TK). We would like to thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sasaki-Honda, M., Kishimoto, T. & Sakurai, H. Diversity challenges and reconciles genetics in facioscapulohumeral muscular dystrophy. J Hum Genet (2025). https://doi.org/10.1038/s10038-025-01401-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s10038-025-01401-6
