
Abstract
Background
Emerging evidence implies a link between high pediatric body mass index (BMI) and an increased risk of multiple sclerosis (MS). However, previous research suggests this association is only present for adolescent obesity and not childhood obesity. The present study aimed to assess the association between pediatric obesity and risk of developing MS, and to investigate if degree of obesity and age at obesity treatment initiation affects the risk. In a subgroup, response to obesity treatment on MS risk was assessed.
Methods
In this cohort study, patients aged 2–19 years from the Swedish Childhood Obesity Treatment Register (BORIS), and matched individuals from the general population were followed prospectively. MS was identified through the National Patient Register. Hazard ratios (HR) adjusted for parental MS were calculated.
Results
The study included 21,652 individuals with pediatric obesity and 102,187 general population comparators. The median age at follow-up was 21 (Q1, Q3 18, 25) years. The adjusted HR (95% CI) for developing MS in the pediatric obesity cohort was 2.28 (1.45–3.58). In stratified analyses, obesity class I was not associated with MS, HR = 1.34 (0.64–2.81), while the association between obesity class II and MS was strengthened, HR = 3.42 (1.89–6.19). MS was associated with both childhood obesity, HR = 3.16 (1.12–8.87), and adolescent obesity, HR = 2.12 (1.28–3.51). A decrease in BMI SDS was not associated with lower likelihood of MS, HR = 1.09 (0.92–1.29) per 0.25 BMI SDS unit decrease.
Conclusions
Both childhood and adolescent obesity are associated with an increased risk of MS. Moreover, a dose-response relationship between the degree of obesity and the risk of future MS was indicated, while response to pediatric obesity treatment did not affect the association, highlighting the importance of preventing high degree of obesity early in life.
Similar content being viewed by others

Introduction
Children and adolescents with obesity face a plethora of complications, including comorbidities, psychosocial challenges, and even premature mortality [1,2,3,4]. Emerging evidence implies a link between high body mass index (BMI) in childhood or adolescence and an increased risk of multiple sclerosis (MS) [5, 6]. MS is a chronic inflammatory condition characterized by demyelination, stemming from an autoimmune response targeting myelin. MS typically manifests between 15 and 55 years of age, affecting women two to three times more often than men [7]. High BMI in young years is estimated to contribute to more than 10% of MS cases [8]. Yet, most studies evaluating the association between pediatric obesity and risk for MS are relatively small cross-sectional [9], have a retrospective design with self-reported weight data [10,11,12], have used solely genetic correlations [13, 14], or use pediatric weight data before the obesity epidemic [5]. In addition, previous research suggest that the association only is present for adolescent obesity and not childhood obesity [15,16,17]. By the age of 20, individuals with a BMI exceeding 27 kg/m² have been found to have twice the risk of developing MS compared to those with a BMI of 18.5–21 kg/m² [6]. Further, there is lacking evidence on whether degree of obesity affects the risk of developing MS.
Therefore, we aimed to prospectively evaluate the risk of developing MS in a large cohort of pediatric patients with obesity and general population comparators, and to investigate if degree of obesity and age at obesity treatment initiation affects the association. Additionally, we aimed to evaluate whether response to obesity treatment affected the risk of MS.
Methods
Study population
In this dynamic cohort study, we included patients aged 2–19 years with obesity [18] enrolled in the Swedish Childhood Obesity Treatment Register (BORIS) between January 1997 and December 2020. BORIS is a national prospective register where children and adolescents receiving obesity treatment in Sweden are recorded [19]. The treatment primarily involves behavioral lifestyle modification. During the obesity treatment period of the current study, the new generation anti-obesity medications was not approved for adolescents in Sweden. Therefore, no patients received pharmacological treatment for obesity. Swedish law and regulations currently allow clinicians to register their patients in BORIS unless the patient chooses to opt-out. For each patient in BORIS, up to five individuals from the general population were contemporary matched on sex, year of birth, and residential area from the Total Population Register, held by Statistics Sweden [20]. Exclusion criteria were secondary obesity (craniopharyngioma), genetic syndromes (e.g. Mb Down, Prader Willi, and Laurence Moon Biedl, see Supplementary Table 1 for full list), and emigrated, deceased, or MS diagnosis before 15 years of age or before obesity treatment initiation. Flowchart of inclusion is presented in Fig. 1. Individuals were followed from obesity treatment initiation, or from 15 years of age if obesity treatment was initiated earlier, until MS diagnosis, death, emigration, or August 2023, whichever came first. Date of death was retrieved from the Cause of Death Register and date of emigration was provided by Statistics Sweden. The study was approved by the regional ethical review board in Stockholm (No. 2016/ 922-31/1 and amendment 2020-02707).

Biological plausible values include BMI 15–90 kg/m2, and height Z-score between −5.0 and 5.5. No available follow-up includes individuals under 15 years of age, deceases or emigrated at start of follow-up.
Variables and definitions
For all included individuals, MS was identified through the Swedish National Patient Register, held by the National Board of Health and Welfare, using International Statistical Classification of Diseases 10th revision code G35. The certainty of MS diagnoses in the National Patient Register is very high (>90%) [21].
Since approximately 30% of MS risk is genetic [22], data of biological parents to individuals in the obesity cohort and the general population comparators were identified and retrieved from Statistics Sweden. Missing data of biological parents, e.g. internationally adopted children, was present in 0.56% in the obesity cohort and 0.92% among the general population comparators. Heredity for MS was deemed if any of the biological parents had MS identified in the National Patient Register.
Age was calculated from the date of birth, which, along with sex, was retrieved from Statistics Sweden. Age at pediatric obesity treatment initiation was categorized into two groups; childhood (2–9.9 years) and adolescence (10–19 years) [23]. Degree of obesity was categorized as class I and class II obesity according to International Obesity Task Force criteria [18]. Obesity treatment response was assessed as body mass index standard deviation score (BMI SDS) [18] reduction (per 0.25 unit).
Statistical analyses
Descriptive characteristics are presented with proportions, mean and standard deviation (SD), or median and quintile 1 (Q1) and quintile 3 (Q3). Unadjusted incidence rate and associated 95% confidence interval (CI) as well as Cox proportional hazard ratio (HR) and 95% CI adjusted for heredity was calculated with attained age as time scale. Proportional hazard assumption was confirmed. Analyses were further stratified for sex, degree of obesity at obesity treatment initiation (obesity class I and obesity class II), and age at obesity treatment initiation. Given the higher validity of repeated MS diagnoses in the National Patient Register [21], the HR was also estimated for repeated diagnosis of MS in sensitivity analyses, requiring at least one diagnosis to be the main diagnosis. Among individuals in the pediatric obesity cohort, treatment response was assessed among those with at least one year (365 days) of treatment data. Covariate in the sub-analysis included parental MS. Missing data was handled using listwise deletion. All analyses were performed in SAS statistical software (version 9.4, Cary, NC, USA).
Results
We included 21,652 individuals from the pediatric obesity cohort and 102,187 general population comparators. In the obesity cohort, 54% were males, the median (Q1, Q3) age at obesity treatment initiation was 11.5 (9.0, 13.9) years, and the distribution of obesity class I and class II were 58% and 42% respectively. The median age at start of follow-up was 15.0 (15.0, 15.0) years and follow-up time was 5.6 (3.0, 9.6) years, corresponding to 20.6 (18.0, 24.6) years of age. Parental MS was present in 0.99% in the obesity cohort and 0.69% in the general population comparators, p < 0.0001.
Over 144,970 person-years, 0.13% (n = 28) developed MS in the pediatric obesity cohort, compared to 0.06% (n = 58) over 698,513 person-years among the general population comparators, p = 0.0002. The mean (SD) age of MS diagnosis was comparable between the groups: 23.4 (5.0) years in the obesity cohort versus 22.8 (5.9) years in the general population comparators, p = 0.64. The distribution of sexes was similar across both groups: In the obesity cohort, females accounted for 64.3% (n = 18) of MS cases, compared to 65.5% (n = 38) the general population comparators, p = 0.91.
The unadjusted incidence rate of MS per 100,000 person years was 19.3 (95% CI: 13.3–28.0) in the obesity cohort and 8.3 (6.4–10.7) among the general population comparators, p = 0.0002. By the age of 30, the estimated cumulative incidence of MS was 0.39% in the obesity cohort versus 0.13% in general population comparators, as illustrated in Fig. 2. Cox proportional hazard regression, adjusted for MS heredity, revealed that the risk of developing MS was more than twice in the pediatric obesity cohort compared to the general population, HR = 2.28 (95% CI 1.45–3.58), p = 0.0003, Table 1.

Number of individuals at each age are displayed per group below the graph.
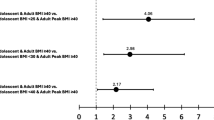
There were no interactions between the exposure (pediatric obesity cohort or general population comparators) and sex (p = 0.90), age at treatment initiation (p = 0.66) on the risk of MS. In stratified analyses, obesity class I was not associated with MS, HR = 1.47 (0.73–2.97), while the association between obesity class II and MS was strengthened, HR = 3.11 (1.85–5.22). Stratified analyses are provided in Fig. 3.

Analyses were adjusted for parental MS. Dashed line- represents HR = 1.0.
In sensitivity analyses, repeated MS diagnosis was assessed. Seven individuals (three in the obesity cohort and four among general population comparators) with single diagnosis of MS were excluded. Of them, two had an MS diagnosis within two months before the end of follow-up. The association between pediatric obesity and risk of MS remained, adjusted HR = 2.18 (1.36–3.51), p = 0.0013.
Among individuals in the pediatric obesity cohort, data of at least one year of treatment was available in 64.6% (n = 13,991), of whom 0.13% (n = 18) was diagnosed with MS, excluding one patient diagnosed with MS prior to end of obesity treatment. The median (Q1, Q3) treatment duration was 3.1 (1.9, 5.0) years with a treatment response of −0.14 (−0.51, 0.13) BMI SDS units. The median time to MS diagnosis after last registered visit to pediatric obesity treatment was 8.1 (5.7, 10.8) years. In the sub-analysis, adjusted for parental MS, a decrease in BMI SDS was not associated with higher likelihood of MS, HR = 1.09 (0.92–1.29) per 0.25 BMI SDS unit decrease. Obesity treatment duration did not affect the outcome. Of those who developed MS, 17% obtained obesity remission, 28% had obesity class I and 55% had obesity class II the last registered visit to obesity treatment.
Discussion
The current prospective nationwide cohort study shows that obesity prior to 10 years of age more than doubles the risk of MS regardless of MS heredity, compared to general population comparators. This study also confirms that obesity in adolescence increases the risk for MS. Additionally, this study suggests a dose-response relationship between degree of obesity and the risk for MS.
In the present study, the estimated risk of obesity on MS was more than double, which is in line with previous studies in adults [6, 15, 17] and studies with retrospective design with self-reported or estimated weight data from pediatric years [10,11,12]. The proportion of males and females that were diagnosed with MS were similar, which was mirrored in the hazard ratio estimates, indicating that obesity affects MS equally in males and females.
This study found that, compared to the general population, class I obesity did not significantly increase the risk for MS, while class II obesity showed a pronounced risk, indicating a dose-response relationship. The underlying mechanism may be attributed to obesity’s role in inducing a persistent, low-grade inflammatory state characterized by elevated levels of proinflammatory cytokines [2]. Type of diet and microbiota may also contribute both to obesity and MS [7]. A study in mice suggest that high-fat diet induced obesity causes a pro-inflammatory micro-environment which promotes CNS inflammation through proliferation and activation of encephalitogenic T cells into the CNS [24]. A cross-sectional study in humans suggest that leptin, typically high in individuals with obesity, promotes autoreactive T-cell proliferation and proinflammatory cytokine secretion [10]. Further, obesity-induced gut microbiota dysregulation has been shown to shift the balance between proinflammatory and anti-inflammatory responses, suggesting that it might act as a link between obesity and MS [22, 25]. Consequently, the proinflammatory condition in obesity most likely amplifies the immune response and thereby increases the risk of developing autoimmune inflammatory disorders, such as MS.
Some case reports have identified individuals developing symptoms of MS shortly after immense weight loss following bariatric surgery [26,27,28]. Conversely, anti-obesity medications based on glucagon-like peptide-1 receptor agonists appear to have a protective effect against the development of MS [29]. This suggests that weigh loss itself may not be a direct contributor to MS. In the present study, modest weigh loss after lifestyle behavioral treatment was not associated with increased risk of MS several year later. Over half of those who developed MS in the present study had class II obesity at their last recorded treatment visit, making direct comparisons to previously published reports challenging. Nevertheless, since obesity itself remains a significant factor contributing to MS risk, as demonstrated in this study, and is associated with MS progression and relapse [30, 31], prevention of high degree of obesity in childhood and adolescence is warranted.
Previous epidemiological research suggest that the association between early life high BMI and MS only is present for adolescent obesity and not childhood obesity [15,16,17]. However, studies using Mendelian randomization with estimated childhood BMI have suggested an effect of childhood BMI on subsequent MS risk [13, 14]. The current study extends these findings by using clinically obtained anthropometric measured in childhood. Consequently, the association between obesity and elevated risk of MS is shown to not be limited to adolescence but extends to childhood. Since treatment for pediatric obesity rarely results in obesity remission [19], it is plausible that early exposure to obesity leads to longer obesity duration. This may lead to a long-term pro-inflammatory response which could act as a catalyst, activating genes that are associated with the development of MS.
Parental MS was more common in the pediatric obesity cohort, which might give rise to a hypothesis of shared predisposition for MS and early obesity onset in the offspring. However, our study design prevents us from drawing any conclusions about causality. A genetic study, using Mendelian randomization, found little evidence of genetic pleiotropy between genes associated with high BMI in childhood and MS later in life [13], which contradicts a shared genetic predisposition. Another plausible explanation for this observation could be the strong hereditary nature of obesity. Consequently, parental BMI may be higher in the obesity cohort, potentially elevating the prevalence of parental MS. Future research should explore the potential synergistic relationship between MS heredity and childhood obesity in increasing MS risk. This research is vital for unraveling the intricate genetic and environmental interactions contributing to MS and obesity.
Despite the large-scale longitudinal study with individual-level data, some limitations should be acknowledged. Firstly, there are several risk factors known to be associated with MS that were not possible to control for, such as smoking, Epstein-Barr viral infection, and low vitamin D levels [16]. Secondly, no genetic data was available, but we were able to adjust for parental MS. Thirdly, as BORIS only includes anthropometric data from clinical visits, obesity duration prior to treatment cannot be determined. Fourthly, obesity status in the control group was unknown, which could cause an underestimation of obesity as a risk factor for MS. Fifthly, the median age at follow-up was 20.8 years of age, which should be kept in mind since MS typically develops between 20 and 40 years of age. Yet, we could observe an effect of pediatric obesity on MS.
Conclusions
Both childhood and adolescent obesity are associated with an increased risk of MS. Moreover, a dose-response relationship between the degree of obesity and the risk of future MS was indicated. Given that weight reduction decreases obesity-induced inflammation, a key factor believed to mediate the link between obesity and MS, our study emphasizes the importance of preventing high degree of obesity early in life.
Data availability
Patient-level data cannot be shared publicly because of third-party data. Given that an ethical approval is obtained, any individual may apply for data from Statistics Sweden via information@scb.se, the Swedish National Board of Health and Welfare via registerservice@socialstyrelsen.se, and the Swedish Childhood Obesity Treatment Register via http://www.e-boris.se/in-english/.
References
Lindberg L, Danielsson P, Persson M, Marcus C, Hagman E. Association of childhood obesity with risk of early all-cause and cause-specific mortality: A Swedish prospective cohort study. PLoS Med. 2020;17:e1003078.
Marcus C, Danielsson P, Hagman E. Pediatric obesity-Long-term consequences and effect of weight loss. J Intern Med. 2022;292:870–91.
Lindberg L, Hagman E, Danielsson P, Marcus C, Persson M. Anxiety and depression in children and adolescents with obesity: a nationwide study in Sweden. BMC Med. 2020;18:30.
Lindberg L, Persson M, Danielsson P, Hagman E, Marcus C. Obesity in childhood, socioeconomic status, and completion of 12 or more school years: a prospective cohort study. BMJ Open. 2021;11:e040432.
Munger KL, Bentzen J, Laursen B, Stenager E, Koch-Henriksen N, Sorensen TI, et al. Childhood body mass index and multiple sclerosis risk: a long-term cohort study. Mult Scler. 2013;19:1323–9.
Hedstrom AK, Olsson T, Alfredsson L. High body mass index before age 20 is associated with increased risk for multiple sclerosis in both men and women. Mult Scler. 2012;18:1334–6.
Mandato C, Colucci A, Lanzillo R, Staiano A, Scarpato E, Schiavo L et al. Multiple Sclerosis-Related Dietary and Nutritional Issues: An Updated Scoping Review with a Focus on Pediatrics. Children. 2023;10:1022.
Pakpoor J, Schmierer K, Cuzick J, Giovannoni G, Dobson R. Estimated and projected burden of multiple sclerosis attributable to smoking and childhood and adolescent high body-mass index: a comparative risk assessment. Int J Epidemiol. 2021;49:2051–7.
Milles P, De Filippo G, Maurey H, Tully T, Deiva K, KidBiosep. Obesity in Pediatric-Onset Multiple Sclerosis: A French Cohort Study. Neurol Neuroimmunol Neuroinflamm. 2021;8:e1044.
Marrodan M, Farez MF, Balbuena Aguirre ME, Correale J. Obesity and the risk of Multiple Sclerosis. The role of Leptin. Ann Clin Transl Neurol. 2021;8:406–24.
Hedstrom AK, Lima Bomfim I, Barcellos L, Gianfrancesco M, Schaefer C, Kockum I, et al. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology. 2014;82:865–72.
Gianfrancesco MA, Acuna B, Shen L, Briggs FB, Quach H, Bellesis KH, et al. Obesity during childhood and adolescence increases susceptibility to multiple sclerosis after accounting for established genetic and environmental risk factors. Obes Res Clin Pr. 2014;8:e435–47.
Harroud A, Mitchell RE, Richardson TG, Morris JA, Forgetta V, Davey Smith G, et al. Childhood obesity and multiple sclerosis: A Mendelian randomization study. Mult Scler. 2021;27:2150–8.
Jacobs BM, Noyce AJ, Giovannoni G, Dobson R. BMI and low vitamin D are causal factors for multiple sclerosis: A Mendelian Randomization study. Neurol Neuroimmunol Neuroinflamm. 2020;7:e662.
Hedstrom AK, Olsson T, Alfredsson L. Body mass index during adolescence, rather than childhood, is critical in determining MS risk. Mult Scler. 2016;22:878–83.
Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. 2017;13:25–36.
Munger KL, Chitnis T, Ascherio A. Body size and risk of MS in two cohorts of US women. Neurology. 2009;73:1543–50.
Cole TJ, Lobstein T. Extended international (IOTF) body mass index cut-offs for thinness, overweight and obesity. Pediatr Obes. 2012;7:284–94.
Hagman E, Danielsson P, Lindberg L, Marcus C, Committee BS. Paediatric obesity treatment during 14 years in Sweden: Lessons from the Swedish Childhood Obesity Treatment Register-BORIS. Pediatr Obes. 2020;15:e12626.
Ludvigsson JF, Almqvist C, Bonamy AK, Ljung R, Michaelsson K, Neovius M, et al. Registers of the Swedish total population and their use in medical research. Eur J Epidemiol. 2016;31:125–36.
Murley C, Friberg E, Hillert J, Alexanderson K, Yang F. Validation of multiple sclerosis diagnoses in the Swedish National Patient Register. Eur J Epidemiol. 2019;34:1161–9.
Montgomery TL, Peipert D, Krementsov DN. Modulation of multiple sclerosis risk and pathogenesis by the gut microbiota: Complex interactions between host genetics, bacterial metabolism, and diet. Immunol Rev. 2024;325:131–51.
Sawyer SM, Azzopardi PS, Wickremarathne D, Patton GC. The age of adolescence. Lancet Child Adolesc Health. 2018;2:223–8.
Ji Z, Wu S, Xu Y, Qi J, Su X, Shen L. Obesity Promotes EAE Through IL-6 and CCL-2-Mediated T Cells Infiltration. Front Immunol. 2019;10:1881.
Samara A, Cantoni C, Piccio L, Cross AH, Chahin S. Obesity, gut microbiota, and multiple sclerosis: Unraveling the connection. Mult Scler Relat Disord. 2023;76:104768.
Alanazy MH, Alomar MA, Aljafen BN, Muayqil TA. Multiple sclerosis and myasthenia gravis following severe weight loss. Neuroscience. 2018;23:158–61.
Asakly S, Magen-Rimon R, Ighbariya A, Marjih-Shallufi M, Ben-Porat T, Ravid S, et al. Bariatric Surgery-Associated Myelopathy. Obes Facts. 2021;14:431–9.
Bitarafan S, Amani K, Sahraian MA, Sarraf P, Soltani D, Moghadasi AN, et al. The first attack of multiple sclerosis presented immediately after voluntary and intensive weight loss: A case series. Iran J Neurol. 2017;16:41–42.
Shirani A, Cross AH, Stuve O. Exploring the association between weight loss-inducing medications and multiple sclerosis: insights from the FDA adverse event reporting system database. Ther Adv Neurol Disord. 2024;17:17562864241241383.
Marrie RA. Comorbidity in multiple sclerosis: implications for patient care. Nat Rev Neurol. 2017;13:375–82.
Tettey P, Simpson S, Taylor B, Ponsonby AL, Lucas RM, Dwyer T, et al. An adverse lipid profile and increased levels of adiposity significantly predict clinical course after a first demyelinating event. J Neurol Neurosurg Psychiatry. 2017;88:395–401.
Acknowledgements
The authors would like to thank all the pediatric obesity treatment units for their contribution to the BORIS register. EH was supported by Åke Wiberg foundation (M22-0183), The Center for Innovative Medicine (FoUI-988950), and Ollie and Elof Ericsson’s foundation. RRP was supported by the Freemason Foundation for Children’s Welfare. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
E Hagman: Project management, literature review, data analysis and interpretation, drafting the initial manuscript. RR Putri, P Danielsson, and C Marcus: Interpretation of results and critical revision of the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki and approved by the regional ethical committee in Stockholm, Sweden (reference numbers 2016/922-31/1 and 2020-02707). The need for patient consent was waived due to anonymized register data.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hagman, E., Putri, R.R., Danielsson, P. et al. Pediatric obesity and the risk of multiple sclerosis: a nationwide prospective cohort study. Int J Obes 49, 1031–1036 (2025). https://doi.org/10.1038/s41366-025-01727-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41366-025-01727-3
This article is cited by
-
Genetic pleiotropy underlying obesity and autoimmune disorders: a large-scale cross-trait gwas analysis in European ancestry populations
Journal of Translational Medicine (2025)
