
Abstract
The role of germline CHEK2 variants in hematopoietic malignancies (HMs) is poorly understood. We examined pathogenic/likely pathogenic (P/LP) CHEK2 variants in patients with hereditary HMs (HHMs), a solid tumor risk cohort, public datasets, and a knock-in mouse model. In the HHM cohort, 57 probands had germline P/LP CHEK2 variants, mostly p.I157T (53%, 30/57). Among CHEK2 p.I157T carriers, 43% (19/44) had myeloid malignancies, 32% (14/44) had lymphoid malignancies, and 2% (1/44) had both. Among those with other germline P/LP CHEK2 alleles, 36% (13/36) had myeloid malignancies, 28% (10/36) had lymphoid malignancies, and 6% (2/36) had both. CHEK2 p.I157T was enriched in HM patients (OR 6.44, 95%CI 3.68–10.73, P < 0.001). In a solid tumor risk cohort, 36% (15/42) of CHEK2 p.I157T patients had a HM family history. A genome wide association study showed enrichment of CHEK2 loss-of-function variants with myeloid leukemia (P = 5.78e−7). In public acute myeloid leukemia (AML) datasets, 1% (16/1348) of patients had P/LP CHEK2 variants. In a public myelodysplastic neoplasms (MDS) dataset, 2% (5/214) had P/LP CHEK2 variants. Chek2 p.I161T mice, homologous to human p.I157T, had worse survival as heterozygotes (P = 0.037) or homozygotes (P = 0.005), with fewer Lin-CD34+ and Lin-cKit+ cells. Our data suggest P/LP CHEK2 variants are HHM risk alleles.
Similar content being viewed by others


Introduction
The CHEK2 gene encodes the checkpoint kinase 2 (CHK2) protein, an integral effector kinase in the ATM-CHK2-TP53 DNA damage response (DDR) pathway [1, 2]. The Mre11 complex recognizes a DNA double-strand break (DSB) and activates ATM, which phosphorylates CHK2, promoting multimerization and autophosphorylation [3, 4]. Activated CHK2 phosphorylates downstream targets, including p53 and BRCA1, driving cell cycle slowing, apoptosis, and autophagy [5, 6]. Deleterious germline CHEK2 variants are considered moderate penetrance risk alleles for breast [7,8,9,10] and prostate [7, 11] cancers, and possibly other solid organ malignancies [12,13,14,15]. These alleles are found at relatively high frequencies in some genetic subgroups, likely due to incomplete penetrance for cancers with onset beyond reproductive age, reducing selective pressure against them [16,17,18].
Small, single-center studies suggest that deleterious germline CHEK2 variants may contribute to the development of non-Hodgkin lymphoma (NHL) [19, 20], myeloproliferative neoplasms (MPNs) [21, 22], and myelodysplastic neoplasms (MDS) [23, 24]. Genome wide association studies (GWAS) implicate deleterious germline CHEK2 alleles as conferring increased risk for clonal hematopoiesis (CH) [25,26,27,28]. Reducing CHEK2 expression by shRNA knockdown promotes the proliferation of Lin-CD34+ cells in long-term culture, a mechanism consistent with a CH phenotype [29]. In addition, deleterious CHEK2 variants are common germline alleles detected in MDS or acute myeloid leukemia (AML) patients undergoing inherited cancer risk testing [30,31,32].
Lacking definitive studies describing the clinical and biological features of deleterious germline CHEK2 variants in patients with hematopoietic malignancies (HMs), we examined the features of patients and populations with deleterious germline CHEK2 variants and studied a knock-in mouse model of the most common such allele seen in HM patients, CHEK2 p.I157T.
Methods
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations, as outlined below. A full list of primers, antibodies, and other reagents utilized is given in Supplemental Table 1, and further details of methods used are provided in the Supplemental Methods. Statistical comparisons were made using unpaired t-tests, and mouse survival was calculated by Kaplan-Meier with significance at P < 0.05, except where otherwise specified.
Written informed consent for all research participants was obtained at the University of Chicago (UC) under Institutional Review Board (IRB)-approved study 11-0014, and at the University of Utah under IRB-approved study 00046740.
Mice were maintained under UC IACUC-approved protocol 71370.
Data collection and methods for clinical cohorts
Data were collected from HM patients with a known inherited cancer risk allele or a suspicion a hereditary hematologic malignancy (HHM) syndrome seen at UC from 2015 through 2022. Detailed inclusion criteria are specified in the Supplemental Methods. At the University of Utah, individuals with CHEK2 p.I157T were identified through testing for solid tumor hereditary cancer risk. All variants were assessed by American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines (Supplemental Table 2). CHEK2 variants were named according to MANE select transcript NM_007194.4. Population frequencies were obtained from gnomAD (v.4.1.0) [33]. Metadata for The Cancer Genome Atlas (TCGA) cohort was obtained from cBioPortal (https://www.cbioportal.org/) [34, 35].
Genome-wide and phenome-wide association studies
Phenome-wide association study (PheWAS) for CHEK2 (ENSG00000183765) with the predicted loss of function (pLoF) burden set and GWAS data for ICD-10 code C92 Myeloid leukaemia were obtained from the UK Biobank (UKBB) through the genebass portal (https://app.genebass.org/, GRCh38, 0.13.0-43c83cc-202402232123) with P-values expressed as optimized nonburden sequence kernel association tests (SKAT-O) [36,37,38].
Identification of CHEK2 variants in public AML datasets
Public RNA-seq data from BEAT AML, Leucegene, and AML PMP consortia were obtained for patients diagnosed with AML or high-grade myeloid neoplasm [39,40,41]. Public RNA-seq data from bone marrow mononuclear or CD34+ cells from MDS patients were obtained from EGAS00001002346 [42]. CHEK2 variants were called as described in Supplemental Methods. Somatic variants and translocations were obtained from the associated metadata. Recurrent fusion events were defined as per European LeukemiaNet (ELN) 2022 definitions [43].
Generation and characterization of a Chek2 mouse model
The Chek2 p.I161T mouse was generated as a constitutional knock-in allele with T8677C/p.I161T in C57BL/6 mice. Mice were followed by twice weekly health checks until 24 months or a humane endpoint. The hematopoietic stem and progenitor cell (HSPC) compartment was examined by flow cytometry following column-based lineage depletion (Miltenyi Biotec). For bulk RNA-seq, Lin-CD34+ cells were flow sorted directly into RLT+ lysis buffer and sequenced on an Illumina NovaSeq PE100 S1 flowcell, followed by transcript quantitation with Salmon (v.1.9.0), and analyzed with DEseq2 (v.1.32.0) and GSEA (v.4.2.2) with the mSigDB m2.all v.2023 gene set [44]. Clonal hematopoiesis was assessed by somatic variant identification on DNA derived from PB collected from 8-9 month-old mice. Whole exome sequencing (WES) with target coverage of 150X was performed on the Illumina NovaSeq X plus with a S4 flowcell and 100 bp paired end reads and analyzed per the genome analysis toolkit (GATK) best practices workflow [45]. Mouse necropsies were performed on all deceased mice, including gross examination, complete blood cell count (CBC), blood smears, tissue sections, immunohistochemistry (IHC), and multicolor flow cytometry. B/T clonotype sequencing was performed with the NEBNext Mouse Immune Sequencing Kit on an Illumina MiSeq with 300 bp paired end reads and analyzed with MiXCR (v.4.4.2).
Results
CHEK2 variants in a HHM testing cohort
Within a total of 1593 research participants in the UC HHM cohort [46], we identified 92 patients with CHEK2 variants from 69 unique families (probands) (Fig. 1). Among all patients, including probands and family members, 87% (80/92) had pathogenic (P) or likely pathogenic (LP) germline CHEK2 variants (CHEK2-path) and 13% (12/92) had variants of uncertain significance (VUS). Among CHEK2 variants observed in probands, 83% (57/69) were CHEK2-path and 17% (12/69) were VUS; benign or likely benign variants were not reported (Supplemental Table 3). The most common CHEK2 variant in the 57 probands with CHEK2-path variants was p.I157T (53%, 30/57) followed by p.T367fs (11%, 6/57), and p.S428F (7%, 4/57) (Fig. 2A). The most common CHEK2-path variant type was missense (68%, 39/57), followed by frameshift (19%, 11/57), and the most affected CHEK2 protein region was the forkhead-associated (FHA) domain (63%, 36/57) followed by the kinase domain (32%, 18/57). The ethnic distribution within this cohort was predominantly European, and the most common specific ancestry for the p.I157T allele was Polish (37%, 11/30) and for p.S428F was Ashkenazi Jewish (100%, 4/4) (Fig. 2B), likely reflecting the underlying population within the greater Chicagoland area.

A A total of 80 patients from 57 families (unique probands) with pathogenic CHEK2 variants (CHEK2-path) were identified from the UC HHM cohort. Amongst these 80 patients, 59 had a HM. In the subset of patients with the CHEK2 p.I157T variant, 34 had a HM. In the subset of patients who were included in the allele burden calculation who had sequential, clinical testing (N = 1085), 851 had a HM and 30 of these had CHEK2-path, with the most common variant being CHEK2 p.I157T (N = 15).

A CHK2 protein schematic showing the locations of the encoded protein variants identified among 69 probands tested for HHMs. CHK2 protein domains are outlined: SQ/TQ (green bar), forkhead-associated (FHA) domain (red bar), and the kinase domain (black bar). Diamonds depict P/LP variants, and circles denote VUS. Red, missense variants; green, intronic and UTR variants; blue, frameshift variants; yellow, nonsense variants; and purple, deletion variants. N gives the number of independent probands/families with a particular allele. B The frequency with which individual alleles were identified amongst probands. Self-reported ethnicities were given by color: Western Europe, red; Polish, blue; Unknown, green; Ashkenazi, purple; Mixed, orange; Eastern Europe, yellow. C Distribution of HMs in those with the CHEK2 p.I157T allele. Color indicates the order of the malignancy for those diagnosed with multiple cancers: first cancer, red; second cancer, blue; third cancer, green. D Cytogenetic and molecular profiles of the myeloid malignancies that developed in those with germline P/LP CHEK2 variants. E–G Representative pedigrees from probands with P/LP germline CHEK2 variants. The proband is indicated by the triangle. The germline CHEK2 variant is indicated for those family members who were genotyped. Cancer diagnoses are given below each pedigree member along with the age at diagnosis (dx.) or death (dc.) when known. Circles indicate women, and squares, men. The generation number is given to the left in Roman numerals. (ns not significant. * P < 0.05, ** P < 0.01, *** P < 0.001).
Among all probands and family members (patients) with CHEK2-path, 74% (59/80) had a diagnosed HM; 54% (32/59) had a myeloid malignancy, 41% (24/59) had a lymphoid malignancy, and 5% (3/59) had both myeloid and lymphoid malignancies. These myeloid malignancies occurred as second malignancies in 34% (12/35) of patients. In the subset of patients with CHEK2 p.I157T (55%, 44/80), 77% (34/44) had a HM; 56% (19/34) had a myeloid malignancy, 41% (14/34) had a lymphoid malignancy, and 3% (1/34) had both myeloid and lymphoid malignancies (Fig. 2C). In the CHEK2 p.I157T subset, (14/44) had ≥2 malignancies diagnosed over their respective lifetimes, versus 39% (14/36) for other P/LP CHEK2 variants (P = 0.638, Fisher’s exact), and 17% (2/12) for CHEK2 VUS (P = 0.475, Fisher’s exact). Less than half of the myeloid malignancies in CHEK2 p.I157T patients were second malignancies (42%, 8/19). In the CHEK2 p.I157T group, the frequency of breast cancer was 9% (4/44) and prostate cancer 11% (5/44). For other P/LP CHEK2 variants, the frequency of breast cancer was 21% (9/36) and prostate cancer 5% (2/36).
Among all diagnosed myeloid malignancies in CHEK2-path patients (N = 35), 34% (12/35) had AML, 26% (9/35) had MDS, 31% (11/35) had MPN, and 9% (3/35) had MDS/MPN (Fig. 2D). In AML patients with CHEK2-path, 25% (4/12) had CBFB-MYH11 [inv(16)(p13.1q22.1)] rearrangement and 42% (5/12) had a normal karyotype. Somatic variants were similar to those seen in de novo AML. When comparing CHEK2-path AML patient cytogenetics with the distribution observed in unselected, de novo AML patients from the TCGA AML dataset, core binding factor (CBF) rearrangements [CBFB-MYH11 or RUNX1-RUNX1T1] were more common in patients with CHEK2-path variants (25%, 4/12) versus TCGA de novo AML (10%, 19/200; P = 0.029, Fisher’s exact; Supplemental Fig. 1A). MDS/MPN was seen in three patients, all of whom had chronic myelomonocytic leukemia (CMML). Among MPN patients, 36% (4/11) had BCR-ABL fusions, and 27% (3/11) had myelofibrosis. Representative pedigrees of CHEK2 p.I157T families with AML and inv(16)/CBFB-MYH11 are shown (Fig. 2E); and with both myeloid and lymphoid malignancies (Fig. 2F, G). Treatment courses for individuals with germline CHEK2 p.I157T and myeloid (Supplemental Fig. 2A) or lymphoid (Supplemental Fig. 2B) malignancies are also demonstrated. Among CHEK2 p.I157T patients with myeloid malignancies, 40% (8/20) had a previous solid organ malignancy; 15% (3/20) had surgical treatment only, and 25% (5/20) had chemotherapy or radiation, one of whom had radiation for an antecedent Hodgkin lymphoma. Among CHEK2 p.I157T patients with lymphoid malignancies, 33% (5/15) had a previous solid organ malignancy; 27% (4/15) had surgical treatment only, and 7% (1/15) had prior chemotherapy treatment.
Within the subset of the sequential probands that received routine clinical testing and had a personal history of myeloid or lymphoid malignancy and were tested for a clinical indication of suspected HHM, the frequency of CHEK2-path variants was 3% (30/851). When compared to the allele frequencies within a control population database (gnomAD v4.1.0, Supplemental Table 4), we observed a significant enrichment of the p.I157T (OR 6.44, 95% CI 3.86-10.73, P < 0.001) and p.S428F (OR 17.16, 95% CI 6.40-46.03, P < 0.001) alleles in patients with HMs, although the p.T367fs (OR 0.68, 0.10–4.87, P = 0.704) allele was not significant (Table 1).
Characteristics of patients with CHEK2 p.I157T in a solid tumor risk cohort
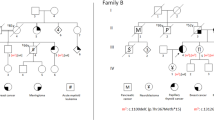
We identified 42 probands with CHEK2 p.I157T at the University of Utah who underwent hereditary testing after referral for solid tumor hereditary cancer risk assessment. Among 42 probands, 71% (30/42) had a personal history of malignancy (Fig. 3A): primarily breast (31%, 13/42) and prostate (12%, 12/42) cancers, with one patient (2%, 1/42) having lymphoma (Fig. 3B). However, a total of 36% (15/42) of these probands reported a family history of HM (Fig. 3C), most commonly leukemia (19%, 8/42) followed by lymphoma (12%, 5/42), with two family histories containing both (5%, 2/42) (Fig. 3D). Representative pedigrees demonstrating diagnoses of both leukemias and lymphomas (Fig. 3E) and both solid and HMs (Fig. 3F, G) are shown.

Characteristics of patients tested for solid tumor risk indications at the University of Utah who have the CHEK2 p.I157T variant. A Personal history of cancer for those with the CHEK2 p.I157T allele. B Primary malignancy type for those with the CHEK2 p.I157T allele who were diagnosed with cancer. C Family HM history for those with the CHEK2 p.I157T allele. D Type of HM within the family for those with the CHEK2 p.I157T allele. E–G Representative pedigrees of patients with the germline CHEK2 p.I157T allele from the University of Utah cohort. The proband is indicated by the triangle. The germline CHEK2 variant is indicated for those family members who were genotyped. Cancer diagnoses are given below each pedigree member along with the age at diagnosis (dx.) or deceased (dc.) when known. Circles indicate women, and squares, men. The generation number is given to the left in Roman numerals.
Phenome-wide and genome-wide association studies
Within the UKBB, we identified 4529 predicted loss of function (pLoF) gene burden associations for CHEK2-path variants by PheWAS (Supplemental Table 5). Among 36 phenotype terms significant at a SKAT-O P-value of <2.5e−6, 36% (13/36) were associated with blood count abnormalities (Fig. 4A). The top phenotype term was platelet crit (P = 2.33e−31, β = 0.008975), followed by age at menopause (P = 2.62e−31, β = 0.019220), and white blood cell (WBC) count (P = 2.30e−26, β = 0.008718). The two specific ICD-10 malignancy codes observed were C50 Malignant Neoplasm of Breast (P = 2.16e−18, β = 0.042475) and C92 Myeloid leukaemia (P = 5.78e−7, β = 0.092176). Within the UKBB, a GWAS for ICD-10 code C92 Myeloid leukaemia showed the strongest association with pLoF variants in the genes TET2 (P = 1.53e−25, β = 0.176629), EZH2 (P = 1.53e−8, β = 0.430923), and CHEK2 (P = 5.78e−7, β = 0.092176) (Supplemental Table 6). Other known myeloid leukemia predisposition genes, including DDX41 (P = 3.68e−4, β = 0.115770) and RUNX1 (P = 3.99e−4, β = 0.176099), were identified at a lower significance level than seen for CHEK2 pLoF variants (Fig. 4B).

A Phenome wide association study (PheWAS) for CHEK2 predicted loss of function (pLoF) variants from the UK Biobank (UKBB). Hematologic phenome terms are indicated by dark red circles and malignancy diagnoses by light green circles. B Genome-wide association study (GWAS) from the UK Biobank for ICD-10 code C92 myeloid malignancy from the UK Biobank, significance lines indicated at -log10(5e-08) and -log10(5e-04) levels. C Cumulative proportion of participants from UKBB with malignant cancer who are CHEK2-wild type (wt) or carry pathogenic/likely pathogenic variants (CHEK2-path) by age of onset. D Cumulative proportion of CHEK2-path UKBB participants with HM diagnosis by age of onset.
In the UKBB population, CHEK2 was the most observed HM risk allele (0.77%, 3867/502,410) followed by BRCA2 (0.30%, 1504/502,410), ATM (0.30%, 1485/502,410), DDX41 (0.15%, 750/502,410), BRCA1 (0.11%, 533/502,410), TP53 (0.01%, 61/502,410), and RUNX1 (0.00%, 1/502,410) (Supplemental Table 9). To assess an association between CHEK2 pLoF variants and HMs, we performed univariate and multivariate logistic regression for sex, age, CHEK2 status, and smoking status after filtering out UKBB participants with non-CHEK2 likely germline cancer risk alleles (Supplemental Fig. 3). For all HMs, CHEK2 status was significant on multivariate analysis (OR 1.91, 95% CI 1.52–2.37, P < 0.001; Supplemental Fig. 4A). For myeloid malignancies, CHEK2 status was also significant on multivariate analysis (OR 2.34, 95% CI 1.31–3.82, P = 0.002) (Supplemental Fig. 4B). CHEK2 status was also significant for all malignant cancers, solid tumors, lymphoid malignancies, breast malignancy, prostate malignancy, and thyroid malignancy, but was not significant for lung or colon malignancy (Supplemental Fig. 5; Supplemental Table 10). In cancer patients with likely germline CHEK2LoF variants, the penetrance for any malignancy was 29.27% by age 90 (Fig. 4C). The penetrance by age 90 for solid tumors was 25.97% (Supplemental Fig. 6A); and for all HMs, was 2.07% (Fig. 4D). Among UKBB participants with AML, 58% (11/19) were de novo, and 42% (8/19) were secondary AML (Supplemental Fig. 6B).
Identification of CHEK2 variants in public MDS/AML datasets
CHEK2 variants were called from bulk RNA-seq reads in three large public AML datasets (BEAT AML, Leucegene, AML PMP) to ensure consistency among studies [39,40,41]. We identified P/LP CHEK2 variants in 1.2% (16/1348) of AML patients in the combined dataset (Fig. 5A). The most common CHEK2 variant was p.I157T in 25% (4/16) followed by p.T367fs in 13% (2/16). Only 6% (1/16) were identified as therapy-related myeloid neoplasms in the metadata. There was no difference in the mean age at sampling between those with P/LP CHEK2 variants (CHEK2-path) (56.33 years) versus wild type (CHEK2-wt) (55.11 years) (P = 0.774) (Fig. 5B). There was no difference in the number of females with CHEK2-path (44%, 7/16) versus CHEK2-wt (44%, 563/1283) (P > 0.999). There was no difference in the mean WBC at sampling between CHEK2-path (37.38×109) versus CHEK2-wt (49.04 × 109) (P = 0.492) (Fig. 5C). We did not observe an enrichment for CBF fusions in CHEK2-path AML patients at 0% (0/16) versus CHEK2-wt at 9% (128/1348) (P = 1, Fisher’s exact). However, we did observe an enrichment for GATA2-MECOM fusions in CHEK2-path at 19% (3/16) versus CHEK2-wt at 1% (17/1348) (P = 0.001, Fisher’s exact) and non-MLLT3 KMT2A rearrangements at 19% (3/16) in CHEK2-path versus CHEK2-wt at 2% (31/1348) (P = 0.006, Fisher’s exact) (Supplemental Fig. 1B). We then examined the frequency of CHEK2-path variants in a genetically distinct cohort of MDS patients with bulk RNA-seq from bone marrow mononuclear or CD34+ cells [42]. We identified CHEK2-path variants in 2% (5/214) of MDS patients in this dataset, and did not observe CHEK2 p.I157T, consistent with the expected population distribution of this allele (Fig. 5D).

A Somatic mutational landscape and disease characteristics of patients with AML with identified P/LP CHEK2 variants. B Median age at time of disease sample in AML patients who are CHEK2-wt (blue, left) vs. those with CHEK2-path variants (red, right). C Burden of bone marrow blasts at time of disease sample in AML patients who are CHEK2-wt (blue, left) vs. CHEK2-path (red, right). D Frequency of CHEK2-path variants in a cohort of MDS patients with bulk RNA-seq from bone marrow mononuclear cells or CD34+ cells. (ns not significant, * P < 0.05, ** P < 0.01, *** P < 0.001).
Generation and characterization of a Chek2 p.I161T mouse model
The human and mouse CHK2 proteins share 83.5% amino acid identity with the mouse CHK2 I161 residue corresponding to the human CHK2 I157 residue (Supplemental Fig. 7). We generated a p.I161T C57BL/6 J knock-in mouse, and confirmed the presence of this allele in tail, bone marrow, and spleen; expression of both wild-type and p.I161T alleles was confirmed in the spleen of Chek2p.I161T/wt mice (Supplemental Fig. 8). Both heterozygotes (Chek2p.I161T/wt) and homozygotes (Chek2p.I161T/p.I161T) were viable and fertile, without developmental defects. Comparable gene and protein expression of Chek2 was identified in bone marrow and spleen for both heterozygotes and homozygotes with the Chek2 p.I161T allele (Supplemental Fig. 9).
Longitudinal characterization of Chek2 p.I161T mice over 24 months demonstrated a progressive increase in WBC for both Chek2p.I161T/wt (P < 0.001, ANOVA) and Chek2p.I161T/p.I161T (P < 0.001, ANOVA) mice versus wt (Supplemental Fig. 10A). There was an increase in absolute neutrophil count (ANC) in Chek2p.I161T/p.I161T (P < 0.001, ANOVA) mice, but not in Chek2p.I161T/wt (P = 0.514, ANOVA) mice versus wt (Supplemental Fig. 10B). There was an increase in absolute lymphocyte count (ALC) for both Chek2p.I161T/wt (P < 0.001, ANOVA) and Chek2p.I161T/p.I161T (P < 0.001, ANOVA) mice versus wt (Supplemental Fig. 10C). At endpoint, although there was no difference in hemoglobin levels among Chek2p.I161T/wt (P = 0.212), Chek2p.I161T/p.I161T (P = 0.163) mice, and wt (Supplemental Fig. 10D), we observed a higher platelet count in Chek2p.I161T/wt (P = 0.018) but not Chek2p.I161T/p.I161T (P = 0.058) as compared to wt mice (Supplemental Fig. 10E). We observed an inferior overall survival for both Chek2p.I161T/wt (P = 0.037) and Chek2p.I161T/p.I161T (P = 0.005) compared to wt mice, although this difference only emerged after the mice had been aged for more than 18 months (Fig. 6A).

A Survival in Chek2wt/wt (blue, N = 13), Chek2p.I161T/wt (red, N = 16), vs. Chek2p.I161T/p.I161T (green, N = 11) mice. B Representative flow plots from lineage depleted marrow demonstrating the Lin-CD34+ and Lin-Sca1+cKit+ compartments in young (2-6 month) Chek2 wt (Chek2wt/wt), heterozygous (Chek2p.I161T/wt), and homozygous (Chek2p.I161T/p.I161T) mice. C Comparison of Lin-CD34+ compartment in young Chek2 mice. D Comparison of Lin-cKit+ compartment in young Chek2 mice. E Comparison of the Lin-Sca1+cKit+ compartment in young Chek2 mice. F Representative flow plots from whole bone marrow nucleated cells (BMNC) demonstrating the CD34+ and cKit+ in aged (24 month) Chek2 wt (Chek2wt/wt), heterozygous (Chek2p.I161T/wt), and homozygous (Chek2p.I161T/p.I161T) mice without pathologic evidence of malignancy. G Comparison of cKit+ compartment in aged Chek2 mice. H Comparison of CD34+ compartment in aged Chek2 mice. I Pre-ranked gene set enrichment analysis (GSEA) for differentially expressed genes from Lin-CD34+ cells from young (aged 5-6 month) Chek2 mice. Highlighted pathways are significant at a false discovery rate (FDR) of <0.25. Pathways from MSigDb m2.all gene set with some titles abbreviated. Full pathway titles and details are in Supplemental Table 7. J–L Summary of consensus diagnosis obtained by analysis of gross features, histology, IHC, and multicolor flow cytometry data from wt (Chek2wt/wt), heterozygous (Chek2p.I161T/wt), or homozygous (Chek2p.I161T/p.I161T) mice. M Comparison of mice with identified pathologic abnormalities at endpoint for Chek2-wt (left) vs. Chek2-mt (right), including heterozygous or homozygous mice. (ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001).
Hematopoietic stem and progenitor cell (HSPC) defects and assessment for clonal hematopoiesis (CH) in the Chek2 p.I161T mouse model
To assess the effects of the Chek2 p.I161T allele on the HSPC compartment, we stained lineage (Lin)-depleted BMNC from Chek2 mice younger than six months with antibodies against Lin, CD34, cKit, and Sca-1 and performed flow cytometry (Fig. 6B). We observed a reduction in Lin-CD34+ cells in mice with Chek2p.I161T/wt (P = 0.030) or Chek2p.I161T/p.I161T (P = 0.045) versus wild-type, but no difference between Chek2p.I161T/wt and Chek2p.I161T/p.I161T (P = 0.604) mice (Fig. 6C). Similarly, we observed a reduction in Lin-cKit+ cells in Chek2p.I161T/wt (P = 0.013) or Chek2p.I161T/p.I161T (P = 0.002) versus wild-type mice, but saw no difference between Chek2p.I161T/wt and Chek2p.I161T/p.I161T (P = 0.204) mice (Fig. 6D). Interestingly, there was no difference in the Lin-Sca1+cKit+ (LSK) compartment between Chek2p.I161T/wt (P = 0.640) or Chek2p.I161T/p.I161T (P = 0.800) versus wild-type mice, nor between Chek2p.I161T/wt and Chek2p.I161T/p.I161T (P = 0.911) mice (Fig. 6E).
To assess these features, mice ~24 months of age were sacrificed without evidence of malignancy, and we examined the proportion of whole blood mononuclear cells (BMNC) that was CD34+ or cKit+ (Fig. 6F). We observed an increase in the cKit+ population in Chek2p.I161T/wt (P = 0.001) and Chek2p.I161T/p.I161T (P = 0.006) versus wild-type mice, with no difference between Chek2p.I161T/wt and Chek2p.I161T/p.I161T (P = 0.056) mice (Fig. 6G). We observed no difference in the CD34+ compartment among Chek2p.I161T/wt (P = 0.106) and Chek2p.I161T/p.I161T (P = 0.436) versus wild-type mice, with no difference between Chek2p.I161T/wt and Chek2p.I161T/p.I161T (P = 0.968) mice (Fig. 6H).
We assessed transcriptional differences and changes to underlying signaling pathways by performing bulk RNA-seq on sorted Lin-CD34 + BMNC from mice aged 5-6 months. We observed differential gene expression (DGE) between Chek2-wt and Chek2-mutant (mt) (combined heterozygotes and homozygotes), with 22 genes being up- or downregulated (log2FC ± 1.5, P < 0.05) (Supplemental Fig. 11A). We observed similar DGE in Chek2p.I161T/wt and Chek2p.I161T/p.I161T separately versus wt (Supplemental Fig. 11B, C). but saw minimal DGE between Chek2p.I161T/wt and Chek2p.I161T/p.I161T (Supplemental Fig. 11D). Unsupervised clustering grouped Chek2wt/wt samples together, with Chek2p.I161T/wt and Chek2p.I161T/p.I161T samples clustering separately (Supplemental Fig. 11E).
We identified ten upregulated gene sets (Supplemental Table 7) and seven downregulated gene sets gene set enrichment analysis (GSEA) at a false discovery rate (FDR) < 0.25. (Fig. 6I) Several hematopoietic-specific transcription factor (TF) networks were upregulated, including CBFA2T3 targets (NES 1.9062, nominal P < 0.001, FDR q-value 0.0199), GATA1 targets (NES 1.8583, nominal P < 0.001, FDR q-value 0.0402), and MLL targets (NES 1.7717, nominal P = 0.002, FDR q-value 0.0864). Some pathways important for epigenetic regulation and differentiation were identified in the downregulated gene list, including PRC2 Methylates Histones and DNA (NES -1.8309, nominal P < 0.001, FDR q-value 0.0152) and Epigenetic Regulation of Gene Expression (NES -1.6826, nominal P < 0.001, FDR q-value 0.1711). Downregulated pathways not significant at FDR < 0.25 included DNA DSB Response (NES -1.4846, nominal P = 0.0119, FDR q-value 0.6892) and NHEJ (NES -1.5063, nominal P 0.0261, FDR q-value 0.6954) (Supplemental Table 7).
To assess for the presence of CH, we performed deep WES on Chek2 mice less than ten months old. After variant calling and filtering (Supplemental Fig. 12), we did not identify differences in the number of somatic variants in Chek2p.I161T/wt (P = 0.252) or Chek2p.I161T/p.I161T (P = 0.552) versus Chek2wt/wt mice, a difference in VAF, or allele type (Supplemental Fig. 13). There was also no difference in the top 100 somatic variants by Chek2 genotype (Supplemental Fig. 14).
The Chek2 p.I161T mouse model develops HMs
We comprehensively profiled aged Chek2 mice that were sacrificed at humane endpoint or 24 months. Several mice demonstrated abnormal necropsy findings (Supplemental Table 8). For example, one mouse had a disorganized hypercellular marrow with megakaryocyte proliferation (Supplemental Fig. 15A) and abnormal circulating monocytoid cells (Supplemental Fig. 15B) and abnormal circulating lymphocytes in another mouse (Supplemental Fig. 15C). Immunohistochemical stains from three separate mice showed diffuse infiltration with PAX5+ cells in a mass (Supplemental Fig. 15D) without CD3+ infiltrate (Supplemental Fig. 15E), diffuse splenic infiltration with lymphocytes that was PAX5- (Supplemental Fig. 15F) and CD3+ (Supplemental Fig. 15G), and a mass with a lymphocytic infiltrate that was PAX5- (Supplemental Fig. 15H) and CD3+ (Supplemental Fig. 15I). Within the Chek2wt/wt group, 85% (11/13) had no overt pathology, 8% (1/13) had no viable tissue, and 8% (1/13) had evidence of a myeloid proliferation (Fig. 6J). For the Chek2p.I161T/wt group, 50% (8/16) had no overt pathology, 13% (2/16) had no viable tissue, 25% (4/16) had evidence of a B-cell lymphoproliferative disorder (LPD), and 13% (2/16) had evidence of a T-cell LPD (Fig. 6K). In the Chek2p.I161T/p.I161T group, 27% (3/11) had no overt pathology, 18% (2/11) had no viable tissue, 9% (1/11) had a myeloid proliferation, 27% (3/11) had a B-cell LPD, and 18% (2/11) had evidence of a T-cell LPD or leukemia (Fig. 6L). More mice had pathologic abnormalities suggestive of a hematopoietic disorder in the Chek2-mt group versus Chek2-wt (P = 0.030, Fisher’s exact; Fig. 6M). There was no difference in the number of mice with abnormalities among males (50%, 7/14) versus females (38%, 5/13; P = 0.704).
Representative flow plots demonstrate: a CD3 + CD4 + T-cell leukemia in a Chek2p.I161T/p.I161T mouse (Supplemental Fig. 16A), transplantable to a CD45.1 recipient (Supplemental Fig. 16B), and clonality demonstrated by the presence of a dominant clone by TCR sequencing (Supplemental Fig. 17A); a CD34 + CD11b + CD117+ proliferation in a Chek2p.I161T/p.I161T mouse (Supplemental Fig. 16C); and a B-cell lymphoproliferative disease (LPD) in a Chek2p.I161T/wt mouse (Supplemental Fig. 16D), with a dominant clone identified by BCR sequencing (Supplemental Fig. 17B).
Discussion
Our findings support deleterious germline CHEK2 variants, and specifically p.I157T, as being a predisposition gene for the development of myeloid and possibly lymphoid malignancies. We observed evidence of a HM phenotype in patients and families with P/LP CHEK2 variants in a HHM cohort, as well as a separate validation cohort that was tested for solid tumor hereditary cancer risk. The presence of strong family histories of leukemia in individuals with deleterious germline CHEK2 variants who were tested for inherited cancer risk based on solid tumor history suggests that it is the P/LP CHEK2 variant driving the observed HM predisposition, rather than being a second order effect arising from the selected nature of our HHM cohort. We also observe that, on a population level, LoF CHEK2 variants associate with hematopoietic abnormalities and myeloid leukemias; although CHEK2 was below the conventional 5e−8 threshold for GWAS analysis, HMs are underrepresented in the UKBB, limiting sensitivity in this context. Interestingly, CHEK2 arises as one of the genes most strongly associated with myeloid leukemias by GWAS, suggesting a link between P/LP CHEK2 P/LP variants and myeloid leukemias, but not implying high penetrance. P/LP variants that are higher penetrance but rarer are likely underrepresented in the UK Biobank, which may explain why we observe RUNX1 and DDX41 at a lower significance level in this analysis. We were unable to assess formally for the penetrance of CHEK2 P/LP variants on the HM phenotype in our cohort. We also observe that CHEK2 P/LP variants do not seem to be highly specific to a specific disease subtype within the family of myeloid diseases, though most patients had MDS or AML. However, we do see that there is an enrichment in some recurrent translocation events; within the original HM cohort CBF rearrangements were enriched, but surprisingly, we did not observe this in the broader AML cohort from public datasets. Instead, we saw enrichment of both GATA2-MECOM and t(v;11q23.3)/KMT2A rearrangements. These findings suggest that CHEK2 P/LP variants may be a proximal risk factor for the development of translocation events, perhaps related to defective homologous recombination pathways, rather than a specific HM disease state. This could also explain the spectrum of HMs we observed, as translocation events are not exclusive to a single HM subtype [47]. Our findings also suggest CHEK2-associated HHM syndromes may be relatively common, based on our population data and the frequency in AML datasets, consistent with previous studies [30,31,32]. Overall, in public datasets, we observed 1-2% of MDS/AML patients had P/LP CHEK2 variants with a comparable age of onset to non-CHEK2 mutated HM patients, with only 6% of these being clearly identified as therapy-related, and a possible enrichment for recurrent translocation events in P/LP CHEK2 carriers with MDS/AML.
Our findings also suggest that CHEK2 likely plays a role in the HSPC compartment. Although we saw Lin-CD34+ and Lin-cKit+ populations were reduced in young Chek2 mice, the LSK compartment was unchanged, suggesting that the impact of Chek2 p.I161T is likely manifested at the level of an oligopotent progenitor (e.g., common myeloid progenitor) rather than a long-term hematopoietic stem cell. Interestingly, in older mice, we observed a reversal in this phenomenon with increased cKit+ cells. There are many possible mechanisms for this, but evolving CH as a compensatory mechanism for a HSPC defect is possible, given a similar phenomenon is seen in patients with germline marrow failure syndromes such as SAMD9/SAMD9L [48]. Although we tested younger mice for the presence of CH by deep WES performed on PB, we did not observe the presence of CH, likely reflecting the young age of the mice; limitations of using PB in mice; and the lack of serial blood draws within this cohort. Many of the differentially enriched pathways in our GSEA relate to hematopoiesis-specific TFs, raising the possibility that CHK2 is either directly or indirectly regulating these TFs. CHEK2 is known to interact with several proteins that affect leukemia-specific pathways, such as KMT2D and FOXM1, but this remains to be explored further [49]. These findings suggest CHEK2 may be relevant to HSPC regulation. Our mouse model also demonstrates that mice carrying Chek2 p.I161T in the heterozygous or homozygous state have worse survival than Chek2 wild-type mice, and that this is likely related to HM development. Future studies will examine the role that cytotoxic and radiation exposures have on the development of HMs in Chek2-mutant mice as deeper understanding regarding the interactions among CHEK2 mutational status, environmental exposures, and HM risk will be crucial. Future in vitro studies should also examine whether certain drugs that are particularly effective in cells with defects in homologous recombination DNA repair (e.g., PARP inhibitors) are effective in eliminating malignant, CHEK2-defective hematopoietic cells.
Limitations of our study include the selected nature of our HHM cohort, being mostly European-derived, and our inability to correct for differences in ethnic composition versus gnomAD. Future studies should examine CHEK2 variants in diverse populations. Sample size limitations also prevented detailed assessment of all disease subtypes. The role for co-inherited modifiers of the CHEK2 phenotype also could not be assessed in this study. We are also limited in knowing the germline status of CHEK2 variants in public datasets, although somatic CHEK2 variants are rare [31]. We were also limited in our mouse studies by the rarity of aged (> 18 mo) Chek2 mice, resulting in a need to perform several experiments on younger mouse cohorts.
Our findings establish that CHEK2 should be routinely tested in all patients being assessed for a HHM syndrome, and likely should be tested for in all patients with a myeloid malignancy. Our observed frequency of 1% in the AML cohort and 2% in the MDS cohort is probably an underestimate, given that any variant not producing a transcript would not have been identified. Identification of CHEK2 variants in HM patients has important implications for counseling and cascade testing, selection of allogeneic hematopoietic stem cell transplant donors, and may inform future clinical trials of targeted treatment approaches for this genetically distinct patient group.
Data availability
Sequencing data are deposited in SRA (PRJNA1119484) and GEO (GSE268966).
Code availability
Code used for UK Biobank data analysis can be found on the Godley Lab github site: https://github.com/LucyGodley/UKB_CHEK2.
References
Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7.
Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–7.
Stracker TH, Usui T, Petrini JH. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair. 2009;8:1047–54.
Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–8.
Stolarova L, Kleiblova P, Janatova M, Soukupova J, Zemankova P, Macurek L, et al. Germline variants in cancer predisposition: stalemate rather than checkmate. Cells 2020;9:2675.
Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–9.
Cybulski C, Górski B, Huzarski T, Masojć B, Mierzejewski M, Debniak T, et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet. 2004;75:1131–5.
Girard E, Eon-Marchais S, Olaso R, Renault AL, Damiola F, Dondon MG, et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int J Cancer. 2019;144:1962–74.
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017;3:1190–6.
Desrichard A, Bidet Y, Uhrhammer N, Bignon YJ. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. 2011;13:R119.
Seppälä EH, Ikonen T, Mononen N, Autio V, Rökman A, Matikainen MP, et al. CHEK2 variants associate with hereditary prostate cancer. Br J Cancer. 2003;89:1966–70.
Kaczmarek-Ryś M, Ziemnicka K, Hryhorowicz ST, Górczak K, Hoppe-Gołębiewska J, Skrzypczak-Zielińska M, et al. The c.470 T > C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered Cancer Clin Pr. 2015;13:8.
Zhunussova G, Afonin G, Abdikerim S, Jumanov A, Perfilyeva A, Kaidarova D, et al. Mutation Spectrum of Cancer-Associated Genes in Patients With Early Onset of Colorectal Cancer. Front Oncol. 2019;9:673.
Obazee O, Archibugi L, Andriulli A, Soucek P, Małecka-Panas E, Ivanauskas A, et al. Germline BRCA2 K3326X and CHEK2 I157T mutations increase risk for sporadic pancreatic ductal adenocarcinoma. Int J Cancer. 2019;145:686–93.
Kinnersley B, Kamatani Y, Labussière M, Wang Y, Galan P, Mokhtari K, et al. Search for new loci and low-frequency variants influencing glioma risk by exome-array analysis. Eur J Hum Genet. 2016;24:717–24.
Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–59.
Oropeza E, Seker S, Carrel S, Mazumder A, Lozano D, Jimenez A, et al. Molecular portraits of cell cycle checkpoint kinases in cancer evolution, progression, and treatment responsiveness. Sci Adv. 2023;9:eadf2860.
Sutcliffe EG, Stettner AR, Miller SA, Solomon SR, Marshall ML, Roberts ME, et al. Differences in cancer prevalence among CHEK2 carriers identified via multi-gene panel testing. Cancer Genet. 2020;246-247:12–17.
Havranek O, Kleiblova P, Hojny J, Lhota F, Soucek P, Trneny M, et al. Association of Germline CHEK2 Gene Variants with Risk and Prognosis of Non-Hodgkin Lymphoma. PLoS ONE. 2015;10:e0140819.
Rudd MF, Sellick GS, Webb EL, Catovsky D, Houlston RS. Variants in the ATM-BRCA2-CHEK2 axis predispose to chronic lymphocytic leukemia. Blood. 2006;108:638–44.
Janiszewska H, Bak A, Pilarska M, Heise M, Junkiert-Czarnecka A, Kuliszkiewicz-Janus M, et al. A risk of essential thrombocythemia in carriers of constitutional CHEK2 gene mutations. Haematologica. 2012;97:366–70.
Janiszewska H, Bąk A, Hartwig M, Kuliszkiewicz-Janus M, Całbecka M, Jaźwiec B, et al. The germline mutations of the CHEK2 gene are associated with an increased risk of polycythaemia vera. Br J Haematol. 2016;173:150–2.
Janiszewska H, Bąk A, Skonieczka K, Jaśkowiec A, Kiełbiński M, Jachalska A, et al. Constitutional mutations of the CHEK2 gene are a risk factor for MDS, but not for de novo AML. Leuk Res. 2018;70:74–8.
Stubbins RJ, Korotev S, Godley LA. Germline CHEK2 and ATM Variants in Myeloid and Other Hematopoietic Malignancies. Curr Hematol Malig Rep. 2022;17:94–104.
Silver AJ, Bick AG, Savona MR. Germline risk of clonal haematopoiesis. Nat Rev Genet. 2021;22:603–17.
Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–8.
Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. 2016;128:1121–8.
Schreurs MAC, Schmidt MK, Hollestelle A, Schaapveld M, van Asperen CJ, Ausems MGEM, et al. Cancer risks for other sites in addition to breast in CHEK2 c.1100delC families. Genet Med 2024;26:101171.
Bao EL, Nandakumar SK, Liao X, Bick AG, Karjalainen J, Tabaka M, et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature. 2020;586:769–75.
Yang F, Long N, Anekpuritanang T, Bottomly D, Savage JC, Lee T, et al. Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult AML patients. Blood. 2022;139:1208–21.
Feurstein S, Churpek JE, Walsh T, Keel S, Hakkarainen M, Schroeder T, et al. Germline variants drive myelodysplastic syndrome in young adults. Leukemia. 2021;35:2439–44.
Feurstein SK, Trottier AM, Estrada-Merly N, Pozsgai MJ, McNeely KE, Drazer MW, et al. Germline predisposition variants occur in myelodysplastic syndrome patients of all ages. Blood. 2022;140:2533–48.
Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024;625:92–100.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–37.
Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30:2375–6.
Juliusdottir T. topr: an R package for viewing and annotating genetic association results. BMC Bioinf. 2023;24:268.
Docking TR, Parker JDK, Jädersten M, Duns G, Chang L, Jiang J, et al. A clinical transcriptome approach to patient stratification and therapy selection in acute myeloid leukemia. Nat Commun. 2021;12:2474.
Burd A, Levine RL, Ruppert AS, Mims AS, Borate U, Stein EM, et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med. 2020;26:1852–8.
Macrae T, Sargeant T, Lemieux S, Hébert J, Deneault E, Sauvageau G. RNA-Seq reveals spliceosome and proteasome genes as most consistent transcripts in human cancer cells. PLoS ONE. 2013;8:e72884.
Shiozawa Y, Malcovati L, Gallì A, Pellagatti A, Karimi M, Sato-Otsubo A, et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood. 2017;130:2642–53.
Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood. 2022;140:1345–77.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinf. 2013;43:11.10.11–11.10.33.
Kraft IL, Basdag H, Koppayi A, Rodgers CV, Saygin C, Haribabu Y, et al. Sequential tumor molecular profiling identifies likely germline variants. Genet Med. 2024;26:101037.
Niroula A, Sekar A, Murakami MA, Trinder M, Agrawal M, Wong WJ, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med. 2021;27:1921–7.
Sahoo SS, Pastor VB, Goodings C, Voss RK, Kozyra EJ, Szvetnik A, et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat Med. 2021;27:1806–17.
Wu Q, Fang C, Wang X, Huang S, Weng G. CHEK2 is a potential prognostic biomarker associated with immune infiltration in clear cell renal cell carcinoma. Sci Rep. 2023;13:21928.
Acknowledgements
The authors thank the patients and families who contributed to the clinical cohorts as well as the UC Transgenic Mouse Facility (RRID:SCR_019171), especially Linda Degenstein and Xiu Chen, for their assistance with the construction and generation of the Chek2 p.I161T knock-in mice. The UC Transgenic Mouse Facility is supported by the UC Cancer Center Support Grant P30CA014599. All animals were cared for under an IACUC-approved protocol according to ethical standards. We also thank Adam Gordon and Masha Kocherginsky from Northwestern University for their statistical support. RJS was supported by the Clinician Investigator Program of BC, Leukemia Lymphoma Society of Canada, and the Canadian Institutes of Health Research. EC was supported by the Victoria Fellowship “veski” in Life Sciences and The Victorian Comprehensive Cancer Centre (VCCC) Picchi Award for Excellence in Cancer Research (Clinical Science). JB was supported by The Terry Fox New Frontiers Program Project Grant (#1074 to AK). Study funding was provided by the Cancer Research Foundation Fletcher Award (LAG), Leukemia and Lymphoma Society Blood Cancer Discoveries Grant Program (LAG), and V Foundation for Cancer Research (LAG). This research has been conducted using the UK Biobank Resource under Application Number 83200.
Author information
Authors and Affiliations
Contributions
RJS designed and performed the research, contributed vital new analytical tools, analyzed data, designed the figures and tables, and wrote and edited the manuscript; SA designed and performed the research, analyzed data, and edited the manuscript; JV compiled a patient cohort, analyzed data, and edited the manuscript; AA performed research with the Chek2 knock-in mice, analyzed data, and edited the manuscript; MP assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; MP assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; IJA performed research with the Chek2 knock-in mice, analyzed data, and edited the manuscript; EC assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; JB performed the research, contributed analytical tools, analyzed data, and edited the manuscript; AKarsan performed the research, contributed analytical tools, analyzed data, and edited the manuscript; CR performed the research, analyzed data, and edited the manuscript; AKoppayi performed the research, contributed analytical tools, analyzed data, and edited the manuscript; HB performed the research, analyzed data, and edited the manuscript; MWD assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; SD assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; JC assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; AEGO assisted in the analysis of a patient cohort, analyzed data, and edited the manuscript; and LAG designed and supervised the research, analyzed the data, edited the figures and tables, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
RJS has received honoraria or participated on advisory boards from AbbVie, Jazz Pharamceuticals, Pfizer, Astellas, Kite/Gilead, BMS, and Sobi. He has received research funding from Jazz Pharmaceuticals. LAG receives royalties from UptoDate, Inc.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stubbins, R.J., Arnovitz, S., Vagher, J. et al. Predisposition to hematopoietic malignancies by deleterious germline CHEK2 variants. Leukemia 39, 1702–1713 (2025). https://doi.org/10.1038/s41375-025-02635-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-025-02635-1
This article is cited by
-
CHEK2 loss endows chemotherapy resistance to hematopoietic stem cells
Leukemia (2026)
-
Frequent and clinically relevant germline DNA repair gene variants in young and familial myeloproliferative neoplasms
Blood Cancer Journal (2026)
-
Impact of Germline DNA Repair Mutations on Clonal Hematopoiesis and Myeloid Neoplasm Development
Current Hematologic Malignancy Reports (2025)
