
Abstract
Thioredoxin-interacting protein (TXNIP), which is also known as thioredoxin-binding protein 2 (TBP2), directly interacts with the major antioxidant protein thioredoxin (TRX) and inhibits its antioxidant function and expression. However, recent studies have demonstrated that TXNIP is a multifunctional protein with functions beyond increasing intracellular oxidative stress. TXNIP activates endoplasmic reticulum (ER) stress-mediated nucleotide-binding oligomerization domain (NOD)-like receptor protein-3 (NLRP3) inflammasome complex formation, triggers mitochondrial stress-induced apoptosis, and stimulates inflammatory cell death (pyroptosis). These newly discovered functions of TXNIP highlight its role in disease development, especially in response to several cellular stress factors. In this review, we provide an overview of the multiple functions of TXNIP in pathological conditions and summarize its involvement in various diseases, such as diabetes, chronic kidney disease, and neurodegenerative diseases. We also discuss the potential of TXNIP as a therapeutic target and TXNIP inhibitors as novel therapeutic drugs for treating these diseases.
Similar content being viewed by others


Introduction
Thioredoxin interacting protein (TXNIP), which is also known as thioredoxin (TRX) binding protein-2 (TBP-2) or vitamin-D3 upregulated protein 1 (VDUP-1), was identified as a binding partner of TRX in a yeast two-hybrid screen1,2. TXNIP was originally discovered in HL-60 cells as a 1,25-dihydroxy vitamin-D3 responsive gene, hence its original name of VDUP-11. TXNIP has an α-arrestin domain that interacts with and reduces the activity of cytosolic TRX and mitochondrial TRX (activated form), thus controlling cellular redox signaling3. TRX helps sustain the activity of the TRX system through a redox mechanism. The TRX system regulates the preservation of a reduced cellular environment and includes TRX reductase, nicotinamide adenine dinucleotide phosphate (NADPH), and TXNIP3,4. TXNIP activates TRX as a negative regulator to maintain the redox balance and is a core mediator of proliferation and apoptosis. Moreover, this factor is also linked to the development of TXNIP-related metabolic diseases such as diabetes3,4,5.
TXNIP plays an essential role in metabolism maintenance by activating inflammatory signaling via the nucleotide-binding oligomerization domain (NOD)-like receptor protein-3 (NLRP3) inflammasome. The inflammasome is composed of three components: NLRP3, the caspase recruitment domain (ASC), and pro-caspase 16. Inflammasome activation is initiated by the interaction between TXNIP and NLRP3. The generation of reactive oxygen species (ROS) induces the interaction of NLRP3, ASC, and pro-caspase 1, resulting in the formation of inflammasomes and the secretion of interleukin 1β (IL-1β) and IL-18, thus facilitating the inflammatory reaction7. TXNIP is considered a critical link between endoplasmic reticulum (ER) stress and inflammation. TXNIP induction by ER stress activates NLRP3 and triggers the formation of the NLRP3 inflammasome complex7,8. Furthermore, ER stress and oxidative stress can induce subcellular shuttling of TXNIP to mitochondria, which triggers ASK1-induced mitochondrial apoptosis9,10.
Several studies have identified TXNIP upregulation in diseases such as kidney disease, neurodegeneration, and type 1 and 2 diabetes. TXNIP modulates the transcription of several genes, indicating the involvement of various mechanisms that implicate a new function of TXNIP. Thus, considering the upregulation of TXNIP in numerous diseases, TXNIP could be a potential drug target for therapeutic development. In this review, we summarize the known roles of TXNIP in the regulatory mechanisms underlying oxidative stress, ER stress-mediated NLRP3 inflammasome activation, pyroptosis, mitochondrial stress-induced apoptosis, and the pathogenesis of diabetes, chronic kidney disease, and neurodegenerative diseases.
Multiple functions of TXNIP in the cellular stress response pathway
The TRX system: TXNIP regulates oxidative stress
TXNIP is a central physiological inhibitor of the TRX redox system, and its transcription and activity levels play a major role in redox regulation. The redox system is composed of TRX, TXNIP, NADPH, and thioredoxin reductase (TRX-R)11. TRX is present as different isoforms, including cytosolic TRX1 (12 kDa) and mitochondrial TRX2 (15.5 kDa). TRX1 is the primary TRX isoform, is widely distributed in the cytosol, nucleus, and plasma membrane and is even secreted into the extracellular space under certain conditions4,11, while TRX2 is specifically localized. TRX reduces oxidized proteins, resulting in the oxidation of 2-cysteine residues and constant alternation between its oxidized (inactivated form) and reduced states (activated form)4. Oxidized TRX is converted to its reduced state by NADPH-dependent TRX-R activity via the catalysis of electron transport from NADPH to oxidized TRX, thus controlling intracellular redox balance12. Compared to wild-type mice, mice with TRX1 overexpression exhibit increased resistance to oxidative stress, cardiomyocyte injury suppression, and lung cancer ablation via the inhibition of ROS production, suggesting protective effects of TRX13.
The essential physiological role of the TRX system is to protect cells from oxidative damage and maintain a reduced intracellular microenvironment by removing intracellular ROS via disulfide reductase activities14. ROS are usually generated by cellular metabolism involving mitochondria, and it is important to regulate optimum levels of ROS in cellular processes involving signal transduction and gene transcription. However, excess accumulation of ROS during cell proliferation can lead to lethal damage to cellular components, such as DNA, RNA, and proteins, which in turn activate cell death programs15. Thus, efficient intracellular antioxidant systems that balance ROS generation and scavenging to sustain redox homeostasis are indispensable for cell metabolism.
TRX1 and TRX2 regulate cytoplasmic and mitochondrial ROS levels, respectively. However, TXNIP suppresses the antioxidant activity of TRX. Mesangial cells from TXNIP-knockout mice (Hcb-19) under high glucose conditions generated two- to threefold less ROS than wild-type cells (C3H), suggesting the inhibitory effect of TXNIP on the redox function of TRX16. In addition, Yodoi et al. showed that transient overexpression of TXNIP in HEK293 and COS-7 cells inhibited the reducing activity of TRX1 and downregulated its expression at the transcript and protein levels17. Thus, TXNIP is a pro-oxidant that increases ROS generation and induces oxidative stress. TXNIP binds specifically to reduced TRX (TRX-(SH)2, activated form) and not to oxidized TRX (TRX-S2, inactivated form) at the catalytic site18. The interaction of TXNIP and reduced TRX via active catalytic sites requires dithiol-disulfide exchange reactions at the redox-related cysteine residues on each protein and induces structural reorganization of TXNIP18. TXNIP has an intramolecular disulfide bond between cysteine residues (Cys) 63 and 247, which is essential for its interaction with Cys 32 of TRX. Studies have identified redox-dependent and redox-independent effects of TXNIP and the production of a C247S (TRX binding site) mutation in TXNIP that cannot bind TRX19. TRX regulates TXNIP function as a lipid inhibitor by improving its stability rather than TXNIP regulating TRX. This distinct complex of TRX and TXNIP, which is also known as the redoxisome, primarily regulates the ROS signaling pathway (cytosolic and mitochondrial)3,4.
In the physiological state, TXNIP is localized to the nucleus and cannot translocate to the cytoplasm10. Under excessive ROS conditions, TXNIP expression is upregulated by inhibition of the phosphorylation of adenosine 5′-monophosphate-activated protein kinase (AMPK) and the translocation of TXNIP from the nucleus to the cytoplasm, thereby resulting in ER and mitochondrial stress20. Nuclear to cytoplasmic translocation of TXNIP promotes the interaction between TXNIP and TRX1, leading to the inhibition of TRX1 activity and resulting in the development of various diseases. Antoine et al. showed that decreased TRX expression due to aortic TXNIP overexpression was associated with endothelial dysfunction related to arterial aging and increased expression of NADPH oxidase21,22. A reduction in aortic TXNIP expression levels increased TRX expression levels, leading to the ablation of NADPH oxidase expression21, which protected against endothelial dysfunction induced by several disorders. Research on diabetes-related mouse models has demonstrated the generation of oxidative stress and excessive ROS production due to the ablation of TRX activity by TXNIP23. In addition, TXNIP regulates the production of ROS through mitochondria and NADPH oxidase under high glucose conditions23,24. These findings demonstrate two-way negative feedback between ROS and TXNIP, and reduced TRX acts as an intermediate20. In response to intracellular ROS accumulation, TXNIP shuttles into mitochondria via unclear mechanisms and binds to the disulfide bond of TRX2, inhibiting the reducing functions of TRX2 and oxidizing TRX2 to form the TXNIP/TRX2 complex. TRX2 is dissociated from apoptotic signal-regulated kinase 1 (ASK1) by TXNIP, and the TXNIP/TRX2 complex triggers the generation of mitochondrial ROS and induces ASK1 phosphorylation. Phosphorylated ASK1 induces the cleaved form of caspase 3 by stimulating cytochrome c (Cyto c) secretion. Thus, oxidative stress-induced TXNIP shuttling promotes the mitochondrial apoptosis pathway, leading to various injuries, including those of mitochondria, cardiac tissues, kidney, and lung4,9,10.
TXNIP induces the ASK1-mediated apoptotic signaling pathway
TXNIP may induce glucocorticoid-related apoptotic signals by antagonizing the antiapoptotic effect of TRX via intracellular ROS scavenging3. TRX promotes antiapoptotic functions by binding to and inhibiting the proapoptotic protein ASK1, which is a member of the mitogen-activated protein (MAP) kinase family25. In response to oxidative stress, TXNIP translocates to the cytosol and/or mitochondria, while it is localized in the nucleus under basal conditions. Cytosolic TXNIP binds to TRX1 and dissociates ASK1 from TXR1, leading to activation of the p38 mitogen-activated protein kinase (p38 MAPK) signaling pathway26. Moreover, in mitochondria, TXNIP binds to mitochondrial TXR2 and prevents the reducing actions of TRX2, resulting in the activation and phosphorylation of ASK110. ASK1 is generally bound to TRX2 under normal conditions; however, under oxidative stress and subsequent translocation of TXNIP to the mitochondria, ASK1 dissociates from TRX2, inducing an apoptotic signal cascade. Disruption of the ASK1-TRX2 interaction induces the phosphorylation of ASK1 and the release of Cyto c from the mitochondria into the cytosol. Cyto c activates Apaf-1 by directly binding to it, leading to its oligomerization into the apoptosome complex, which recruits pro-caspase 9 and initiates autocatalytic cleavage prior to pro-caspase 9 separation from the apoptosome and becoming inactive27. Cleaved caspase 9 can directly activate caspase 3 by cleaving pro-caspase 3, which leads to a programmed apoptosis pathway4,28 (Fig. 1).

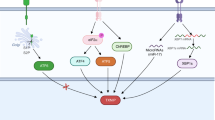
Under normal conditions, TXNIP is primarily localized in the nucleus. This allows TRX2 in the mitochondria to remain bound to ASK1, inhibiting the activation of ASK1 and promoting cellular survival. In response to oxidative stress, TXNIP shuttles to the mitochondria, where it competes with ASK1 for TRX2 binding, generating a TXNIP/TRX2 complex and leading to ASK1 phosphorylation. p-ASK1 stimulates Cyto c release into the cytosol, which activates Apaf-1. Apaf-1 oligomerizes into an apoptosome complex. Pro-caspase 9 is recruited to the apoptosome and is activated by autocleavage. Activated caspase 9 cleaves pro-caspase 3 and leads to a programmed apoptosis pathway.
TXNIP regulates the survival of pancreatic β-cells, which are considerably sensitive to oxidative stress due to their low expression levels of antioxidant enzymes such as TRX and manganese superoxide dismutase (MnSOD), and β-cell apoptosis plays a key role in type 1/2 diabetes29. TXNIP overexpression in β-cells activates apoptosis primarily by stimulating the mitochondrial death signaling pathway involving caspase 3 cleavage, Cyto c release, and phosphorylation/activation of ASK110,29. On the other hand, TXNIP depletion has protective effects and enhances β-cell survival even under high glucose conditions by improving AKT/Bcl-xL signaling30. Recently, Chen et al. identified a novel regulatory mechanism of the CCAAT/enhancer binding protein homologous protein (CHOP)-dependent TXNIP signaling pathway in nephrotic syndrome (NS). Albuminuria-induced ER stress promotes the CHOP-TXNIP signaling axis, leading to activation of the mitochondrial p-ASK1-mediated intrinsic apoptosis pathway9. Several in vitro models have demonstrated the contribution of TXNIP to apoptosis, including retinal pericytes exposed to high glucose and endothelial cells stressed by palmitate31,32. Thus, TXNIP-related apoptosis is considered a potential target in models of various disorders13.
TXNIP activates the NLRP3 inflammasome by mediating ER stress
TXNIP plays an essential role in cellular metabolism by activating inflammatory signaling via NLRP36. The initiation of inflammatory signaling requires the formation of inflammatory sensors (known as inflammasomes). Inflammasomes are multiprotein complexes consisting of NLRP3 oligomers, ASC, and pro-caspase 130. NLRP3 contains three domains: the pyrin domain (PYD), leucine-rich repeat domain (LRR), and nucleotide binding/oligomerization domain (NACHT)33. ASC contains two domains: the caspase recruitment domain (CARD) and PYD. Pro-caspase 1 is composed of the CARD and the p10/30 domain33. NLRP3 inflammasome activation is initiated by the stimulation of nuclear factor-κB (NF-κB), which translocates to the nucleus under oxidative stress conditions. NF-κB, which controls the transcriptional induction of NLRP3 and proinflammatory cytokines, including pro-IL-1β and pro-IL-18, was shown to provide the first step in the activation of the NLRP3 inflammasome33. The second step of NLRP3 inflammasome activation is initiated by the direct interaction between TXNIP and NLRP3 in a redox-related manner. Proinflammatory pathway activation and ROS accumulation induce the interaction of TXNIP with NLRP3 and ASC via the pyrin domain of NLRP3, and subsequently, the CAR domain of ASC recruits and interacts with pro-caspase 132. In addition, these stress conditions promote the separation of TXNIP from TRX2, which enables TXNIP to bind directly with NLRP3 in the mitochondria9,34. These interactions lead to the formation of the NLRP3 inflammasome and induce the cleavage of pro-caspase 1, which results in the production of activated caspase 1. Caspase 1 processes pro-IL-18 and pro-IL-1β into their mature forms (IL-18 and IL-1β) and mediates their secretion into the cytosol, which facilitates the inflammatory reaction. Active caspase 1 cleaves gasdermin D (GSDMD) within the linker between the N- and C-terminal domains35. The N-terminal fragment shuttles to the plasma membrane and oligomerizes to form membrane pores with diameters of 10–20 nm35. The membrane pores promote the release of the inflammatory cytokines IL-1β/IL-18, membrane rupture, and cell swelling, eventually leading to pyroptosis7 (Fig. 2).

The UPR is induced by the accumulation of misfolded proteins in the ER. Disassembly of BiP from IRE1α, PERK, and ATF6 during ER stress activates each domain. Activated IRE1α induces the nonconventional splicing of X-box binding protein-1 (xbp1u; unspliced form) mRNA and forms xbp1s mRNA, which encodes XBP1s (a transcriptional activator). PERK is phosphorylated by ER stress, causing the phosphorylation of eIF2α. This process prevents ribosome assembly, which leads to translational inhibition and allows the cell to deal with ER stress. However, ATF4 dissociates translation inhibition during stress conditions and induces CHOP activation. Under stress conditions, ATF6 is translocated to the Golgi compartment and cleaved by S1P/S2P enzymes. Cleaved ATF6 (ATF6c) induces CHOP and promotes the expression of XBP1s. CHOP induces TXNIP shuttling from the nucleus to the mitochondria, which is required for the generation of mitochondrial ROS. The induction of CHOP/TXNIP signaling activates the NLRP3 inflammasome. Ultimately, active caspase 1 stimulates the maturation of IL-1β/IL-18 and GSDMD cleavage, leading to inflammation and pyroptosis-associated pathways.
Several studies have reported that the induction of TXNIP by ER stress promotes the activation of the NLRP3 inflammasome7. The unfolded protein response (UPR) is initiated by the stimulation of three ER transmembrane proteins located in the ER: activating transcription factor 6 (ATF6), protein kinase RNA (PKR)-like ER kinase (PERK), and inositol-requiring enzyme-1α (IRE1α)36 (Fig. 2).
ATF6 is a type 2 transmembrane protein that contains cytosolic and luminal domains. The activation of ATF6 by ER stress is dependent on binding immunoglobulin protein (BiP) (also known as glucose-regulated protein (GRP78)) binding36. Under normal conditions, BiP is located in the luminal domain of ATF637. UPR activation releases ATF6 and results in its Golgi localization. ATF6 disulfide bonds are remodeled under ER stress, which induces ATF6 translocation to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P and S2P)37. ATF6c, the cytosolic domain of ATF6, translocates into the nucleus and stimulates the transcription of UPR-related genes by directly binding to the ER stress response component or by interacting with nuclear transcription factor Y (NF-Y)38. ATF6 mostly encodes ER stress-related transcription factors such as X-box binding protein 1 (XBP1), ER chaperones, and ER-associated protein degradation (ERAD) components36. It is thought that because ATF6 directly encodes X-box binding protein-1, the activation of XBP1 can enhance CHOP expression39. In addition, ER stress induced by albuminuria can stimulate ATF6 and lead to increased CHOP levels9. Elevated expression of CHOP results in the overexpression of TXNIP, which ultimately activates the NLRP3 inflammasome, similar to PERK and IRE1α signaling.
IRE1α is a type 1 transmembrane protein with a kinase and endoribonuclease domain. After being activated, IRE1α phosphorylates and dimerizes, activating its RNase domain, which catalyzes the deletion of a 26-nucleotide intron with the mRNA sequence of XBP140. This splicing step is accomplished by the RtcB protein, which ligates spliced XBP1 mRNA, allowing the translation of XBP1s40. XBP1s is a major regulator of genes involved in ERAD and protein folding. Additionally, the kinase domain of IRE1α can couple ER stress with inflammation. The kinase IRE1α activates TNF receptor-associated factor 2 (TRAF2) and c-Jun N-terminal kinase (JNK) signaling modules, which can initiate inflammatory responses41. IRE1α can thus regulate TXNIP/NLRP3 inflammasome activity. ER stress-induced IRE1α overexpression activates CHOP by stimulating XBP1s or JNK phosphorylation by ASK19,41. In addition, an increase in IRE1α can activate TXNIP by reducing the activity of microRNA-17 (miR-17), which is a TXNIP-destabilizing miRNA.
PERK, a type I transmembrane kinase, is activated by ER stress. PERK activation induces phosphorylation of the eukaryotic initiation factor 2α (eIF2α) subunit, which inhibits protein synthesis42. This process decreases the excess proteins entering the ER in stressed cells and permits the selective translation of the mRNA encoding transcription factor 4 (ATF4)38. Sustained ATF4 expression by eIF2α phosphorylation under ER stress allows the induction of CHOP and CHOP-dependent TXNIP/NLRP3 inflammasome activation42,43. Thus, the novel regulatory mechanism of TXNIP/NLRP3 inflammatory signaling via PERK activation represents a convergence point of different stress pathways governed by a specific regulator.
Overall, the stimulation of these three signal transducers by ER stress typically regulates the activity of CHOP and TXNIP. The induction of CHOP/TXNIP signaling activates the NLRP3 inflammasome and ultimately caspase 1 to stimulate the maturation and release of IL-1β and IL-1841. Thus, CHOP/TXNIP signaling is a core regulator of NLRP3 inflammasome activation and the apoptosis/pyroptosis pathway and may act as a key mediator linking an enormous variety of deleterious stimuli such as oxidative stress and inflammation.
Role of TXNIP in the pathogenesis of diseases
Diabetes
Diabetes is a common metabolic disorder characterized by hyperglycemia, loss of functional pancreatic β-cell mass, and insufficient production of the glucose-lowering hormone insulin44,45. Moreover, diabetes can cause secondary complications, including kidney failure, nerve damage, blindness, and cardiovascular disease46. Pancreatic β-cells play an important role in maintaining glucose levels by sensing blood glucose levels and secreting insulin26.
TXNIP is the most upregulated gene in human pancreatic islet cells exposed to high levels of glucose47,48. Glucose leads to dose- and time-dependent recruitment of carbohydrate response element-binding protein (ChREBP) to the TXNIP promoter, which regulates TXNIP transcription in vivo in human islets, as well as INS-1 β-cells49. In contrast, forkhead transcription factor O1 (FOXO1) competes with ChREBP to bind to the TXNIP promoter in vivo in human islets and INS-1 cells. FOXO1 significantly decreases glucose-induced TXNIP expression by blocking glucose-induced ChREBP binding to the TXNIP promoter48. Mammalian target of rapamycin (mTOR) also decreases β-cell TXNIP expression by inhibiting ChREBP transcriptional activity. β-cell-specific mTOR deficiency increases the expression levels of TXNIP and ChREBP, leading to severe reductions in β-cell survival, β-cell mass, islet size, and insulin secretion in streptozotocin (STZ)-induced diabetic mice50.
The induction of TXNIP in the islets of human diabetic patients and diabetic mice causes β-cell apoptosis47,50,51. TXNIP overexpression by high glucose exposure was accompanied by caspase 3 pathway-mediated β-cell apoptosis in INS-1 cells and the isolated islets of mice and humans. Glucose toxicity-induced β-cell apoptosis was attenuated in TXNIP-deficient mouse islets51. Furthermore, TXNIP deficiency prevented mitochondrial β-cell death by activating antiapoptotic AKT/Bcl-xL signaling and protected against type 1 and type 2 diabetes by maintaining β-cell mass and function30. Therefore, TXNIP plays a pivotal role in β-cell survival, function, and glucose homeostasis.
The proinflammatory cytokine IL-1β is associated with β-cell dysfunction and death, leading to insulin deficiency in both type 1 and type 2 diabetes8,47. Glucose-induced TXNIP is essential for activation of the NLRP3 inflammasome pathway. TXNIP binds to NLRP3 and activates the NLRP3 inflammasome, resulting in caspase 1 activation and IL-1β production8,34. Furthermore, TXNIP is a critical link between ER stress and inflammation in β-cell death. High glucose-induced ER stress increases TXNIP expression, which promotes ER stress-mediated NLRP3 inflammasome activation, IL-1β production, and β-cell death8. TXNIP deficiency impairs NLRP3 inflammasome activation and IL-1β secretion. Txnip-/- and Nlrp3-/- mice have similar phenotypes with improvements in glucose tolerance and insulin sensitivity compared to wild-type mice after eight weeks of a high-fat diet feeding34. Therefore, the inhibition of TXNIP is a reasonable therapeutic strategy against diabetes.
Chronic kidney disease
Chronic kidney disease (CKD) is characterized by irreversible functional or structural kidney damage and has emerged as one of the most prominent causes of death worldwide52,53. Patients with progressive CKD can have common symptoms such as proteinuria/albuminuria, glomerulosclerosis, and interstitial fibrosis6,9,53,54. Interestingly, heavy proteinuria is not only a symptom but also a cause of glomerulosclerosis, interstitial fibrosis, and loss of kidney function9. TXNIP expression is significantly increased in human proteinuric kidney disease, including focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN), and diabetic nephropathy (DN)9. Furthermore, upregulation of the ER stress-induced transcription factor CHOP due to albuminuria drives TXNIP shuttling from the nucleus to the mitochondria, which is required for mitochondrial ROS production in Lamb2-/- mice, which is an NS model. Mitochondrial-specific TRX2 oxidation by mitochondrial ROS dissociates TXNIP, activates the NLRP3 inflammasome complex and releases ASK 1 to induce mitochondria-dependent apoptosis. Thus, TXNIP inhibition by CHOP deletion suppresses NLRP3 inflammasome activation and p-ASK1-dependent mitochondrial apoptosis, decreasing albuminuria and improving kidney function in NS9.
Renal interstitial fibrosis is the final common pathway of progressive CKD53,54,55. TXNIP deletion attenuates renal fibrosis and damage by regulating SMAD3 and p38/ERK MAPK phosphorylation in a unilateral ureteral obstruction (UUO) mouse model of renal interstitial fibrosis. UUO-induced renal inflammation is suppressed by inhibiting the activation of NFκB and NLRP3 inflammasomes and the induction of proinflammatory cytokines IL-1β, IL-18, and MCP1 in the kidneys of TXNIP-/- mice56. In addition, kidney aging is accompanied by renal fibrosis, which drives CKD53,55,57. TXNIP overexpression in tubular epithelial cells increases the expression levels of the cellular senescence markers p16INK4a and γH2AX and the profibrotic factors α-SMA, TGF-β1/SMAD3, and collagen I, which ultimately lead to aging-related renal fibrosis and a decline in kidney function. Furthermore, a study showed that TXNIP directly interacts with signal transducer and activator of transcription 3 (STAT3) and promotes the STAT3 signaling pathway-activated profibrotic response, which was markedly reduced by the STAT3 inhibitor HY-N0174, as well as genetic TXNIP deletion57.
DN is a major clinical complication of diabetes and the most common cause of CKD58,59. STZ-induced diabetic mice exhibit significant increases in functional and structural kidney damage, including albuminuria, glomerular fibrosis, podocyte foot process effacement, glomerular basement membrane thickening, Nox4-mediated oxidative stress, and IL-1β-mediated inflammation, which are attenuated by TXNIP deletion58. Furthermore, high glucose-induced TXNIP upregulation is a key mediator of intracellular ROS generation and apoptosis in cultured human podocytes58 and HK-2 human proximal tubular cells60. The overexpression of FOXO1, a positive regulator of antioxidant genes, prevents high glucose-induced TXNIP upregulation, TRX downregulation, and ROS accumulation, resulting in reduced apoptosis in proximal tubular cells60. Moreover, TXNIP silencing mitigates Bax/Caspase 3 pathway-induced apoptosis by suppressing mTOR and p38 MAPK signaling activation in high glucose-induced cultured podocytes and STZ-induced diabetic kidneys. High glucose-induced podocyte apoptosis is also alleviated by treatment with Raptor and Rictor shRNAs, the mTOR-specific inhibitor KU-0063794, and the p38 MAPK inhibitor SB20358059.
Overall, the upregulation of TXNIP was observed and implicated in pathological pathways in vivo and in vitro in the NS model, UUO-induced renal fibrosis model, aging-related renal fibrosis model, DN model, and human proteinuric kidney diseases. Thus, emerging evidence suggests that TXNIP is a key pathological regulator of CKD.
Neurodegenerative diseases
TXNIP has been implicated in neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD)6,26. AD is the most common age-related neurodegenerative disease among the elderly population. In the AD brain, clinical symptoms are accompanied by pathological features, including extracellular amyloid β (Aβ) plaque deposition and abnormal intracellular accumulation of hyperphosphorylated tau26,61,62. Continuous accumulation of Aβ and hyperphosphorylated tau in the brain triggers ER stress, oxidative stress, and inflammation, ultimately contributing to neurodegeneration61. Several studies have reported a significant increase in TXNIP in the cortex and hippocampus of postmortem human AD brains and an in vivo AD mouse model61,62,63,64. The upregulation of TXNIP in the AD brain is mostly related to inflammation6,26. ER stress induced by phospho-tau and Aβ promotes the TXNIP-induced NLRP3 inflammasome pathway, which leads to IL-1β-induced neuroinflammation in the human AD brain61,62. Recently, TXNIP is closely associated with Aβ and receptor for advanced glycation end products (RAGE), which are counterreceptors of proinflammatory ligands such as Aβ in the hippocampus of 5xFAD AD mice. The RAGE-TXNIP axis induces Aβ translocation to mitochondria, leading to mitochondrial dysfunction and oxidative stress, which induce NLRP3 inflammasome activation, IL-1β secretion, and the cleavage of the pyroptotic protein GSDMD. Thus, TXNIP silencing blocks NLRP3 inflammasome-induced IL-1β secretion and GSDMD activation in the hippocampus of 5xFAD AD mice63.
PD is the second most common neurodegenerative disease and is characterized by α-synuclein accumulation and dopaminergic neuron loss65,66. TXNIP was significantly increased in human α-synuclein-overexpressing A53T mice and α-synuclein-transfected HEK293 cells. TXNIP overexpression impairs lysosomes by downregulating the critical lysosomal membrane protein ATP13A2, resulting in α-synuclein accumulation. Increased TXNIP in the mouse substantia nigra causes dopaminergic neuron loss65. Treatment with MPP+ , a dopaminergic neurotoxin, leads to TXNIP-mediated NLRP inflammasome activation and pyroptosis66. Collectively, these studies demonstrate that TXNIP plays a role as a pathological contributor and accelerator in the progression of AD and PD.
Therapeutic potential of targeting TXNIP in diseases
TXNIP is a key pathological regulator of several diseases, including CKD, diabetes, and neurodegenerative diseases, and is an attractive therapeutic target for the development of novel drugs. Several studies have reported that some small-molecule compounds and phytochemicals can inhibit TXNIP expression and TXNIP-induced signaling pathways. Here, we review a variety of TXNIP inhibitors and discuss the mechanisms by which they inhibit TXNIP directly or indirectly (Table 1).
Verapamil, an anti-hypertensive calcium channel blocker, inhibits TXNIP transcription by decreasing the binding of ChREBP to the TXNIP promoter45,46,67. Oral administration of verapamil prevents β-cell apoptosis and improves endogenous β-cell function, glucose homeostasis, and insulin sensitivity in diabetic mouse models45 and in adults with recent-onset type 1 diabetes46. Additionally, verapamil treatment reduces tau phosphorylation at Ser202/Thr205 by blocking the TXNIP/p38 MAPK pathway and preventing Aβ-induced NLRP3 inflammasome activation and IL-1β secretion in the hippocampus of 5xFAD AD model mice63,68.
Metformin is widely used as a first-line treatment for type 2 diabetes67,69,70,71. Treatment with metformin prevents TXNIP/NLRP3 inflammasome activation by suppressing ER stress in the adipose tissues of STZ-injected diabetic mice, thereby blocking diabetes-induced adipose dysfunction69. Metformin inhibits high glucose-induced TXNIP/NLRP3 inflammasome activation through AMPK activation in macrophages, which suppresses diabetes-mediated atherosclerosis70. In addition, metformin suppresses TXNIP expression and the TXNIP-NLRP3 interaction, which protects against intestinal ischemia‒reperfusion injury and pyroptosis via the TXNIP-NLRP3-GSDMD pathway71.
SRI-37330 was recently identified as an orally bioavailable, nontoxic, small molecule that suppresses the mRNA and protein expression of TXNIP by inhibiting TXNIP promoter activity in INS-1 cells and mouse and human islets. Oral treatment with SRI-37330 decreased glucagon secretion and activity, reduced hepatic glucose production, and reversed hepatic steatosis in STZ- and obesity-induced diabetic mice44.
The antioxidant Dl‐3‐n‐butylphthalide (Dl‐NBP) has recently been used clinically for the treatment of cerebral ischemia. Dl‐NBP treatment enhances TRX activity and reduces TXNIP expression, the TXNIP-NLRP3 interaction, and NLRP3 inflammasome activation by upregulating Nrf2, a major transcription factor that responds to oxidative stress. The Dl‐NBP-mediated signaling pathway mitigates neuronal apoptosis and Aβ plaque-associated microglial dystrophy in the brains of APP/PS1 AD model mice64.
Oral administration of the small-molecule W2476, which is a modulator of TXNIP expression, rescues STZ-induced diabetic mice by promoting β-cell survival and insulin secretion72. Similarly, inhibiting TXNIP expression with the natural flavonoid quercetin and antioxidant allopurinol improves liver inflammation and lipid accumulation in STZ-induced diabetic rats73.
The phytochemical salidroside, which is isolated from the herbal plant Rhodiola rosea, alleviates high glucose-induced cell proliferation, oxidative stress, and ECM accumulation in HBZY-1 rat glomerular mesangial cells by inhibiting TXNIP-NLRP3 inflammasome pathway activation74. Salidroside administration also attenuates hepatic steatosis and hepatic inflammation in high-fat diet-fed mice by inhibiting the AMPK-dependent TXNIP/NLRP3 pathway75.
In addition, several other small molecule compounds, such as diltiazem45 and dimethyl fumarate (DMF)76, as well as other phytochemicals, such as resveratrol69 and fisetin77, have shown inhibitory effects on TXNIP expression and TXNIP-induced signaling pathways.
Conclusion and future perspective
We reviewed currently available information on the roles of the multifunctional protein TXNIP in the pathogenesis of diseases such as diabetes, chronic kidney disease, and neurodegenerative disease, and summarized a variety of TXNIP inhibitors. Emerging evidence suggests that TXNIP is a promising therapeutic target, and its inhibition protects against the development of various diseases. According to previous studies, CHOP inhibition, FOXO1 overexpression, and ChREBP inhibition for targeting the TXNIP promoter to inhibit TXNIP expression could be used to develop effective TXNIP inhibitors and novel drug candidates. Moreover, targeting TXNIP could have a secondary effect through pharmacological inhibition of the NLRP3 inflammasome for inflammatory disease treatment because TXNIP is essential for activation of the NLRP3 inflammasome pathway. This review may be helpful to better understand the pathogenic roles of TXNIP in various diseases to develop effective therapeutic strategies.
References
Junn, E. et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J. Immunol. 164, 6287–6295 (2000).
Nishiyama, A. et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 274, 21645–21650 (1999).
Tsubaki, H., Tooyama, I. & Walker, D. G. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21249357 (2020).
Yoshihara, E. et al. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 4, 514 (2014).
Parikh, H. et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 4, e158 (2007).
Qayyum, N., Haseeb, M., Kim, M. S. & Choi, S. Role of thioredoxin-interacting protein in diseases and its therapeutic outlook. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22052754 (2021).
He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 25, 1285–1298 (2015).
Oslowski, C. M. et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 16, 265–273 (2012).
Park, S. J. et al. Blocking CHOP-dependent TXNIP shuttling to mitochondria attenuates albuminuria and mitigates kidney injury in nephrotic syndrome. Proc. Natl Acad. Sci. USA 119, e2116505119 (2022).
Saxena, G., Chen, J. & Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 285, 3997–4005 (2010).
Spindel, O. N., World, C. & Berk, B. C. Thioredoxin interacting protein: redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal 16, 587–596 (2012).
Gao, P. et al. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J. Diabetes Res. 2015, 504761 (2015).
Du, Y., Zhang, H., Zhang, X., Lu, J. & Holmgren, A. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J. Biol. Chem. 288, 32241–32247 (2013).
He, L. et al. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol. Biochem. 44, 532–553 (2017).
Rowe, L. A., Degtyareva, N. & Doetsch, P. W. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 45, 1167–1177 (2008).
Shah, A. et al. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J. Biol. Chem. 288, 6835–6848 (2013).
Masutani, H., Yoshihara, E., Masaki, S., Chen, Z. & Yodoi, J. Thioredoxin binding protein (TBP)-2/Txnip and alpha-arrestin proteins in cancer and diabetes mellitus. J. Clin. Biochem. Nutr. 50, 23–34 (2012).
Biaglow, J. E. & Miller, R. A. The thioredoxin reductase/thioredoxin system: novel redox targets for cancer therapy. Cancer Biol. Ther. 4, 6–13 (2005).
Jiang, N. et al. Thioredoxin-interacting protein: a new therapeutic target in bone metabolism disorders. Front. Immunol. 13, 955128 (2022).
Cao, X., He, W., Pang, Y., Cao, Y. & Qin, A. Redox-dependent and independent effects of thioredoxin interacting protein. Biol. Chem. 401, 1215–1231 (2020).
Bedarida, T. et al. Resveratrol decreases TXNIP mRNA and protein nuclear expressions with an arterial function improvement in old mice. J. Gerontol. A Biol. Sci. Med Sci. 71, 720–729 (2016).
Bedarida, T. et al. Reduced endothelial thioredoxin-interacting protein protects arteries from damage induced by metabolic stress in vivo. FASEB J. 32, 3108–3118 (2018).
Miyahara, H. et al. Thioredoxin interacting protein protects mice from fasting induced liver steatosis by activating ER stress and its downstream signaling pathways. Sci. Rep. 12, 4819 (2022).
Fang, S. et al. High glucose condition upregulated Txnip expression level in rat mesangial cells through ROS/MEK/MAPK pathway. Mol. Cell. Biochem. 347, 175–182 (2011).
Masutani, H., Ueda, S. & Yodoi, J. The thioredoxin system in retroviral infection and apoptosis. Cell Death Differ. 12, 991–998 (2005).
Pan, M., Zhang, F., Qu, K., Liu, C. & Zhang, J. TXNIP: a double-edged sword in disease and therapeutic outlook. Oxid. Med. Cell. Longev. 2022, 7805115 (2022).
Wurstle, M. L., Laussmann, M. A. & Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 318, 1213–1220 (2012).
Nasoohi, S., Ismael, S. & Ishrat, T. Thioredoxin-interacting protein (TXNIP) in cerebrovascular and neurodegenerative diseases: regulation and implication. Mol. Neurobiol. 55, 7900–7920 (2018).
Minn, A. H., Hafele, C. & Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 146, 2397–2405 (2005).
Chen, J. et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 22, 3581–3594 (2008).
Perrone, L., Devi, T. S., Hosoya, K. I., Terasaki, T. & Singh, L. P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 1, e65 (2010).
Panse, M. et al. Palmitate and insulin counteract glucose-induced thioredoxin interacting protein (TXNIP) expression in insulin secreting cells via distinct mechanisms. PLoS ONE 13, e0198016 (2018).
Luo, B. et al. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front. Physiol. 8, 519 (2017).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Wang, Y. Y., Liu, X. L. & Zhao, R. Induction of Pyroptosis and Its Implications in Cancer Management. Front. Oncol. 9, 971 (2019).
Ye, J. et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 (2000).
Shen, J., Snapp, E. L., Lippincott-Schwartz, J. & Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 25, 921–932 (2005).
Yoshida, H. et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767 (2000).
Hu, H., Tian, M., Ding, C. & Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 9, 3083 (2018).
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. & Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001).
Hetz, C. & Papa, F. R. The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 (2018).
Rozpedek, W. et al. The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 16, 533–544 (2016).
Yan, W. et al. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl Acad. Sci. USA 99, 15920–15925 (2002).
Thielen, L. A. et al. Identification of an anti-diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metab. 32, 353–365 e358 (2020).
Xu, G., Chen, J., Jing, G. & Shalev, A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 61, 848–856 (2012).
Ovalle, F. et al. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat. Med. 24, 1108–1112 (2018).
Thielen, L. & Shalev, A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr. Opin. Endocrinol. Diabetes Obes. 25, 75–80 (2018).
Kibbe, C., Chen, J., Xu, G., Jing, G. & Shalev, A. FOXO1 competes with carbohydrate response element-binding protein (ChREBP) and inhibits thioredoxin-interacting protein (TXNIP) transcription in pancreatic beta cells. J. Biol. Chem. 288, 23194–23202 (2013).
Cha-Molstad, H., Saxena, G., Chen, J. & Shalev, A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J. Biol. Chem. 284, 16898–16905 (2009).
Chau, G. C. et al. mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress. J. Cell Biol. 216, 2091–2105 (2017).
Chen, J., Saxena, G., Mungrue, I. N., Lusis, A. J. & Shalev, A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 57, 938–944 (2008).
Kovesdy, C. P. Epidemiology of chronic kidney disease: an update 2022. Kidney Int. Suppl. (2011) 12, 7–11 (2022).
Djudjaj, S. & Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 65, 16–36 (2019).
Liu, Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 69, 213–217 (2006).
Weinstein, J. R. & Anderson, S. The aging kidney: physiological changes. Adv. Chronic Kidney Dis. 17, 302–307 (2010).
Wu, M. et al. Thioredoxin-interacting protein deficiency ameliorates kidney inflammation and fibrosis in mice with unilateral ureteral obstruction. Lab Invest. 98, 1211–1224 (2018).
He, Q. et al. Role and mechanism of TXNIP in ageing-related renal fibrosis. Mech. Ageing Dev. 196, 111475 (2021).
Shah, A. et al. Thioredoxin-interacting protein deficiency protects against diabetic nephropathy. J. Am. Soc. Nephrol. 26, 2963–2977 (2015).
Song, S. et al. TXNIP deficiency mitigates podocyte apoptosis via restraining the activation of mTOR or p38 MAPK signaling in diabetic nephropathy. Exp. Cell Res. 388, 111862 (2020).
Ji, L. et al. FOXO1 overexpression attenuates tubulointerstitial fibrosis and apoptosis in diabetic kidneys by ameliorating oxidative injury via TXNIP-TRX. Oxid. Med. Cell Longev. 2019, 3286928 (2019).
Ismael, S. et al. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer’s disease. Neurochem. Int. 148, 105104 (2021).
Li, L. et al. Thioredoxin-interacting protein (TXNIP) associated NLRP3 inflammasome activation in human Alzheimer’s disease brain. J. Alzheimers Dis. 68, 255–265 (2019).
Sbai, O. et al. Correction to: RAGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Abeta to mitochondria in microglia. Cell Death Dis. 13, 368 (2022).
Wang, C. Y. et al. Dl-3-n-Butylphthalide Inhibits NLRP3 Inflammasome and Mitigates Alzheimer’s-Like Pathology via Nrf2-TXNIP-TrX Axis. Antioxid. Redox Signal 30, 1411–1431 (2019).
Su, C. J. et al. Thioredoxin-interacting protein induced alpha-synuclein accumulation via inhibition of autophagic flux: implications for Parkinson’s disease. CNS Neurosci. Ther. 23, 717–723 (2017).
Zeng, R. et al. MicroRNA-135b alleviates MPP(+)-mediated Parkinson’s disease in in vitro model through suppressing FoxO1-induced NLRP3 inflammasome and pyroptosis. J. Clin. Neurosci. 65, 125–133 (2019).
Masutani, H. Thioredoxin-interacting protein in cancer and diabetes. Antioxid. Redox Signal. 36, 1001–1022 (2022).
Melone, M. A. B. et al. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm. Res. 35, 44 (2018).
Li, A. et al. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell Endocrinol. 434, 36–47 (2016).
Tang, G. et al. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE(-/-) mice. Biomed. Pharmacother. 119, 109410 (2019).
Jia, Y. et al. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 32, 101534 (2020).
Li, T. et al. W2476 ameliorates beta-cell dysfunction and exerts therapeutic effects in mouse models of diabetes via modulation of the thioredoxin-interacting protein signaling pathway. Acta Pharmacol. Sin. 38, 1024–1037 (2017).
Wang, W. et al. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to alleviate inflammation and lipid accumulation in diabetic rats. Br. J. Pharmacol. 169, 1352–1371 (2013).
Wang, S., Zhao, X., Yang, S., Chen, B. & Shi, J. Salidroside alleviates high glucose-induced oxidative stress and extracellular matrix accumulation in rat glomerular mesangial cells by the TXNIP-NLRP3 inflammasome pathway. Chem. Biol. Interact. 278, 48–53 (2017).
Zheng, T. et al. Salidroside attenuates high-fat diet-induced nonalcoholic fatty liver disease via AMPK-dependent TXNIP/NLRP3 pathway. Oxid. Med. Cell Longev. 2018, 8597897 (2018).
Amin, F. M., Abdelaziz, R. R., Hamed, M. F., Nader, M. A. & Shehatou, G. S. G. Dimethyl fumarate ameliorates diabetes-associated vascular complications through ROS-TXNIP-NLRP3 inflammasome pathway. Life Sci. 256, 117887 (2020).
Li, P., Chen, D. & Huang, Y. Fisetin administration improves LPS-induced acute otitis media in mouse in vivo. Int. J. Mol. Med. 42, 237–247 (2018).
Acknowledgements
We would like to thank Dr. Ying Maggie Chen and Dr. Kyoung-Jin Min for their contributions to this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, EH., Park, SJ. TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Exp Mol Med 55, 1348–1356 (2023). https://doi.org/10.1038/s12276-023-01019-8
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-023-01019-8
This article is cited by
-
Organelle stress in NLRP3 inflammasome: a central mediator of neurodegenerative diseases
Molecular Neurodegeneration (2026)
-
SATB1 regulates TXNIP inhibiting invasion and infiltration in T-cell acute lymphoblastic leukemia
Cancer Gene Therapy (2026)
-
Thioredoxin-Binding protein 2 and apoptosis Signal-Regulating kinase 1: redox stress sensors and therapeutic targets in hepatocellular carcinoma
Medical Oncology (2026)
-
Effect of Verapamil on Blood Glucose in Type 1 and Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis
Cardiovascular Drugs and Therapy (2026)
-
Chronic Oxaliplatin Treatment Induces CIPN in Mice via Activation of the TXNIP Pathway
Molecular Neurobiology (2026)
