
Abstract
Obesity promotes a range of associated conditions, including hearing impairment; however, mechanisms are lacking. Self-evidently, an insult on any cellular constituent of the auditory organ can disrupt hearing. Here, using the mouse auditory cell line, HEI-OC1, we provide insights into adipose-associated ototoxicity. Adipose extracts from mice with obesity, diet- or genetically induced, suppress HEI-OC1’s survival and ATP generation. Proteomic profiling shows an upregulation of the inflammatory response pathway and proteins such as Podoplanin and Low-density lipoprotein receptor. Likewise, the Programmed cell death 4 (PDCD4) protein was induced. These results correspond to a downregulation of glycolysis and oxidative phosphorylation but an upregulation of the G2/M checkpoint. Additionally, pathways such as IL6-JAK-STAT3, IL2-STAT5, interferon gamma response, cholesterol response, bile acid metabolism, RAS, Apoptosis, and TGF-β were upregulated. Furthermore, the adipose extracts cause cellular morphological changes consistent with cells under stress. Functional assays point to alterations in levels of proteins related to calcium and ER homeostasis/stress. The ER-resident protein SARAF, an inhibitor of calcium overfilling, is among the proteins markedly downregulated. GRP78 protein levels increased, suggesting ER/calcium stress. Finally, Thapsigargin impairs HEI-OC1 survival, reminiscent of the effect of the adipose tissue extracts. Our analyses warrant further exploration of inflammation and ER/calcium stress in connection to obesity-associated ototoxicity.
Similar content being viewed by others


Introduction
Ototoxicity precedes Hearing loss (HL), a debilitating disease affecting individuals across all ages, and is characterized by a disruption of sound transmission through the cochlea. Globally, there are currently ~1.5 billion people with HL, and the World Health Organization (WHO) projects nearly 2.5 billion people to have some degree of HL by 2050, and 700 million to need hearing rehabilitation. The difficulty in expanding the currently limited knowledge about HL remains compounded by the lack of understanding of its relationship with potential risk factors such as obesity. The latter is one of the top global health crises of our time, causing more than 2.8 million deaths each year, per statistics from the WHO. Animal studies suggest an association of obesity with auditory decline [1]. Likewise, population-based studies linked obesity to a range of otorhinolaryngological diseases, including HL [2,3,4]. Yet, the molecular mechanisms underlying the ontogeny and progression of obesity-associated ototoxicity and HL are lacking. Furthermore, HL contributes to other uncharacterized conditions, such as cognitive decline and imbalance, and thus carries enormous socioeconomic impacts. Here, we explore mechanistic insights into adipose-associated ototoxicity in a murine auditory cell line, the House Ear Institute-Organ of Corti 1 (HEI-OC1), widely used in research related to ototoxicity [5].
Materials and methods
Cell culture and reagents
The HEI-OC1 cell line was a gift from Dr. Dominic Cosgrove’s lab. Cells were maintained in DMEM/F-12 medium (cat# 30-2006; ATCC) supplemented with 5%–10% fetal bovine serum (FBS, cat# 16000044; Thermo Scientific), 100U/ml penicillin, 0.1 mg/ml streptomycin, and 0.29 mg/ml L-glutamine. To propagate HEI-OC1, the cells were maintained at 33 °C and the culture medium supplemented with 10 U/ml mouse recombinant γ-interferon (cat# RD-8617, Invitrogen). Sub-confluent cultures were dissociated with 0.05% trypsin, 0.02% EDTA. TNF-α (cat# 315-01A-5UG) was from Peprotech, and Oleic acids (cat#: 031997.06) were from Thermo Scientific. Palmitic acids (cat# P0500-10G) and Linoleic acids (cat# L1376-10G) were purchased from Sigma, while Thapsigargin (cat# 12758S) was from Cell Signaling Technologies.
Animal studies
20-week-old lean C57bl6/6J mice (Stock# 380056) or 20-week old C57BL/J6 mice with diet-induced obesity, DIO, (Stock# 380050) or 10-week old B6.Cg-Lepob/J mice, herein referred to as mice with genetically induced obesity, ob/ob (Stock# 000632), were purchased from Jackson Labs. The lean mice were fed a low-fat diet (10% fat) with the same protein content as the DIO mice fed a high-fat diet (60% fat). The mice were briefly held at the animal facility at Boys Town National Research Hospital under IACUC protocol #23-05. The goal was to use tissues from age-matched male and female mice. However, due to the unavailability of female mice for each group above (control, DIO, and ob/ob), the mice used were all male.
Adipose tissue conditioned media preparation
Retroperitoneal and epididymal adipose tissues were collected from each group above, washed with PBS, and cut into small pieces. Serum-free media was added to the tissues (1 ml of media/100 mg of tissue) and incubated for ~22 h in a cell culture incubator. The conditioned media (CM) was collected, centrifuged at 1900 × g for 30 min, filtered through a 0.22 µm filter, aliquoted into 1.5 ml Eppendorf tubes, and stored at −80 °C until use. For treatment, CM was diluted and contained 3% FBS.
Survival assay by Alamar Blue reduction assay
Alamar Blue (cat#: BUF012A; Bio-Rad, Hercules, CA) was used. Briefly, after treating the cells as indicated, the media was replaced with maintenance media containing 10% Alamar Blue for 2–4 h, and the absorbance was read at 570 nm and 600 nm. Then, the following formula was used to calculate the change in Alamar Blue reduction, a measure of cytotoxicity according to the manufacturer’s protocol:
O1 = molar extinction coefficient (E) of oxidized alamarBlue (Blue) at 570 nm; O2 = E of oxidized alamarBlue at 600 nm; R1 = E of reduced alamarBlue (Red) at 570 nm; R2 = E of reduced alamarBlue at 600 nm; A1 = absorbance of test wells at 570 nm; A2 = absorbance of test wells at 600 nm; N1 = absorbance of negative control well (media plus alamarBlue but no cells) at 570 nm; N2 = absorbance of negative control well (media plus alamarBlue but no cells) at 600 nm.
ATP assay
The ATPlite (cat# 6016941) from Revvity (Waltham, MA) was used. In brief, 50 μL of mammalian cell lysis solution was added to cells in 100 μL per well in a microplate and shaken for 5 min on an orbital shaker at 700 rpm. Then, 50 μL substrate solution was added to the wells and shaken for 5 min in an orbital shaker at 700 rpm. The plate was dark-adapted for 10 min and the luminescence was measured using the Spectramax iD5 Plate Reader.
Proteomics and analyses
Cells, seeded in triplicate, were treated with adipose tissue CM from lean mice or mice with obesity, collected by trypsinization and washed in PBS. Aliquots from each replicate were lysed in 1X RIPA buffer (Cat# 9806S, Cell Signaling) for western blot (below). Proteomic profiling was assessed on the remaining cell pellets (3 replicates each) using the Proteome Discoverer 2.3 software at the Mass Spectrometry Services branch of the Auditory and Vestibular Technology Core at Creighton University, Omaha, NE, supported by Creighton University and grants GM103427, GM139762 from the National Institute of General Medical Science (NIGMS). Gene set enrichment was carried out using the GSEA software, and a volcano plot was performed using the “ggplot2” package in R. All statistical analyses were performed using Graphpad Prism 10.4.0. Student’s t-test was used to test the significance between two groups.
Western blot
Cell lysate was quantified using the Pierce BCA Protein Assay (cat# 23227, Thermo Fisher). Thirty micrograms of proteins were separated on an SDS-PAGE gel, transferred to a PVDF membrane (cat# 1620174, Bio-Rad), blocked in 5% milk, and incubated with primary antibodies overnight, followed by washing in TBS buffer and incubation in appropriate secondary antibodies for 1 h at RT. The blot was washed and imaged on an Azure 600 imager, using Clarity Western ECL Substrate (cat# 1705060, Bio-Rad). Antibodies against β-actin (cat# 4967S; 1:3K dilution), GRP78/Bip (cat# 3177S; 1:1K dilution), and PDCD4 (cat# 9535S; 1:1K dilution) were purchased from Cell Signaling Technologies. Antibodies against mouse podoplanin, PDPN (cat# AF3244-SP; 1:4K), and mouse low-density lipoprotein receptor (LDLR) (cat# AF2255-SP; 1:4K) were from R&D Systems, along with the Rabbit Anti-Goat IgG HRP Affinity (cat# HAF017). The lyophilized PDPN and LDLR antibodies were reconstituted in PBS as per the vendor’s instructions, with no vortexing.
Droplet digital PCR (ddPCR)
Total RNA was extracted from the treated HEI-OC1 cells using the QIAzol reagents (cat# 79306, Qiagen). ddPCR was carried out as previously described [6]. The following primer sets were used:
Mouse SARAF: CTGGAACGACCCTGACAGAAT (forward primer); AACACACTTCAACTGTGGGATAG (reverse primer).
Mouse β-actin: GGCTGTATTCCCCTCCATCG (forward primer); CCAGTTGGTAACAATGCCATGT (reverse primer).
Statistical analysis
One-way ANOVA or Student’s t-test was used where appropriate.
Results and discussion
Obesity is an inflammatory disorder featuring an accumulation of lipids in adipocytes, leading to adipocyte enlargement and adipose tissue biomass accrual. Accordingly, obesity is commonly associated with hypercholesterolemia and hypertriglyceridemia, which drive a range of human diseases. Circulating lipids are signaling molecules that alter cell and tissue function in the environment of obesity. Their pro-inflammatory response induces tissue remodeling or injury. Still, the environment with obesity is a hotbed of pathologies such as cancer, whereby secretion of the pro-inflammatory cytokines and lipids from the adipose tissue creates an inflamed microenvironment permissive of aggressive tumor cell growth. However, the mechanisms by which the same environment might impair other cell/tissue types, such as cochlear cells, remain largely unexplored. The cellular pathophysiology of cochlear dysfunction and hearing loss (HL) is emerging. It is widely accepted that genetic mutations or environmental factors such as noise or drug exposure can induce a dysfunction of the cochlea, leading to HL. Several cellular processes underlying cochlear cell dysfunction/death and HL have been elucidated, such as reactive oxygen species release [7], calcium overload [8], and endoplasmic reticulum (ER) stress [9,10,11,12]. However, there are currently very limited studies with mechanistic insights into obesity-related ototoxicity.
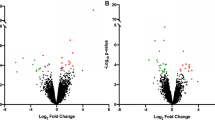
The murine auditory cell line, HEI-OC1, has been extensively used as a model cell line in studies related to auditory research and ototoxic agents [5], with currently close to 450 studies employing that cell model. Using those cells, we presented here analyses suggesting a correlation of adipose-associated auditory cell dysfunction with inflammation-related pathways in vitro. Treatment of HEI-OC1 cells with adipose tissue extracts from mice with diet- or genetically induced obesity reduced cell survival (Fig. 1A) and impaired ATP generation (Fig. 1A), hallmarks of dysfunction in cellular metabolism. Proteomic profiling of the treated cells revealed an upregulation of the inflammatory response pathway (Fig. 1C) as the second most significantly altered pathway. Podoplanin (PDPN), a cell surface glycoprotein implicated in chronic inflammatory diseases, and the LDLR involved in lipid signaling, showed the highest fold increase in that pathway (Fig. 1D, E) and were validated by western blot (Fig. 1F). These effects correspond with a downregulation of glycolysis (Fig. 1G) and oxidative phosphorylation (Fig. 1H), two major cellular bioenergetic pathways, but an upregulation of the G2M checkpoint pathway (Fig. 1I) associated with cell growth inhibition. PDPN was reported to be stimulated by circulating cytokines [13] such as those in obesity. Likewise, as a receptor, LDLR helps internalize circulating low-density lipoproteins (LDL) known as the “bad cholesterol,” as well as other free fatty acids [14]. These lipids can induce ototoxicity, especially in the context of obesity-driven hypercholesterolemia and hypertriglyceridemia, where high lipid/cholesterol internalization could lead to lipid/cholesterol overload, alteration in cell lipid metabolism, lipo-ototoxicity, and tissue dysfunction. For instance, an oxidized form of LDL was reported to be toxic to vascular endothelial cells [15, 16], suggesting that elevated LDL, such as that found in obesity, might also lead to tissue injury, including cochlear/auditory tissue injury. Our data indicate that free fatty acids impair HEI-OC1 cell function (Fig. 1J–L), aligning with the growing evidence that abnormal lipid metabolism predisposes to HL [17, 18]. Consistent with cell dysfunction, the proteomic dataset showed an induction of the Programmed cell death 4 (PDCD4) protein (Fig. 1E), validated by western blot (Fig. 1F). Additional upregulated pathways include IL6-JAK-STAT3 signaling, IL2-STAT5 signaling, interferon gamma response, cholesterol response, bile acid metabolism, RAS signaling, Apoptosis, and TGF-β signaling (Supplementary Fig. A). These pathways are related to cytokines, lipid signaling, MAPK activation, stress response, and cell death. Like the effect of free fatty acids (Fig. 1J–L), cytokines also impair HEI-OC1 cells (Fig. 1M).

Cells were treated with diluted extracts from the indicated adipose tissues for 24 h, followed by a survival assay by Alamar Blue (A), or ATP assay (A) or cell visualization under a microscope (B). DIO is short for diet-induced obesity; ob/ob is a reference to the genetically induced obesity. C–I Proteomic analysis following treatment with adipose extracts (DIO). GSEA showing key pathways alterations (C, D, G–I), Volcano plot (E) showing relevant proteins, validated by western blot (F). J–M HEI-OC1 cells were treated with Linoleic acids (LA), or Oleic acids (OA), or Palmitic acids (PA), 150 µM, or TNF-α (150 ng/ml) for 24 h, followed by a survival assay. N HEI-OC1 cells were exposed to adipose tissue extracts, followed by ddPCR with primers for the SARAF gene. O HEI-OC1 cells were treated with Thapsigargin (Tg, 150 nM) for 24 h, followed by a survival assay.
Exposure of the HEI-OC1 cells to the adipose extracts from the mice with obesity also induces cell morphological changes consistent with cells under stress (i.e., rounding) (Fig. 1B). Post-proteomics functional studies revealed that the adipose tissue extracts might impair HEI-OC1 cells by altering proteins related to calcium and ER homeostasis/stress. The proteomic analyses indicated that the ER-resident protein, SARAF (Store-Operated Calcium Entry Associated Regulatory Factor), is among the proteins markedly downregulated (Fig. 1E), which was monitored by digital droplet PCR (Fig. 1N) due to difficulty with antibody-based detection. The protein is a regulator of cytosolic calcium levels [19]. Deficiency in the SARAF protein was reported to induce metabolic changes that led to sarcopenic obesity [20], suggesting not just a negative association of SARAF protein levels with fat accumulation/obesity, but also a potential connection of SARAF deficiency to cell energy metabolism. The marked downregulation of SARAF upon exposure of the adipose tissue extract suggests an alteration in calcium signaling upon exposure of the adipose tissue extracts. Evaluation of GRP78, the putative ER-resident and a calcium-sensitive protein known to be induced by elevated cytosolic calcium levels [21], showed an increased level (Fig. 1F), further suggesting a disruption of calcium homeostasis in the HEI-OC1 cells upon exposure to the adipose tissue extracts from mice with obesity. Using a pharmacological agent (thapsigargin) widely employed to induce cytosolic calcium accumulation [22], we validated that the HEI-OC1 cells’ calcium disruption impairs cell survival (Fig. 1O), reminiscent of the effect of the adipose tissue extracts from the mice with diet- or genetically induced obesity (Fig. 1A). While prior research has established that acute ER stress contributes to ototoxicity and HL [10,11,12], co-treatment of the cells with adipose extracts and 4-phenylbutyric acid (4-PBA), an ER stress mitigator, did not indicate a significant improvement in ATP levels (Supplementary Fig. B). This may be due to the relatively weak induction of GRP78 expression (Fig. 1F), suggesting that the level of ER stress was not sufficiently robust to affect ATP production or to be mitigated by 4-PBA. Nonetheless, the well-documented link between acute ER stress, inflammation, and auditory cell dysfunction provides a solid foundation for future studies to further explore the mechanistic link between inflammation and ER/calcium to obesity-associated ototoxicity.
Data availability
All supporting data are available upon reasonable request to the corresponding author(s).
References
Kim SJ, Gajbhiye A, Lyu AR, Kim TH, Shin SA, Kwon HC, et al. Sex differences in hearing impairment due to diet-induced obesity in CBA/Ca mice. Biol Sex Differ. 2023;14:10.
Lalwani AK, Katz K, Liu YH, Kim S, Weitzman M. Obesity is associated with sensorineural hearing loss in adolescents. Laryngoscope. 2013;123:3178–84.
Lee JS, Kim DH, Lee HJ, Kim HJ, Koo JW, Choi HG, et al. Lipid profiles and obesity as potential risk factors of sudden sensorineural hearing loss. PLoS ONE. 2015;10:e0122496.
Krajewska Wojciechowska J, Krajewski W, Zatonski T. The association between ENT diseases and obesity in pediatric population: a systemic review of current knowledge. Ear Nose Throat J. 2019;98:E32–43.
Kalinec G, Thein P, Park C, Kalinec F. HEI-OC1 cells as a model for investigating drug cytotoxicity. Hear Res. 2016;335:105–17.
Tom WA, Chandel DS, Jiang C, Krzyzanowski G, Fernando N, Olou A, et al. Genotype characterization and MiRNA expression profiling in Usher syndrome cell lines. Int J Mol Sci. 2024;25:9993.
Bottger EC, Schacht J. The mitochondrion: a perpetrator of acquired hearing loss. Hear Res. 2013;303:12–9.
Fridberger A, Flock A, Ulfendahl M, Flock B. Acoustic overstimulation increases outer hair cell Ca2+ concentrations and causes dynamic contractions of the hearing organ. Proc Natl Acad Sci USA. 1998;95:7127–32.
Herranen A, Ikaheimo K, Lankinen T, Pakarinen E, Fritzsch B, Saarma M, et al. Deficiency of the ER-stress-regulator MANF triggers progressive outer hair cell death and hearing loss. Cell Death Dis. 2020;11:100.
Fujinami Y, Mutai H, Mizutari K, Nakagawa S, Matsunaga T. A novel animal model of hearing loss caused by acute endoplasmic reticulum stress in the cochlea. J Pharmacol Sci. 2012;118:363–72.
Hu J, Li B, Apisa L, Yu H, Entenman S, Xu M, et al. ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23(erl/erl) mutant mice. Cell Death Dis. 2016;7:e2485.
Li J, Akil O, Rouse SL, McLaughlin CW, Matthews IR, Lustig LR, et al. Deletion of Tmtc4 activates the unfolded protein response and causes postnatal hearing loss. J Clin Investig. 2018;128:5150–62.
Honma M, Minami-Hori M, Takahashi H, Iizuka H. Podoplanin expression in wound and hyperproliferative psoriatic epidermis: regulation by TGF-beta and STAT-3 activating cytokines, IFN-gamma, IL-6, and IL-22. J Dermatol Sci. 2012;65:134–40.
Bucci C, Seru R, Annella T, Vitelli R, Lattero D, Bifulco M, et al. Free fatty acids modulate LDL receptor activity in BHK-21 cells. Atherosclerosis. 1998;137:329–40.
Fei X, Cen X, Zhao R, Wang J, Cui H. PRMT5 knockdown enhances cell viability and suppresses cell apoptosis, oxidative stress, inflammation and endothelial dysfunction in ox-LDL-induced vascular endothelial cells via interacting with PDCD4. Int Immunopharmacol. 2023;122:110529.
Hodis HN, Kramsch DM, Avogaro P, Bittolo-Bon G, Cazzolato G, Hwang J, et al. Biochemical and cytotoxic characteristics of an in vivo circulating oxidized low density lipoprotein (LDL-). J Lipid Res. 1994;35:669–77.
Kojima Y, Ito S, Furuya N. Hearing improvement after therapy for hyperlipidemia in patients with chronic-phase sudden deafness. Ann Otol Rhinol Laryngol. 2001;110:105–8.
Gopinath B, Flood VM, Teber E, McMahon CM, Mitchell P. Dietary intake of cholesterol is positively associated and use of cholesterol-lowering medication is negatively associated with prevalent age-related hearing loss. J Nutr. 2011;141:1355–61.
Palty R, Raveh A, Kaminsky I, Meller R, Reuveny E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell. 2012;149:425–38.
Gataulin D, Kuperman Y, Tsoory M, Biton IE, Nataniel T, Palty R, et al. Store-operated Ca(2+) entry regulatory factor alters murine metabolic state in an age-dependent manner via hypothalamic pathways. PNAS Nexus. 2023;2:pgad068.
Chen LY, Chiang AS, Hung JJ, Hung HI, Lai YK. Thapsigargin-induced grp78 expression is mediated by the increase of cytosolic free calcium in 9L rat brain tumor cells. J Cell Biochem. 2000;78:404–16.
Jones KT, Sharpe GR. Thapsigargin raises intracellular free calcium levels in human keratinocytes and inhibits the coordinated expression of differentiation markers. Exp Cell Res. 1994;210:71–6.
Funding
The work was supported by funds from the Ryan Foundation to MRF.
Author information
Authors and Affiliations
Contributions
AAO: conceptualization, methodology, formal analysis, investigation, visualization, supervision; project administration, writing; WT: formal analysis; software; GK: investigation; CJ: investigation; DC: investigation; NF: investigation; RT: investigation; D Cosgrove: resource (cell line), review/editing; MRF: conceptualization, methodology, visualization, supervision; project administration, funding acquisition, review/editing. All authors have read and agreed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The animal studies were approved by Boys Town National Research Hospital under IACUC protocol #23-05. All methods were performed in accordance with the IACUC guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Olou, A.A., Tom, W., Krzyzanowski, G. et al. Ototoxic impacts of adipose-derived extracts on a murine auditory cell line: molecular insights from proteomic analyses. Int J Obes 49, 2549–2553 (2025). https://doi.org/10.1038/s41366-025-01912-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41366-025-01912-4
