
Abstract
Leukemia, although most likely starts as a monoclonal genetic/epigenetic anomaly, is a polyclonal disease at manifestation. This polyclonal nature results from ongoing evolutionary changes in the genome/epigenome of leukemia cells to promote their survival and proliferation advantages. We discuss here how genetic and/or epigenetic aberrations alter intracellular microenvironment in individual leukemia clones and how extracellular microenvironment selects the best fitted clones. This dynamic polyclonal composition of leukemia makes designing an effective therapy a challenging task especially because individual leukemia clones often display substantial differences in response to treatment. Here, we discuss novel therapeutic approach employing single cell multiomics to identify and eradicate all individual clones in a patient.
Similar content being viewed by others

Carcinogenesis is a phenomenon where Darwinian principles are applicable to elucidate mechanisms responsible for intratumoral heterogeneity [1]. Cancer evolution is a process in which tumor cells adapt to the environment to promote the survivability and expansion of the best fitted clones [2,3,4,5]. This process depends on intracellular and extracellular factors [6,7,8,9,10,11,12,13]. Among intracellular factors, high demand for energy (ATP) and building materials (nucleic acids, amino acids, lipids, sugars), deregulated signaling pathways, accelerated cell cycle and protection from apoptosis generate metabolic and replication stress resulting in accumulation of spontaneous DNA damage and mutagenesis. Extracellular factors include, among other, hypoxia, stress, interaction with the microenvironment, microbiota, infection, inflammation, and antitumor immune response. The extra- and intra- cellular environmental pressure generates genetic and epigenetic alterations leading to clonal selection. Also, therapeutic regimens are powerful factors forcing genetic and epigenetic aberrations and clonal selection [14,15,16]. Tumor cells must adapt to these dynamic extra- and intra- cellular challenges resulting in expansion of the best fitted clones [17,18,19,20].
Hallmarks of clonal evolution
Single cell “multiomics” approach [21] supported by computational power [22] revolutionized our perception of tumors [3, 6, 17], including myeloid malignancies [2, 5], which are highly dynamic polyclonal diseases. The interplay between genetic alterations (occurring spontaneously because of ongoing genomic instability, under the microenvironmental pressure, and after the treatment), heritable epigenetic modifications (occurring under the microenvironmental pressure and after the treatment) and immune responses produces malignant clones designed to thrive under specific conditions [23, 24].
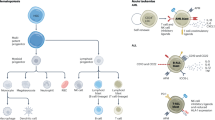
Genetic, epigenetic, proteomic, methylome and metabolomic aberrations define clonal functional heterogeneity and cooperativity which regulate clone-specific phenotypes such as selective microenvironmental advantages, resistance to immunological challenges and sensitivity to therapies [20, 24,25,26]. These ongoing fitness challenges are the hallmarks of malignant clonal expansion (Fig. 1A).

A The interactions between extracellular factors (microenvironment, immune response/inflammation, infection, microbiota, and treatment) and intracellular factors (epigenetics, signaling, metabolism, DNA damage response = DDR, transcription, splicing, cell cycle, and other) generate unique clonal composition in individual tumors. B Standard treatment removes and/or reduces some clones, while others survive eventually facilitating the emergence of new clone(s). Conversely, clonal attack targets all clones at the same time thus eradicating the disease.
Clonal hematopoiesis with indeterminate potential (CHIP) is accompanied by emergence of clones carrying mutations in several genes such as DNMT3A, TET2, JAK2, SF3B1, ASXL1, TP53, CBL, GNB1, BCOR, U2AF1, CREBBP, CUX1, SRSF2, MLL2, SETD2, SETDB1, GNAS, PPM1D, and BCORL1 and by increased rate of progression to hematologic malignancy [27]. CHIP is associated with aging, infections, chemotherapy and/or cigarette smoking, which alter tissue microenvironments to facilitate the selection and expansion of CHIP clones harboring specific mutations [28, 29]. For example, aging favors mutations in SF3B1, SRSF2, ATM, and TET2 among others [28]. Inflammatory stress promotes expansion and further malignant transformation of hematopoietic clones carrying TET2, DNMT3A, and ASXL1 mutations regulating DNA methylation and chromatin remodeling [29], and certain microbiota favor clones harboring DNMT3A, FLT3, and NPM1 mutations [30]. Smoking promotes clones with ASXL1, SRSF2, SF3B1, and JAK2 mutations [28]. On the other hand, expansion of the clones with TP53 and PPM1D mutations which regulate DNA damage response is triggered by genotoxic stress and PARP inhibitor [14, 31, 32].
Remarkably, specific clonal advantages are often encoded by two, or more co-existing mutations. For example, Flt3(ITD) mutation generating an oncogenic tyrosine kinase (OTK) when combined mutations in Dnmt3a or Tet2 in murine hematopoietic cells alter gene expression to an extend not seen with either mutation alone [20, 33, 34]. In addition, FLT3(ITD);NRASmut clone was selected under the treatment with FLT3 kinase inhibitor gilteritinib, whereas these mutations are usually mutually exclusive without the treatment [35]. Moreover, KRAS and NRAS mutations emerged in JAK2 and CALR mutant clones during the treatment of myelofibrosis with JAK1/2 kinase inhibitor ruxolitinib [36].
It appears that the order of mutations acquisition is not random and some mutations in epigenetic modifiers like DNMT3A, ASXL1, TET2 or IDH1/2 are more likely to happen at the early stages, but FLT3(ITD) and mutations in RAS genes affecting intracellular signaling are more likely to happen at the late stages of transformation and disease progression [2, 37]. In addition, comprehensive study on 1540 AML patient samples showed a distinct, reoccurring co-mutation patterns. For instance, NRAS(G12/G13) but not NRAS(Q61) co-occurs with NPM1 mutations. This confirms that the effect of mutations (even within the same gene) and their combinations might not have comparable effect [37]. Leukemogenesis is therefore an ordered and specific evolutionary process rather than an accidental chain of events.
Tumor cells and their surrounding microenvironment form a dynamic ecosystem [8]. Hypoxia, a hallmark of most tumors including leukemia in bone marrow niche, is a key factor in tumor microenvironment and can promote the outgrowth of tumor clones evading immune response and modulating their response to antitumor drugs [38, 39].
Genetic and epigenetic aberrations regulate the response to treatment
There are numerous reports about how genetic and epigenetic aberrations regulate the sensitivity to a variety of drugs; the examples are summarized in Table 1. These discoveries led to successful clinical trials resulting in paradigm shifting on anti-tumor therapies.
For example, OTKs generated by abnormal activation due to mutations, translocations or amplifications are implicated in the induction, maintenance, malignant progression of cancer [40]. Therefore, OTKs have emerged as major targets for drug discovery which generated numerous FDA approved therapeutic modalities [41]. Another example of specific oncogene associated with particular treatment are acute promyelocytic leukemia cells expressing PML::RARA fusion protein which are very sensitive to all-trans retinoic acid and arsenic trioxide [42].
Exploring synthetic lethality as a therapeutic approach paved the way for new discoveries focused on cancer-related genomic and epigenomic abnormalities associated with the sensitivity to specific drugs [43]. In this field, synthetic lethality triggered by PARP inhibitors in BRCA1/BRCA2-mutated tumors revolutionized the treatment of breast and ovarian carcinomas, and also of other cancers carrying mutations in genes, which products are involved in homologous recombination (HR) [44]. However, mutations in BRCA1/BRCA2 genes were detected only in 1-2% adult acute myeloid leukemia (AML) [45]. Moreover, genomic alterations in other DNA double strand breaks (DSBs) repair genes such as ATM, PRKDC, ATR, RAD51, and RAD54 were reported in <1% acute leukemias.
Despite that mutations in DSBs repair genes are rare in hematological malignancies, PARP inhibitors found the application in triggering synthetic lethality in leukemias [46]. For example, gene expression and mutation analysis (GEMA) identified a cohort of acute leukemias displaying downregulated BRCA1/BRCA2 expression levels which were sensitive to PARP inhibitor [47]. In addition, RUNX1::RUNX1T1, PML::RARA, IGH::MYC, TC3::HLF translocations and KDM6A, IDH1/2, TET2, and WT1 mutations compromised BRCA1/BRCA2-dependent HR-mediated repair of DSBs thus making leukemia cells sensitive to PARP inhibitor [34, 48,49,50,51,52,53,54]. These results provided foundation for numerous completed and ongoing clinical trials for PARP inhibitors in hematological malignancies [45, 46].
Moreover, BRCA1/BRCA2-deficiency and sensitivity to PARP inhibitor could be induced by FLT3 and JAK1/2 kinase inhibitors in AML and myeloproliferative neoplasm (MPN) cells expressing FLT3(ITD) and JAK2(V617F) OTKs, respectively [55]. The latter observation led to recent new clinical trial combining JAK2 inhibitor pacritinib and PARP inhibitor talazoparib in ruxolitinib-refractory MPN (NCT06218628).
In addition to DSB repair defects, dysregulation of other DNA damage response (DDR) mechanisms with potential therapeutic applications have been detected in leukemias. For example, mismatch repair (MMR) deficiencies due to somatic deletions of the genes that regulate MSH2 degradation were found in a cohort of newly diagnosed acute lymphoblastic leukemia (ALL) [56]. DNA polymerase beta inhibitor oleanolic acid treatment resulted in synthetic lethality in ALL with MMR deficiency through increased cellular apurinic/apyrimidinic sites, DNA strand breaks and apoptosis [57]. Also, AML cells displaying downregulation of the base excision repair (BER) glycosylase OGG1 were more sensitive to cytarabine [58]. Moreover, the odds of achieving complete remission in secondary AML patients were higher for the XPD(D312N) and XPD(K751Q) genotypes, previously associated with suboptimal nucleotide excision repair (NER) of more complex DNA lesions such as bulky DNA adducts [59, 60].
Epigenetics is dynamic and heritable modifications in DNA and histones that regulate gene activity independently of DNA sequence and can promote carcinogenesis and modulate drug response [61,62,63]. For example, loss of MLH1 and MGMT expression due to methylation resulted in resistance to platinum cytotoxic drugs and temozolomide, respectively [64, 65]. On the other hand, elevated expression of KDM5A caused resistance to cisplatin [66]. Thus, inhibitors of DNA methyltransferases (DNMTs), histone acetyltransferases and deacetylases (HATs and HDACs, respectively), and histone methyltransferases and demethylases (HMT and HDMT) have been developed and are used to treat cancer [67].
Potential clinical application of drugs targeting epigenetic modification such as DNMT and HDAC inhibitors has been explored in leukemias with limited success [68, 69]. The combinations of DNMT and/or HDAC inhibitors with other drugs, e.g., PARP inhibitors are under development [70, 71], but still lacking clear guidance about patient selection depended on genetic and epigenetic aberrations in leukemia. Recent studies with dual EZH1/EZH2 inhibitor valemelostat in adult T cell leukemia/lymphoma shed some light on mutation-dependent response [72]. For example, malignant clones with poor prognostic variations, including PRKCB, TP53, IRF4 and PDL1 responded well to the inhibitor, whereas these carrying FN1, CCR4, MYO7B, and TET2 mutations were less sensitive/resistant.
The complexity of anti-tumor drug responses is multiplied by co-existence of the genetic and epigenetic aberrations which might generate mutational cooperativity linked to specific gain of functions [20, 73]. In addition, polyclonal composition of the majority of leukemias adds another challenge because individual clones may inherit a unique and overlapping genetic and epigenetic aberrations which regulate drug response in individual clones [74, 75]. Therefore, it is paramount the develop clonal biomarkers of the response to treatment.
Clonal biomarkers defining the response to drugs
Genomic profiling identified pre-existing trackable sensitive and resistant tumor clones and revealed aberrations involved in drug response [76]. For example, clone carrying amplified MET was resistant to EGFR kinase inhibitor gefitinib and a clone with CDKN2A loss appeared sensitive to the combination of trametinib (MEK1/MEK2 kinase inhibitor) and palbociclib (CDK4/6 kinase inhibitor).
Regarding leukemia, AML clones with IDH1 mutations seemed more sensitive to ABL1 kinase inhibitor ponatinib; conversely these with NRAS mutations were resistant to EGFR kinase inhibitor pelitinib [77]. Moreover, AML clones with DNMT3A mutations were resistant to anthracyclines, such as daunorubicin which combined with cytarabine is a part of 7 + 3 regimen [78]. In addition, AML cells carrying RUNX1::RUNX1T1 and CBFB::MYH11 translocations seemed more sensitive to the cytarabine [58]. Mutations in SF3B1 and TP53 were a risk factor for rapid clonal evolution and disease progression in chronic lymphocytic leukemia (CLL) patients treated with chemotherapy [79].
Malignant clones usually carry more than one mutation which may interfere with each other. For example, while AML clones expressing FLT3(ITD) kinase seemed more responsive to the kinase inhibitor crizotinib, co-occurrence of DNMT3A mutation predicted resistance not only to the kinase inhibitor, but also to axitinib (P-glycoprotein efflux transporter inhibitor), cediranib (VEGFR inhibitor), ponatinib (ABL1 kinase inhibitor) and tofacitinib (JAK1/3 kinase inhibitor) [77].
AML and MPN cells usually accumulate spontaneous DNA damage, including highly toxic DSBs, induced by metabolic products and replication stress [55, 80, 81]. AML and MPN clones activate DDR mechanisms which are regulated by clone-specific mutations to survive endogenous genotoxic stress suggesting that DDR is a legitimate therapeutic target. For example, FLT3(ITD), JAK2(V617F), MPL(W515L), CALR(del52), TETmut, DNMT3Amut, RUNX1::RUNX1T1, RML::RARA, SRSF2mut, IDH1mut, and KITmut regulate DDR and affect the response of leukemia cells to PARP, RAD52 and Polθ inhibitors by modulating the expression of DNA repair proteins such as BRCA1 and BRCA2 (involved in HR) and Polθ (a key factor in microhomology-mediated end-joining) [34, 47, 49, 81,82,83].
Again, these mutations might interfere with each other to modulate DSB repair [34, 55, 81, 84, 85]. While TET2mut clones carrying FLT3(ITD), NRAS, JAK2(V617), CALRmut or MPLmut were sensitive to PARP inhibitors, the counterparts carrying DNMT3Amut were resistant [34]. Remarkably, FLT3(ITD);DNMT3Amut;TET2mut clone regained the sensitivity to PARP inhibitors. Moreover, RUNX1-RUNX1T1;FLT3(ITD) clones were sensitive to PARP inhibitor, conversely RUNX1::RUNX1T1; KITmut were resistant [84, 85]. CBFB::MYH11; KITmut and CBFB::MYH11; NRASmut were also associated with clonal resistance and sensitivity, respectively, to PARP inhibitor but not to doxorubicin [85].
Altogether, these results highlight the complexity of genetic signatures regulating clonal response to genotoxic agents, kinase inhibitors and DDR inhibitors. Single cell “omics” (e.g., transcriptomics, genomics, epigenomics, proteomics, metabolomics) and more recently single cell “multiomics” simultaneously integrating various “singleomics” should help to better understanding of cancer polyclonal composition and therapeutic vulnerabilities [26, 86]. Recently, combined analyses of numerous scRNAseq databases precipitated in the generation of artificial intelligence (AI) -powered PERCEPTION approach to predict responses to targeted therapies if multiple myeloma and breast cancer [87].
Clonal attack
Personalized medicine is an approach to tailoring disease prevention and treatment that considers differences in patient’s tumor genetic/epigenetic/metabolomic/immunologic characteristics. Clonal medicine is an innovative approach to tailoring the treatment that eliminates tumor clones based on clone’s unique genetic/epigenetic/metabolomic/immunologic makeup.
Tumor evolution is responsible for cancer heterogeneity which has to be accounted for when planning a therapeutic approach [88, 89]. Variabilities in tumor clonal response to the treatment has been suggested more than 45 years ago [90]. Tumor clones respond differently to various drugs due to their unique and overlapping “multiomics” properties. Thus, a successful therapeutic regimen must simultaneously eradicate all malignant clones (Fig. 1B).
For example, AML and MPN are heterogenous diseases, and clonal heterogeneity hampers the effectiveness of therapeutics against these hematological malignancies [2, 5]. Clones within a leukemia sample demonstrate differences in response to current drugs leading to disease relapse from the pre-existing clones selected by the treatment and/or those induced under the therapeutic pressure due to continuous clonal evolution [5, 91, 92]. The genetic landscape of malignant clones in a patient may be complicated since individual clones can carry multiple mutations, both overlapping and unique for the individual clones [93].
Recently, we designed a successful multiclonal attack to eradicate AML and MPN clones which accumulate DNA damage due to continuous metabolic stress [94]. We showed that a combination of DDR inhibitors simultaneously attacking all clones in a patient sample eradicated the disease in vitro and in vivo. The “clonal attack” by DDR inhibitors shifts the paradigm of genotoxic therapies from those using non discriminative cytotoxic drugs to those selectively attacking DDR vulnerabilities in AML and MPN clones. Therapeutic potential of the combinatorial effects of FDA approved drugs (e.g., tyrosine kinase inhibitors, and hypomethylating, genotoxic and pro-apoptotic agents) with DDR inhibitors need to be explored to evaluate if standard treatment enhance the effectiveness of DDR inhibitors in combating malignant clones.
Since clonal heterogeneity and DNA damage are hallmarks of cancer, the “clonal attack” may open new era in cancer treatment and be broadly applicable to the quest for cure [3, 17]. However, integration of the “clonal attack” into everyday clinical practice would require two steps. First, an extensive experimental single cell “multiomics” data must be gathered to generate enough information for artificial intelligence (AI) to effectively resolve tumor clonal composition and predict individual clones’ sensitivity to therapeutic interventions [95]. Second, hospitals must have the personnel and equipment to be able to perform “multiomic” analysis and interpret the data on daily basis.
References
Vendramin R, Litchfield K, Swanton C. Cancer evolution: Darwin and beyond. EMBO J. 2021;40:e108389.
Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587:477–82.
Dentro SC, Leshchiner I, Haase K, Tarabichi M, Wintersinger J, Deshwar AG, et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021;184:2239–54.e39.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Morita K, Wang F, Jahn K, Hu T, Tanaka T, Sasaki Y, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun. 2020;11:5327.
McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–28.
Grockowiak E, Korn C, Rak J, Lysenko V, Hallou A, Panvini FM, et al. Different niches for stem cells carrying the same oncogenic driver affect pathogenesis and therapy response in myeloproliferative neoplasms. Nat Cancer. 2023;4:1193–209.
Seferbekova Z, Lomakin A, Yates LR, Gerstung M. Spatial biology of cancer evolution. Nat Rev Genet. 2023;24:295–313.
Rodriguez-Meira A, Norfo R, Wen S, Chédeville AL, Rahman H, O’Sullivan J, et al. Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat Genet. 2023;55:1531–41.
Anderson AR, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–15.
Caprioli C, Nazari I, Milovanovic S, Pelicci PG. Single-Cell Technologies to Decipher the Immune Microenvironment in Myeloid Neoplasms: Perspectives and Opportunities. Front Oncol. 2021;11:796477.
Lythgoe MP, Mullish BH, Frampton AE, Krell J. Polymorphic microbes: a new emerging hallmark of cancer. Trends Microbiol. 2022;30:1131–4.
Ma Y, Kroemer G. The cancer-immune dialogue in the context of stress. Nat Rev Immunol. 2024;24:264–81.
Wong TN, Miller CA, Jotte MRM, Bagegni N, Baty JD, Schmidt AP, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun. 2018;9:455.
Wong TN, Miller CA, Klco JM, Petti A, Demeter R, Helton NM, et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood. 2016;127:893–7.
Calleja A, Yun S, Moreilhon C, Karsenti JM, Gastaud L, Mannone L, et al. Clonal selection in therapy-related myelodysplastic syndromes and acute myeloid leukemia under azacitidine treatment. Eur J Haematol. 2020;104:488–98.
Black JRM, McGranahan N. Genetic and non-genetic clonal diversity in cancer evolution. Nat Rev Cancer. 2021;21:379–92.
Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22:792–9.
de Boer B, Prick J, Pruis MG, Keane P, Imperato MR, Jaques J, et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell. 2018;34:674–89.e8.
Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27:502–15.
Dimitriu MA, Lazar-Contes I, Roszkowski M, Mansuy IM. Single-Cell Multiomics Techniques: From Conception to Applications. Front Cell Dev Biol. 2022;10:854317.
Adossa N, Khan S, Rytkönen KT, Elo LL. Computational strategies for single-cell multi-omics integration. Comput Struct Biotechnol J. 2021;19:2588–96.
Cobaleda C, Godley LA, Nichols KE, Wlodarski MW, Sanchez-Garcia I. Insights into the Molecular Mechanisms of Genetic Predisposition to Hematopoietic Malignancies: The Importance of Gene-Environment Interactions. Cancer Discov. 2024;14:396–405.
Riley PA. Epimutation and Cancer: Carcinogenesis Viewed as Error-Prone Inheritance of Epigenetic Information. J Oncol. 2018;2018:2645095.
Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25:379–92.
Baysoy A, Bai Z, Satija R, Fan R. The technological landscape and applications of single-cell multi-omics. Nat Rev Mol Cell Biol. 2023;24:695–713.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16.
Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52:1219–26.
Florez MA, Tran BT, Wathan TK, DeGregori J, Pietras EM, King KY. Clonal hematopoiesis: Mutation-specific adaptation to environmental change. Cell Stem Cell. 2022;29:882–904.
Woerner J, Huang Y, Hutter S, Gurnari C, Sánchez JMH, Wang J, et al. Circulating microbial content in myeloid malignancy patients is associated with disease subtypes and patient outcomes. Nat Commun. 2022;13:1038.
Arends CM, Kopp K, Hablesreiter R, Estrada N, Christen F, Moll UM, et al. Dynamics of clonal hematopoiesis under DNA-damaging treatment in patients with ovarian cancer. Leukemia. 2024;38:1378–89.
Ushijima Y, Naruse S, Ishikawa Y, Kawashima N, Sanada M, Nakashima M, et al. Initiating-clone analysis in patients with acute myeloid leukemia secondary to essential thrombocythemia. Sci Rep. 2024;14:15906.
Meyer SE, Qin T, Muench DE, Masuda K, Venkatasubramanian M, Orr E, et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discov. 2016;6:501–15.
Maifrede S, Le BV, Nieborowska-Skorska M, Golovine K, Sullivan-Reed K, Dunuwille WMB, et al. TET2 and DNMT3A Mutations Exert Divergent Effects on DNA Repair and Sensitivity of Leukemia Cells to PARP Inhibitors. Cancer Res. 2021;81:5089–101.
McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019;9:1050–63.
Mylonas E, Yoshida K, Frick M, Hoyer K, Christen F, Kaeda J, et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat Commun. 2020;11:73.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374:2209–21.
Terry S, Engelsen AST, Buart S, Elsayed WS, Venkatesh GH, Chouaib S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020;492:1–10.
Bruno S, Mancini M, De Santis S, Monaldi C, Cavo M, Soverini S. The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities. Int J Mol Sci. 2021;22:6857.
Paul MK, Mukhopadhyay AK. Tyrosine kinase - Role and significance in Cancer. Int J Med Sci. 2004;1:101–15.
Hussain S, Mursal M, Verma G, Hasan SM, Khan MF. Targeting oncogenic kinases: Insights on FDA approved tyrosine kinase inhibitors. Eur J Pharm. 2024;970:176484.
Rérolle D, Wu HC, de Thé H. Acute Promyelocytic Leukemia, Retinoic Acid, and Arsenic: A Tale of Dualities. Cold Spring Harb Perspect Med. 2024;a041582.
Setton J, Zinda M, Riaz N, Durocher D, Zimmermann M, Koehler M, et al. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021;11:1626–35.
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5:387–93.
Padella A, Ghelli Luserna Di Rorà A, Marconi G, Ghetti M, Martinelli G, Simonetti G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J Hematol Oncol. 2022;15:10.
Boila LD, Sengupta A. Unifying targeted therapy for leukemia in the era of PARP inhibition. Exp Hematol. 2023;124:1–14.
Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest. 2017;127:2392–406.
Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9:eaal2463.
Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21:1481–90.
Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Invest. 2003;112:1751–61.
Maifrede S, Martin K, Podszywalow-Bartnicka P, Sullivan-Reed K, Langer SK, Nejati R, et al. IGH/MYC Translocation Associates with BRCA2 Deficiency and Synthetic Lethality to PARP1 Inhibitors. Mol Cancer Res. 2017;15:967–72.
Piao J, Takai S, Kamiya T, Inukai T, Sugita K, Ohyashiki K, et al. Poly (ADP-ribose) polymerase inhibitors selectively induce cytotoxicity in TCF3-HLF-positive leukemic cells. Cancer Lett. 2017;386:131–40.
Boila LD, Chatterjee SS, Banerjee D, Sengupta A. KDM6 and KDM4 histone lysine demethylases emerge as molecular therapeutic targets in human acute myeloid leukemia. Exp Hematol. 2018;58:44–51.e7.
Gbyli R, Song Y, Liu W, Gao Y, Biancon G, Chandhok NS, et al. In vivo anti-tumor effect of PARP inhibition in IDH1/2 mutant MDS/AML resistant to targeted inhibitors of mutant IDH1/2. Leukemia. 2022;36:1313–23.
Nieborowska-Skorska M, Maifrede S, Dasgupta Y, Sullivan K, Flis S, Le BV, et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood. 2017;130:2848–59.
Diouf B, Cheng Q, Krynetskaia NF, Yang W, Cheok M, Pei D, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298–303.
Teng J-Y, Yang D-P, Tang C, Fang H-S, Sun H-Y, Xiang Y-N, et al. Targeting DNA polymerase β elicits synthetic lethality with mismatch repair deficiency in acute lymphoblastic leukemia. Leukemia. 2023;37:1204–15.
Owen N, Minko IG, Moellmer SA, Cammann SK, Lloyd RS, McCullough AK. Enhanced cytarabine-induced killing in OGG1-deficient acute myeloid leukemia cells. Proc Natl Acad Sci USA. 2021;118:e2016833118.
Kuptsova N, Kopecky KJ, Godwin J, Anderson J, Hoque A, Willman CL, et al. Polymorphisms in DNA repair genes and therapeutic outcomes of AML patients from SWOG clinical trials. Blood. 2007;109:3936–44.
Kuptsova-Clarkson N, Ambrosone CB, Weiss J, Baer MR, Sucheston LE, Zirpoli G, et al. XPD DNA nucleotide excision repair gene polymorphisms associated with DNA repair deficiency predict better treatment outcomes in secondary acute myeloid leukemia. Int J Mol Epidemiol Genet. 2010;1:278–94.
Lu Y, Chan Y-T, Tan H-Y, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020;19:79.
Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer. 2014;14:747–53.
Yuan H, Lu Y, Feng Y, Wang N. Epigenetic inhibitors for cancer treatment. Int Rev Cell Mol Biol. 2024;383:89–144.
Zeller C, Dai W, Steele NL, Siddiq A, Walley AJ, Wilhelm-Benartzi CS, et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene. 2012;31:4567–76.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003.
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80.
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62.
Duchmann M, Itzykson R. Clinical update on hypomethylating agents. Int J Hematol. 2019;110:161–9.
Pal D, Raj K, Nandi SS, Sinha S, Mishra A, Mondal A, et al. Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies. Cancers. 2023;15:2808.
Baer MR, Kogan AA, Bentzen SM, Mi T, Lapidus RG, Duong VH, et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin Cancer Res. 2022;28:1313–22.
Valdez BC, Yuan B, Murray D, Ramdial JL, Nieto Y, Popat U, et al. Synergistic cytotoxicity of fludarabine, clofarabine, busulfan, vorinostat and olaparib in AML cells. Front Oncol. 2023;13:1287444.
Yamagishi M, Kuze Y, Kobayashi S, Nakashima M, Morishima S, Kawamata T, et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature. 2024;627:221–8.
You JS, Jones PA. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell. 2012;22:9–20.
Meir Z, Mukamel Z, Chomsky E, Lifshitz A, Tanay A. Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat Genet. 2020;52:709–18.
Sadida HQ, Abdulla A, Marzooqi SA, Hashem S, Macha MA, Akil ASA-S, et al. Epigenetic modifications: Key players in cancer heterogeneity and drug resistance. Transl Oncol. 2024;39:101821.
Acar A, Nichol D, Fernandez-Mateos J, Cresswell GD, Barozzi I, Hong SP, et al. Exploiting evolutionary steering to induce collateral drug sensitivity in cancer. Nat Commun. 2020;11:1923.
Benard BA, Leak LB, Azizi A, Thomas D, Gentles AJ, Majeti R. Clonal architecture predicts clinical outcomes and drug sensitivity in acute myeloid leukemia. Nat Commun. 2021;12:7244.
Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. 2016;22:1488–95.
Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26.
Chen E, Ahn JS, Massie CE, Clynes D, Godfrey AL, Li J, et al. JAK2V617F promotes replication fork stalling with disease-restricted impairment of the intra-S checkpoint response. Proc Natl Acad Sci USA. 2014;111:15190–5.
Vekariya U, Toma M, Nieborowska-Skorska M, Le BV, Caron MC, Kukuyan AM, et al. DNA polymerase theta protects leukemia cells from metabolically induced DNA damage. Blood. 2023;141:2372–89.
Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer cell. 2016;30:337–48.
Liu ZS, Sinha S, Bannister M, Song A, Arriaga-Gomez E, McKeeken AJ, et al. R-Loop Accumulation in Spliceosome Mutant Leukemias Confers Sensitivity to PARP1 Inhibition by Triggering Transcription-Replication Conflicts. Cancer Res. 2024;84:577–97.
Maifrede S, Nieborowska-Skorska M, Sullivan-Reed K, Dasgupta Y, Podszywalow-Bartnicka P, Le BV, et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood. 2018;132:67–77.
Nieborowska-Skorska M, Paietta EM, Levine RL, Fernandez HF, Tallman MS, Litzow MR, et al. Inhibition of the mutated c-KIT kinase in AML1-ETO-positive leukemia cells restores sensitivity to PARP inhibitor. Blood Adv. 2019;3:4050–4.
Liang A, Kong Y, Chen Z, Qiu Y, Wu Y, Zhu X, et al. Advancements and applications of single-cell multi-omics techniques in cancer research: Unveiling heterogeneity and paving the way for precision therapeutics. Biochem Biophys Rep. 2024;37:101589.
Sinha S, Vegesna R, Mukherjee S, Kammula AV, Dhruba SR, Wu W, et al. PERCEPTION predicts patient response and resistance to treatment using single-cell transcriptomics of their tumors. Nat Cancer. 2024;5:938–52.
Amirouchene-Angelozzi N, Swanton C, Bardelli A Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017;7:805–17.
Saito Y, Mochizuki Y, Ogahara I, Watanabe T, Hogdal L, Takagi S, et al. Overcoming mutational complexity in acute myeloid leukemia by inhibition of critical pathways. Sci Transl Med. 2017;9:eaao1214.
Pályi I, Oláh E, Sugár J. Drug sensitivity studies on clonal cell lines isolated from heteroploid tumour cell populations. I. Dose response of clones growing in monolayer cultures. Int J Cancer. 1977;19:859–65.
Parkin B, Ouillette P, Li Y, Keller J, Lam C, Roulston D, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121:369–77.
Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer. 2019;58:839–49.
Ediriwickrema A, Gentles AJ, Majeti R. Single-cell genomics in AML: extending the frontiers of AML research. Blood. 2023;141:345–55.
Toma MM, Karami A, Nieborowska-Skorska M, Chirtala KN, Pepek M, Hadzijusufovic E, et al. Clonal medicine targeting DNA damage response eradicates leukemia. Leukemia. 2024;38:671–5.
He X, Liu X, Zuo F, Shi H, Jing J. Artificial intelligence-based multi-omics analysis fuels cancer precision medicine. Semin Cancer Biol. 2023;88:187–200.
Morra F, Luise C, Visconti R, Staibano S, Merolla F, Ilardi G, et al. New therapeutic perspectives in CCDC6 deficient lung cancer cells. Int J Cancer. 2015;136:2146–57.
Tsujino T, Takai T, Hinohara K, Gui F, Tsutsumi T, Bai X, et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat Commun. 2023;14:252.
Klimovich B, Merle N, Neumann M, Elmshäuser S, Nist A, Mernberger M, et al. p53 partial loss-of-function mutations sensitize to chemotherapy. Oncogene. 2022;41:1011–23.
Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–30.
Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61:6185–93.
Huang Y, Liu N, Liu J, Liu Y, Zhang C, Long S, et al. Mutant p53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA. Cell Cycle. 2019;18:3442–55.
Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci USA. 1993;90:5742–6.
Stahl M, Menghrajani K, Derkach A, Chan A, Xiao W, Glass J, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021;5:1552–64.
Long J, Parkin B, Ouillette P, Bixby D, Shedden K, Erba H, et al. Multiple distinct molecular mechanisms influence sensitivity and resistance to MDM2 inhibitors in adult acute myelogenous leukemia. Blood. 2010;116:71–80.
Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene. 2006;25:304–9.
Zoumpoulidou G, Alvarez-Mendoza C, Mancusi C, Ahmed RM, Denman M, Steele CD, et al. Therapeutic vulnerability to PARP1,2 inhibition in RB1-mutant osteosarcoma. Nat Commun. 2021;12:7064.
Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2018;29:640–5.
Wang Q, Yu T, Ke ZH, Wang FF, Yin JN, Shao Y, et al. RB1 aberrations predict outcomes of immune checkpoint inhibitor combination therapy in NSCLC. Front Oncol. 2023;13:1172728.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl J Med. 2015;373:1697–708.
Martino C, Pandya D, Lee R, Levy G, Lo T, Lobo S, et al. ATM-Mutated Pancreatic Cancer: Clinical and Molecular Response to Gemcitabine/Nab-Paclitaxel After Genome-Based Therapy Resistance. Pancreas. 2020;49:143–7.
Choi M, Kipps T, Kurzrock R. ATM Mutations in Cancer: Therapeutic Implications. Mol Cancer Ther. 2016;15:1781–91.
Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87.
Kim KH, Kim HS, Kim SS, Shim HS, Yang AJ, Lee JJB, et al. Increased Radiosensitivity of Solid Tumors Harboring ATM and BRCA1/2 Mutations. Cancer Res Treat. 2022;54:54–64.
Shapiro GI, Tibes R, Gordon MS, Wong BY, Eder JP, Borad MJ, et al. Phase I studies of CBP501, a G2 checkpoint abrogator, as monotherapy and in combination with cisplatin in patients with advanced solid tumors. Clin Cancer Res. 2011;17:3431–42.
Dillon KM, Bekele RT, Sztupinszki Z, Hanlon T, Rafiei S, Szallasi Z, et al. PALB2 or BARD1 loss confers homologous recombination deficiency and PARP inhibitor sensitivity in prostate cancer. NPJ Precis Oncol. 2022;6:49.
Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N Engl J Med. 2021;384:2394–405.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat. 2014;147:401–5.
Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun. 2021;12:3636.
Wang J, Ding Q, Fujimori H, Motegi A, Miki Y, Masutani M. Loss of CtIP disturbs homologous recombination repair and sensitizes breast cancer cells to PARP inhibitors. Oncotarget. 2016;7:7701–14.
Postel-Vinay S, Bajrami I, Friboulet L, Elliott R, Fontebasso Y, Dorvault N, et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene. 2013;32:5377–87.
Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49.
Czyż M, Toma M, Gajos-Michniewicz A, Majchrzak K, Hoser G, Szemraj J, et al. PARP1 inhibitor olaparib (Lynparza) exerts synthetic lethal effect against ligase 4-deficient melanomas. Oncotarget. 2016;7:75551–60.
Kuhmann C, Li C, Kloor M, Salou M, Weigel C, Schmidt CR, et al. Altered regulation of DNA ligase IV activity by aberrant promoter DNA methylation and gene amplification in colorectal cancer. Hum Mol Genet. 2014;23:2043–54.
Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair. 2007;6:1079–99.
Chen SH, Huang WT, Kao WC, Hsiao SY, Pan HY, Fang CW, et al. O6-methylguanine-DNA methyltransferase modulates cisplatin-induced DNA double-strand breaks by targeting the homologous recombination pathway in nasopharyngeal carcinoma. J Biomed Sci. 2021;28:2.
Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–4.
Papouli E, Cejka P, Jiricny J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004;64:3391–4.
Koppensteiner R, Samartzis EP, Noske A, von Teichman A, Dedes I, Gwerder M, et al. Effect of MRE11 loss on PARP-inhibitor sensitivity in endometrial cancer in vitro. PLoS One. 2014;9:e100041.
Gruber JJ, Afghahi A, Timms K, DeWees A, Gross W, Aushev VN, et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat Cancer. 2022;3:1181–91.
Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nature structural &. Mol Biol. 2010;17:1247–54.
Smith MA, Hampton OA, Reynolds CP, Kang MH, Maris JM, Gorlick R, et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr Blood Cancer. 2015;62:91–8.
Min A, Im SA, Yoon YK, Song SH, Nam HJ, Hur HS, et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther. 2013;12:865–77.
Setton J, Selenica P, Mukherjee S, Shah R, Pecorari I, McMillan B, et al. Germline RAD51B variants confer susceptibility to breast and ovarian cancers deficient in homologous recombination. NPJ Breast Cancer. 2021;7:135.
Liu P, Lin C, Liu L, Lu Z, Tu Z, Liu H. RAD54B mutations enhance the sensitivity of ovarian cancer cells to poly(ADP-ribose) polymerase (PARP) inhibitors. J Biol Chem. 2022;298:102354.
Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci USA. 2012;109:15030–5.
Hurley RM, Wahner Hendrickson AE, Visscher DW, Ansell P, Harrell MI, Wagner JM, et al. 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol Oncol. 2019;153:127–34.
Feng Y, Li X, Cassady K, Zou Z, Zhang X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front Oncol. 2019;9:210.
Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32:783–90.
SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z, et al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012;103:269–73.
Okita Y, Narita Y, Miyakita Y, Ohno M, Matsushita Y, Fukushima S, et al. IDH1/2 mutation is a prognostic marker for survival and predicts response to chemotherapy for grade II gliomas concomitantly treated with radiation therapy. Int J Oncol. 2012;41:1325–36.
Tran AN, Lai A, Li S, Pope WB, Teixeira S, Harris RJ, et al. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol. 2014;16:414–20.
An X, Tiwari AK, Sun Y, Ding PR, Ashby CR Jr., Chen ZS. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34:1255–68.
Wildschut MHE, Mena J, Dördelmann C, van Oostrum M, Hale BD, Settelmeier J, et al. Proteogenetic drug response profiling elucidates targetable vulnerabilities of myelofibrosis. Nat Commun. 2023;14:6414.
Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17:875–82.
Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J Biol Chem. 2014;289:9247–53.
Growney JD, Clark JJ, Adelsperger J, Stone R, Fabbro D, Griffin JD, et al. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood. 2005;106:721–4.
Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–5.
Li Y, Mao T, Wang J, Zheng H, Hu Z, Cao P, et al. Toward the next generation EGFR inhibitors: an overview of osimertinib resistance mediated by EGFR mutations in non-small cell lung cancer. Cell Commun Signal. 2023;21:71.
Plo I, Nakatake M, Malivert L, de Villartay JP, Giraudier S, Villeval JL, et al. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood. 2008;112:1402–12.
Apostolidou E, Kantarjian HM, Verstovsek S. JAK2 inhibitors: A reality? A hope? Clin Lymphoma Myeloma. 2009;9:S340–5.
Pratz KW, Koh BD, Patel AG, Flatten KS, Poh W, Herman JG, et al. Poly (ADP-Ribose) Polymerase Inhibitor Hypersensitivity in Aggressive Myeloproliferative Neoplasms. Clin Cancer Res. 2016;22:3894–902.
Kitazawa M, Miyagawa Y, Koyama M, Nakamura S, Hondo N, Miyazaki S, et al. Drug sensitivity profile of minor KRAS mutations in colorectal cancer using mix culture assay: The effect of AMG-510, a novel KRAS G12C selective inhibitor, on colon cancer cells is markedly enhanced by the combined inhibition of MEK and BCL-XL. Mol Clin Oncol. 2021;15:148.
Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021;11:142–57.
Yuan X, Bu H, Zhou J, Yang CY, Zhang H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J Med Chem. 2020;63:11368–96.
Zhang H, Nakauchi Y, Köhnke T, Stafford M, Bottomly D, Thomas R, et al. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer. 2020;1:826–39.
Ohashi K, Sequist LV, Arcila ME, Lovly CM, Chen X, Rudin CM, et al. Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res. 2013;19:2584–91.
Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22.
Yokoyama D, Hisamori S, Deguchi Y, Nishigori T, Okabe H, Kanaya S, et al. PTEN is a predictive biomarker of trastuzumab resistance and prognostic factor in HER2-overexpressing gastroesophageal adenocarcinoma. Sci Rep. 2021;11:9013.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27.
Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, et al. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018;78:5363–74.
Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell. 2019;36:194–209.e9.
Torgersen ML, Engedal N, Bøe SO, Hokland P, Simonsen A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood. 2013;122:2467–76.
Maifrede S, Martinez E, Nieborowska-Skorska M, Di Marcantonio D, Hulse M, Le BV, et al. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017;1:1467–72.
Byrd JC, Ruppert AS, Mrózek K, Carroll AJ, Edwards CG, Arthur DC, et al. Repetitive cycles of high-dose cytarabine benefit patients with acute myeloid leukemia and inv(16)(p13q22) or t(16;16)(p13;q22): results from CALGB 8461. J Clin Oncol. 2004;22:1087–94.
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78.
Yoshida H, Kitamura K, Tanaka K, Omura S, Miyazaki T, Hachiya T, et al. Accelerated degradation of PML-retinoic acid receptor alpha (PML-RARA) oncoprotein by all-trans-retinoic acid in acute promyelocytic leukemia: possible role of the proteasome pathway. Cancer Res. 1996;56:2945–8.
Zhu J, Koken MH, Quignon F, Chelbi-Alix MK, Degos L, Wang ZY, et al. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc Natl Acad Sci USA. 1997;94:3978–83.
Adnan Awad S, Dufva O, Ianevski A, Ghimire B, Koski J, Maliniemi P, et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia. 2021;35:1087–99.
Tothova Z, Valton AL, Gorelov RA, Vallurupalli M, Krill-Burger JM, Holmes A, et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight. 2021;6:e142149.
Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature. 2018;557:580–4.
Acknowledgements
TS has been sponsored by NCI R01s: CA237286, CA244044, CA244179, CA244707, CA186238, CA247707 and CA283396, and by the grant from the Leukemia and Lymphoma Society TRP-6628-21.
Author information
Authors and Affiliations
Contributions
MMT helped write the manuscript, TS wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toma, M.M., Skorski, T. Star wars against leukemia: attacking the clones. Leukemia 38, 2293–2302 (2024). https://doi.org/10.1038/s41375-024-02369-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-024-02369-6
