
Abstract
Diffuse large B-cell lymphoma (DLBCL), the most common B-cell non-Hodgkin lymphoma (B-NHL), is characterized by strong aggression, high heterogeneity, and poor prognosis. Consequently, there is an urgent need to identify crucial therapeutic targets. Here, we found that the transcription factor zinc-finger and homeobox 2 (ZHX2) was highly expressed in DLBCL. Subsequently, ZHX2 was proven to be critical for promoting DLBCL cell proliferation by inhibiting ferroptosis. Mechanistically, ZHX2 bound to the promoter region of the solute carrier family 3-member 2 (SLC3A2) gene through liquid-liquid phase separation (LLPS) and activated its function to negatively regulate ferroptosis. Furthermore, we constructed lipid nanoparticles ZHX2-siRNA@LNP targeting DLBCL, which effectively inhibited the growth of the tumors in vivo. In summary, our study indicated that the LLPS of ZHX2 protected DLBCL against ferroptosis through induction of SLC3A2, and disturbing it with ZHX2-siRNA@LNP could significantly repress DLBCL, providing a promising therapeutic strategy for DLBCL.

Similar content being viewed by others


Introduction
Diffuse large B-cell lymphoma (DLBCL), accounting for 30–40% of the B-cell non-Hodgkin lymphoma (B-NHL), is a highly aggressive and heterogeneous type of tumor [1]. In the past few years, with the advent of new targeted therapies based on rituximab, the treatment of most patients with DLBCL has made remarkable progress [2, 3]. However, approximately one-third of DLBCL patients still remain refractory or relapse and eventually die from disease progression [4, 5]. Therefore, identifying novel therapeutic targets and exploring their mechanisms are critical for DLBCL treatment.
Ferroptosis is a nonapoptotic cell death characterized by iron-dependent lipid peroxidation, glutathione consumption, and mitochondrial shrinkage [6]. It has gained increasing attention in DLBCL research. Specifically, the overexpression of glutathione peroxidase 4 (GPX4), a crucial ferroptosis regulator, weakens the sensitivity of DLBCL cells to ferroptosis [7], whereas bromodomain protein 4 promotes ferroptosis suppressor protein 1 (FSP1) expression to protect germinal center B-cell (GCB) DLBCL cells from ferroptosis [8]. Moreover, glutamine α-ketoglutarate inhibits DLBCL growth by inducing reactive oxygen species (ROS) production and TP53-mediated ferroptosis [9]. In conclusion, driving ferroptosis may provide an effective anticancer strategy for DLBCL-targeted therapy.
Zinc finger and homeobox 2 (ZHX2), a transcription factor from the ZHX family with zinc finger and homeobox domains (HD) [10], regulates diverse cellular processes including mitochondrial oxidative phosphorylation, cell maturation, and polarization [11,12,13]. Importantly, ZHX2 is also involved in tumorigenesis and cancer progression; for example, it inhibits hepatocellular carcinoma by blocking lipid lipase-mediated lipid uptake and repressing Notch1 via the NF-YA interaction [14, 15], while its downregulation promotes Hodgkin lymphoma pathogenesis [16] and correlates with poor prognosis in multiple myeloma [17]. In contrast, ZHX2 overexpression drives clear cell renal cell carcinoma (ccRCC) progression by activating NF-κB [18, 19] and enhances triple-negative breast cancer metastasis through HIF1α signaling and the suppression of mesenchymal-epithelial transition [20, 21]. These findings demonstrate that ZHX2 is a potentially promising regulator of tumors. However, the role and mechanism of ZHX2 in DLBCL remain unclear.
In this study, we systematically showed that ZHX2 inhibits ferroptosis and promotes DLBCL development. Mechanistically, the HD3 of ZHX2 drives liquid-liquid phase separation (LLPS) and consequently strengthens its interaction with the promoter region of solute carrier family 3-member 2 (SLC3A2), a ferroptosis negative regulator. Furthermore, we designed ZHX2-siRNA@LNP, which targets ZHX2 and effectively inhibits tumor growth in vivo. Overall, these findings highlight that ZHX2 is a promising target and that targeting it could significantly repress the progression of DLBCL, which provides a novel idea for the treatment of DLBCL.
Materials and methods
Patients and clinical samples
Freshly excised lymph node samples from 60 newly diagnosed DLBCL patients (30 GCB and 30 non-GCB subtypes) and 30 human tonsil tissues were collected from Qilu Hospital of Shandong University. All patients who provided clinical specimens signed a written informed consent form. This study complied with the Declaration of Helsinki and its later amendments or comparable ethical standards and was approved by the Qilu Hospital of Shandong University Ethics Committee (NO. KYLL-2021(KS)-366).
In vivo xenografts
This study was approved by the Animal Ethics and Welfare Society of the Qilu Hospital of Shandong University (NO. KYLL-2021(KS)-366). For xenograft experiments, 6-week-old female NOD.Cg-PrkdcscidIl2rgem1Smoc (NSG) mice (Cat. NO. NM-NSG-001) were purchased from Shanghai Model Organisms. NSG mice were housed in specific pathogen-free environments (sex was not restricted in this experiment).
Results
ZHX2 is overexpressed in DLBCL and promotes DLBCL proliferation
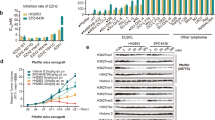
To clarify the characteristics of ZHX2 in DLBCL, immunohistochemistry (IHC) showed elevated expression in DLBCL (n = 60) compared to normal tonsils (n = 30; mean optical density (MOD) mean = 0.01305), regardless of the GCB type (n = 30; MOD mean = 0.05743, p < 0.0001) or non-GCB (n = 30; MOD mean = 0.04922; p < 0.0001, all Student’s t-test) subtype (Fig. 1A, Supplementary Fig. 1A). Then, ZHX2 mRNA (n = 30/group, DLBCL tissues: p = 0.0002/0.0003; n = 3/group, cell lines: p < 0.005; all Student’s t-test) and protein levels were also significantly elevated in DLBCL tissues and cell lines compared to tonsillar germinal center (GC) B cells (Fig. 1B, C, Supplementary Fig. 1B, C). Taken together, these results demonstrate that ZHX2 is highly expressed in DLBCL.

A IHC staining revealed ZHX2 expression in GCB DLBCL (n = 30), non-GCB DLBCL tissues (n = 30), and tonsil tissues (n = 30). B ZHX2 mRNA expression levels in GCB DLBCL (n = 30), non-GCB DLBCL (n = 30), and GC B cells from tonsil tissues (n = 30). C Western blot analysis of ZHX2 protein expression in GCB DLBCL (n = 3), non-GCB DLBCL (n = 3), and GC B cells from tonsil tissues (n = 6). For (A), scale bar, 50 μm (upper); 10 μm (lower). Data are mean ± SD values.
To determine whether the high expression of ZHX2 promotes DLBCL proliferation, we constructed lentiviruses with ZHX2 knockdown (KD) to transfect three DLBCL cell lines, including SU-DHL-4, DB, and U2932 cells (Supplementary Fig. 1D, E). We observed that ZHX2 KD significantly inhibited proliferation by CCK-8 assay (n = 3; shCtrl vs. sh#1/sh#2, SU-DHL-4: p = 0.0121, p < 0.0001; DB: p < 0.0001, respectively; U2932: p < 0.0001, respectively; Two-way ANOVA analysis) and colony formation (n = 3; shCtrl vs. sh#1/sh#2, SU-DHL-4: p = 0.0003/0.0001; DB: p = 0.0018/0.0012; U2932: p = 0.0003/0.0002; all Student’s t-test.) (Fig. 2A, B, Supplementary Fig. 1F). Conversely, ZHX2 overexpression enhanced proliferation (n = 3; CCK-8: SU-DHL-4, p < 0.0001; DB, p < 0.0001; U2932, p < 0.0001; Two-way ANOVA analysis) (n = 3; Colony formation: SU-DHL-4, p = 0.0021; DB, p = 0.0002; U2932, p = 0.0074; all Student’s t-test) of DLBCL cells (Supplementary Fig. 2A–D). To clarify the oncogenic role of ZHX2 in vivo, we established DLBCL xenograft models. As expected, knockdown of ZHX2 obviously reduced the tumor size (n = 5; mean: shCtrl vs. sh#1/sh#2, 0.7975 vs. 0.1460/0.08917; p = 0.0004/0.0002; all Student’s t-test) and volume (n = 5; shCtrl vs. sh#1/sh#2, p < 0.0001, respectively; Two-way ANOVA analysis) of the SU-DHL-4 (GCB subtype) xenograft model (Fig. 2C, Supplementary Fig. 2E), consistent with the results in vitro. Additionally, IHC revealed that ZHX2 knockdown significantly reduced tumor infiltration (n = 5; hematoxylin and eosin (HE), cell number/100*100 μm2, shCtrl vs. sh#1/sh#2, mean: 104 vs. 67/49) and proliferation (n = 5; Ki-67 index: shCtrl vs. sh#1/sh#2: 52.7% vs. 29.5%/24.4%) (Supplementary Fig. 2F) [22, 23]. Similarly, ZHX2 knockdown inhibited tumor growth (n = 5; tumor weight, mean: shCtrl vs. sh#1/sh#2, 0.5779 vs. 0.08458/0.06967, p = 0.001/0.0008; all Student’s t-test) (tumor volume: shCtrl vs. sh#1/sh#2, p < 0.0001, respectively; Two-way ANOVA analysis) in the U2932 (non-GCB subtype) xenograft mouse model (Supplementary Fig. 3A, B). Collectively, these findings indicate that ZHX2 overexpression promotes DLBCL proliferation.

A CCK-8 assay of SU-DHL-4, DB, and U2932 cells (30,000 cells/mL) in control and ZHX2 knockdown group for 0, 24, 48, 72, and 96 h (n = 3). B Colony formation assay of SU-DHL-4, DB, and U2932 cells in control and ZHX2 knockdown group (20,000 cells/mL). After 2 weeks, colonies were counted (n = 3). C Representative image of tumors from SU-DHL-4 cell xenograft models (n = 5 in each group). For (B), scale bar, 2 mm. Data are mean ± SD values.
ZHX2 inhibits ferroptosis to maintain DLBCL malignant progression
To identify the mechanism by which ZHX2 facilitates DLBCL proliferation, RNA-sequencing (RNA-seq) of ZHX2 knockdown SU-DHL-4 cells identified 753 upregulated and 496 downregulated differentially expressed genes (DEGs, q < 0.05, |log₂(fold-change) |≥1) (Supplementary Fig. 4A). In addition, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses confirmed significant enrichment of DEGs in ferroptosis-related functional categories (Supplementary Fig. 4B, C, Supplementary Tables 1, 2). To confirm this hypothesis, RT-qPCR demonstrated that ZHX2 knockdown affects mRNA levels of ferroptosis-related molecules (n = 3; p < 0.005, all Student’s t-test), including SLC7A11, SLC3A2, GPX4, ACSL4, and LPCAT3 (Supplementary Fig. 4D). These results suggest that ZHX2 may regulate DLBCL by repressing ferroptosis.
Ferroptosis is an iron-dependent cell death driven by lipid peroxidation, mitochondrial shrinkage, ROS accumulation, and glutathione (GSH) depletion [24]. To determine whether ZHX2 regulates DLBCL development by influencing ferroptosis, transmission electron microscopy (TEM) was used to detect mitochondrial shrinkage in the ZHX2 KD group compared to the control group (Fig. 3A). Given that Fe2+ is essential for promoting ferroptosis in cells, we used FerroOrange probes to measure the intracellular Fe2+ concentration and demonstrated that its concentration in ZHX2 KD DLBCL cells increased significantly (Fig. 3B). Moreover, knockdown of ZHX2 elevated ROS levels (n = 3; Relative mean fluorescence intensity (MFI) mean, shCtrl vs. sh#1/sh#2, SU-DHL-4, 1.00 vs. 2.07/2.62, p = 0.0003/0.0001; DB: 1.00 vs. 2.09/2.41, p = 0.0005/0.0007; U2932, 1.00 vs. 2.00/1.80, p = 0.0001, p < 0.0001; all Student’s t-test) (Fig. 3C, Supplementary Fig. 5A). Considering that the levels of GSH and malondialdehyde (MDA) are critical for the assessment of ferroptosis [25, 26], we detected their changes in DLBCL cells, finding ZHX2 KD significantly decreased GSH levels (n = 4; mean: SU-DHL-4, shCtrl vs. sh#1/sh#2, 23.58 vs. 9.504/6.70, p = 0.0007/0.0002; DB: shCtrl vs. sh#1/sh#2, 28.40 vs. 18.13/21.76, p = 0.0036/0.0253; U2932, shCtrl vs. sh#1/sh#2, 28.49 vs. 16.18/14.40, p = 0.0234/0.0087; all Student’s t-test) and increased MDA levels (n = 3; mean: SU-DHL-4, shCtrl vs. sh#1/sh#2, 4.346 vs. 15.22/20.16; p = 0.0157/0.0015; DB: shCtrl vs. sh#1/sh#2, 5.550 vs. 21.43/18.57; p = 0.0025/0.0038; U2932, shCtrl vs. sh#1/sh#2, 3.906 vs. 17.66 /13.35; p = 0.0213/0.0233; all Student’s t-test) (Supplementary Fig. 5B, C). Conversely, ZHX2 overexpression inhibited ferroptosis, evidenced by reduced ROS levels (n = 3; relative MFI mean: SU-DHL-4, 1.00 vs. 0.67, p = 0.0183; DB, 1.00 vs. 0.53, p = 0.0200; U2932, 1.00 vs. 0.60, p = 0.0039; all Student’s t-test)and elevated GSH levels (n = 3; mean: SU-DHL-4, 25.45 vs. 47.72, p = 0.0019; DB, 29.94 vs. 46.38, p = 0.0099; U2932, 24.32 vs. 41.49, p = 0.0066; all Student’s t-test) (Supplementary Fig. 5D, E). Taken together, these results confirm that ZHX2 inhibits ferroptosis to promote DLBCL development.

A TEM images of ZHX2 knockdown and control in DLBCL cells, including SU-DHL-4, DB, and U2932. White arrows indicate mitochondria. B Levels of intracellular Fe2+ measured by FerroOrange probes. C Flow cytometry analysis of the ROS levels in control or ZHX2 KD cells. For (A), scale bars, 1 μm. For (B), scale bar, 10 μm. Data are mean ± SD values.
ZHX2 undergoes LLPS
LLPS is a newly identified intracellular form that mainly manifests as the formation of protein granules and plays a crucial role in ferroptosis [27, 28]. To determine whether ZHX2-mediated ferroptosis involves LLPS, we analyzed the amino acid sequence of ZHX2 and found that it contains intrinsically disordered regions that could drive LLPS [29] (Fig. 4A). Then, we purified the EGFP-labeled ZHX2 protein to evaluate whether it underwent LLPS in vitro (Supplementary Fig. 6A). As expected, in contrast to the purified EGFP protein, the EGFP-ZHX2 protein was turbid after fully mixing (Supplementary Fig. 6B, C). Additionally, we showed that the purified EGFP-ZHX2 protein, but not EGFP alone, formed several micrometer-sized spherical droplets, which were abolished after 1 min of treatment with 10% 1,6-hexanediol (1,6-Hex) (Fig. 4B). To further illustrate the biophysical properties of the EGFP-ZHX2 droplets, Fluorescence recovery after photobleaching (FRAP) confirmed that the EGFP-ZHX2 droplets were dynamic and exhibited rapid fluorescence recovery (~40 s, 80% recovery) after bleaching, indicating that the droplets were in a liquid-like state (n = 8) (Supplementary Fig. 6D). In addition, the ZHX2 phase-separated droplets simultaneously underwent rapid fusion in both differential interference contrast (DIC) and fluorescence imaging (Supplementary Fig. 6E).

A The amino acid sequence of the ZHX2 protein was analyzed via the disorder region prediction tool PONDR (http://www.pondr.com/). B Upon treatment with 300 mM NaCl and 10% PEG 8000, EGFP-ZHX2 (30 μM) formed microdroplets, which disappeared 10% 1, 6-Hex was added for 1 min, while EGFP did not. C HEK293T cells transfected with EGFP or ZHX2-EGFP plasmids (2 μg/mL) for 48 h were treated with 0.1 M mannitol, showing no aggregation in EGFP-transfected cells, while punctate aggregates appeared and then disappeared within 3 min of 10% 1, 6-Hex treatment in ZHX2-EGFP-transfected cells (n = 3). For (B), scale bar, 5 μm. For (C), scale bar, 2 μm.
Next, to investigate whether ZHX2 drives LLPS in cells, we first constructed HEK293T cells overexpressing ZHX2 (Supplementary Fig. 7A). After the addition of 0.1 M mannitol, the green granules began to appear in HEK293T cells transfected with ZHX2-EGFP and the particles became more aggregated with time, which was not observed in HEK293T-EGFP cells (Fig. 4C). Subsequently, the granules were eliminated after treatment with 10% 1,6-Hex (Fig. 4C). Furthermore, we confirmed the dynamics of the ZHX2 droplets using the FRAP assay, and noticed that the fluorescence recovery reached up to ~80% within 60 s after bleaching (n = 8, Supplementary Fig. 7B), and the fusion of ZHX2-EGFP puncta were also observed (Supplementary Fig. 7C), indicating that ZHX2 undergoes LLPS in HEK293T cells. To verify whether ZHX2 drives LLPS in DLBCL cells, we detected endogenous ZHX2 and found that it exhibited granule aggregation (n = 3, Supplementary Fig. 7D). Meanwhile, we overexpressed ZHX2-EGFP and EGFP in those cells and found that ZHX2-EGFP had obvious particles (n = 3, Supplementary Fig. 7E). In summary, ZHX2 undergoes LLPS.
ZHX2 inhibits LLPS through the HD3 to regulate ferroptosis
To elucidate the specific mechanism of LLPS in ZHX2, we analyzed the functional domains of ZHX2 using the Animal TFDB database combined with previous reports [10]. ZHX2 contains four DNA-binding domains: C2H2 (107–157), HD1 (447–495), HD2 (537–581) and HD3 (636–681) (Supplementary Fig. 8A). To determine the specific structure of the induced LLPS, we constructed four corresponding mutant plasmids tagged with EGFP: ZHX2-ΔC2H2-EGFP, ZHX2-ΔHD1-EGFP, ZHX2-ΔHD2-EGFP, and ZHX2-ΔHD3-EGFP (Supplementary Fig. 8B). We observed that the ZHX2 LLPS with truncated C2H2, HD1, or HD2 was not affected, whereas the truncated HD3 domain resulted in the disappearance of LLPS, compared to ZHX2-EGFP, suggesting that the HD3 domain was crucial region to undergo the LLPS (Supplementary Fig. 8C). Similar results were validated in DLBCL cells transfected with ZHX2-ΔHD3-EGFP (Z-ΔHD3-EGFP) (Fig. 5A). Additionally, we purified the EGFP-tagged ZHX2-ΔHD3 (EGFP-Z-ΔHD3) protein and did not observe the absence of micrometer-sized spheres and turbidity, compared to EGFP-ZHX2 (Supplementary Fig. 8D–F). Overall, these data suggest that the LLPS of ZHX2 depends on its HD3 domain.

A No puncta were observed in DLBCL cells transfected with Z-ΔHD3-EGFP plasmid (2 μg/mL) for 48 h, whereas the ZHX2-EGFP group showed puncta (n = 3). B Cell viability of control, ZHX2 with LLPS, and Z-ΔHD3 without LLPS treated DLBCL cells (30,000 cells/mL) at 48 h (n = 3). C Representative photograph of tumors, tumor weights, and volumes of SU-DHL-4 cell xenograft models (n = 5 in each group). For (A), scale bar, 2 μm. Data are mean ± SD values.
Next, we clarified whether ZHX2 regulates ferroptosis through HD3 domain-mediated LLPS. Specifically, we transfected three DLBCL cell lines with EGFP-labeled ZHX2-ΔHD3 (Z-ΔHD3) overexpressing lentivirus to disturb the occurrence of phase separation, and then detected key indicators of ferroptosis. As expected, similar to the control group, no phase-separated Z-ΔHD3 had little effect on the ROS(n = 3; relative MFI mean, Ctrl vs. Z-ΔHD3: SU-DHL-4, 1.00 vs. 0.97, p = 0.8380; DB: 1.00 vs. 0.89, p = 0.4256; U2932, 1.00 vs. 0.97; p = 0.7509; all Student’s t-test), GSH(n = 3; Ctrl vs. Z-ΔHD3: SU-DHL-4, 26.83 vs. 29.18, p = 0.4144; DB: 30.19 vs. 36.67, p = 0.2179; U2932, 22.20 vs. 20.72, p = 0.5051; all Student’s t-test), and MDA levels (n = 3; Ctrl vs. Z-ΔHD3: SU-DHL-4, 4.568 vs. 4.063, p = 0.3344; DB: 8.144 vs. 7.126, p = 0.5323; U2932, 8.324 vs. 8.477, p = 0.9382; all Student’s t-test) (Supplementary Fig. 9A–C). Furthermore, the CCK-8 assay showed that Z-ΔHD3 without LLPS had no influence on promoting the growth of DLBCL cells (n = 3; Ctrl vs. Z-ΔHD3, SU-DHL-4, p = 0.4395; DB: p = 0.2979; U2932, p = 0.1961; all Student’s t-test) (Fig. 5B). To investigate whether the function of ZHX2 depends on the HD3 in vivo, we established xenografts using SU-DHL-4 and U2932 cells expressing ZHX2 or Z-ΔHD3. The results showed that the Z-ΔHD3 group, similar to the control group, significantly inhibited the promotive effect of ZHX2 on tumor growth in mice (n = 5; SU-DHL-4: tumor weight, mean: ZHX2 vs. Z-ΔHD3, 1.445 vs. 0.8285, p = 0.0016, all Student’s t-test; tumor volume: ZHX2 vs. Z-ΔHD3, p < 0.0001; Two-way ANOVA analysis) (n = 5; U2932: tumor weight, mean: ZHX2 vs. Z-ΔHD3, 1.180 vs. 0.625, p = 0.0026; all Student’s t-test; tumor volume: ZHX2 vs. Z-ΔHD3, p < 0.0001; Two-way ANOVA analysis) (Fig. 5C, Supplementary Fig. 9D–F). In summary, ZHX2 undergoes LLPS via its HD3 domain, and further promotes DLBCL cell proliferation by inhibiting ferroptosis.
LLPS of ZHX2 represses ferroptosis by activating SLC3A2
To investigate how ZHX2 regulates ferroptosis through HD3-mediated LLPS, we performed chromatin immunoprecipitation-sequencing (ChIP-seq) to detect the differences between ZHX2 with LLPS and Z-ΔHD3 with the disappearance of LLPS in DB cells, identifying 273 DEGs (q < 0.05, |log₂FC | ≥ 1) (Supplementary Fig. 10A). GO and KEGG enrichment analyses of the 273 DEGs revealed significant enrichment in ferroptosis-related functional categories or pathways (Supplementary Fig. 10B). Subsequently, integration of the ChIP-seq and RNA-seq data identified ferroptosis-related genes (SLC3A2, ATG5, HSPA5, and ACSL1) as potential downstream targets of ZHX2 (Supplementary Fig. 10C). Among the four genes, SLC3A2 was considered for further verification based on the following considerations: (1) ZHX2 binds to the SLC3A2 gene with a high binding peak near the transcription initiation site (Fig. 6A); (2) SLC3A2 has been reported to play a pivotal role in ferroptosis [30, 31]. To verify the regulatory role of ZHX2 on SLC3A2 expression, we transiently transfected ZHX2 and Z-ΔHD3 as well as a reporter plasmid driven by the wild-type/mutant SLC3A2 promoter in DB cells. As expected, overexpression of ZHX2 increased SLC3A2 promoter activity in dual-luciferase assays (n = 4; ZHX2/PLG4.26 vs. ZHX2/SLC3A2 WT, p = 0.0044; ZHX2/PLG4.26 vs. ZHX2/SLC3A2 Mut, p = 0.1418) (Fig. 6B). However, overexpression of Z-ΔHD3 without LLPS failed to activate this activity (n = 4; Z-ΔHD3/PLG4.26 vs. Z-ΔHD3/ SLC3A2 WT, p = 0.1033; ZHX2/ SLC3A2 WT vs. Z-ΔHD3/ SLC3A2 WT, p = 0.0081; all Student’s t-test) (Fig. 6B). Similarly, the LLPS of ZHX2 also increased its binding to SLC3A2 promoter in HEK293T-ZHX2-EGFP cells after stimulation with mannitol (n = 4; ZHX2/SLC3A2 WT vs. ZHX2/SLC3A2 WT+ mannitol, p = 0.0039; Student’s t-test) but it had no change in HEK293T-ZHX2-EGFP cells without mannitol treatment (Supplementary Fig. 10D). Moreover, we also found that ZHX2 overexpression in DLBCL cell lines increased SLC3A2 protein levels, while Z-ΔHD3 overexpression did not affect (Supplementary Fig. 10E). Overall, ZHX2 binds to the SLC3A2 promoter via HD3-mediated LLPS and activates its expression.

A The IGV diagram showing that phase-separated ZHX2 bound near the TSS of the SLC3A2 gene promoter from ChIP-seq data, while Z-ΔHD3 without LLPS did not. B ZHX2 or Z-ΔHD3 overexpression plasmids (1 μg/mL) were co-transfected into DB cells with 1 μg/mL wild-type or mutant SLC3A2 luciferase reporter vectors. Dual-luciferase reporter assay showed the effects of ZHX2 and Z-ΔHD3 on wild-type and mutant SLC3A2 reporter genes (n = 4). C Cell viability of DB cells (30000 cells/mL) treated with SLC3A2 KD in the control, ZHX2 with LLPS, and Z-ΔHD3 without LLPS groups at 48 h (n = 3). Data are mean ± SD values.
Recent studies have shown that SLC3A2 can inhibit ferroptosis in laryngeal carcinomas and lung adenocarcinoma [30, 31]; its function in DLBCL remains unclear. First, we identified a positive correlation between ZHX2 and SLC3A2 expression utilizing tissue microarrays (n = 30, respectively; p = 0.0024, R2 = 0.2838, Pearson correlation analysis) (Supplementary Fig. 10F). Moreover, the expression of SLC3A2 in DLBCL tissues was significantly higher than that in GC B cells derived from normal tonsils by RT-qPCR (n = 30, each; GC B vs. GCB DLBCL/non-GCB DLBCL, p = 0.0006/0.0002, all Student’s t-test) and western blot (Supplementary Fig. 11A, B). Next, to clarify whether SLC3A2 inhibits ferroptosis in DLBCL, we transfected DB cells with SLC3A2 knockdown (KD) lentiviruses (n = 5) (Supplementary Fig. 11C, D). Expectedly, TEM showed mitochondrial shrinkage in SLC3A2 KD cells (Supplementary Fig. 11E). Then, SLC3A2 KD DB cells presented increased intracellular Fe²⁺ concentrations (Supplementary Fig. 11F) and ROS levels (n = 5; relative MFI mean: 1.00 vs. 2.16, p < 0.0001; Student’s t-test), MDA levels (n = 5; mean: 6.303 vs. 21.56, p < 0.0001; Student’s t-test), and decreased GSH levels (n = 5; mean: 15.75 vs. 27.81, p = 0.0007; Student’s t-test) (Supplementary Fig. 11G–I). In addition, SLC3A2 KD inhibited DB cells' viability (n = 3; p = 0.0231; Student’s t-test) (Supplementary Fig. 11J). Collectively, SLC3A2 can inhibit ferroptosis and promote DLBCL cell growth.
To further investigate whether the liquid-liquid phase-separated HD3 of ZHX2 regulates DLBCL proliferation through SLC3A2, we downregulated SLC3A2 on the basis of transfection of ZHX2 or Z-ΔHD3, respectively, in DLBCL cells (Supplementary Fig. 11K). Subsequently, CCK-8 assay revealed that ZHX2 overexpression significantly promoted the proliferation of DLBCL cells (n = 3; Ctrl/shCtrl vs. ZHX2/shCtrl, p = 0.0008), and this effect could be counteracted by knocking down SLC3A2 (n = 3; Ctrl/shCtrl vs. ZHX2/shSLC3A2, p = 0.4981), whereas cell proliferation in the Z-ΔHD3 group was no significantly different from that in the control group (n = 3; Ctrl/shCtrl vs. Z-ΔHD3/shCtrl, p = 0.8824) (Fig. 6C). Interestingly, SLC3A2 knockdown in the Z-ΔHD3 group without LLPS more obviously suppressed cell viability (n = 3; ZHX2/shSLC3A2 vs. Z-ΔHD3/shSLC3A2, p = 0.0021; all Student’s t-test) (Fig. 6C). Taken together, these results suggest that ZHX2 activates SLC3A2 through LLPS mediated by its HD3 domain, thus inhibiting ferroptosis and promoting DLBCL cell proliferation.
Nanoparticle tumor-targeted delivery of ZHX2-siRNA attenuates DLBCL progression
LNP-mRNA technology is widely used for targeting tumor cells in various cancer models [32]. In view of this, we designed DiR modified LNP called cRGD (fc)-ZHX2-siRNA@LNP (ZHX2-siRNA@LNP) for targeted DLBCL delivery (Fig. 7A). Specifically, cRGD (fc) is a cyclic RGD peptide fused to an Fc fragment that selectively targets DLBCL cells with high contents of αv-integrin and β3-integrin [33]. Our results also confirmed that both of them were highly expressed in DLBCL cells (Supplementary Fig. 12A). Subsequently, we observed that ZHX2-siRNA@LNP entered SU-DHL-4 cells by TEM (Supplementary Fig. 12B). Furthermore, the ZHX2-siRNA@LNP inhibited cell proliferation (n = 3; SU-DHL-4, p < 0.0001; U2932, p < 0.0001; Two-way ANOVA analysis), suggesting that it plays an anti-tumor role in vitro. (Supplementary Fig. 12C). Interestingly, compared to SU-DHL-4 and U2932, which exhibited high ZHX2 expression, inhibition was weaker in low-ZHX2-expressing SU-DHL-2 cells (n = 3; p = 0.0117; Two-way ANOVA analysis), suggesting that the effectiveness of ZHX2-siRNA@LNP is mainly dependent on ZHX2 expression levels (Supplementary Fig. 12C).

A Schematic of the preparation process of ZHX2-siRNA@LNP. B For the SU-DHL-4 xenograft models, the load and distribution of LNP in tumors and organs were photographed with an IVIS (n = 4 in each group). C Representative HE and Ki-67 images of tumors from SU-DHL-4 cell xenograft models, including the PBS, ZHX2-siCtrl@LNP, and ZHX2-siRNA@LNP groups. For (C), scale bar, 50 μm. Data are mean ± SD values.
To evaluate LNP delivery efficacy in vivo, we established SU-DHL-4 (GCB DLBCL) xenograft model. After multiple intravenous injections of LNP, in vivo imaging system (IVIS) imaging measured ZHX2-siRNA@LNP distribution in tumors and organs, showing the highest fluorescence at tumor sites, followed by the liver and spleen (Fig. 7B). Moreover, the calculation of tumor weight (n = 4; PBS vs. ZHX2-siCtrl@LNP/ ZHX2-siRNA@LNP: 0.7678 vs. 0.7826/0.1102, p = 0.9085/0.0003; Student’s t-test) and volume (n = 4; PBS vs. ZHX2-siCtrl@LNP/ZHX2-siRNA@LNP: p = 0.7842, p < 0.0001; Two-way ANOVA analysis) showed that ZHX2-siRNA@LNP significantly inhibited tumor growth (Supplementary Fig. 12D). Additionally, the analysis of HE and Ki-67 analyses revealed decreased infiltration and proliferation (n = 4; PBS vs. ZHX2-siCtrl@LNP/ZHX2-siRNA@LNP, HE, cell number/100*100 μm2, mean:101 vs. 99/51; Ki-67 index, 56.09 vs. 54.10/25.9) (Fig. 7C). The above results demonstrated that ZHX2-siRNA@LNP could exert targeted anti-tumor effects in vivo. To further verify whether ZHX2-siRNA@LNP repressed tumor growth by targeting ZHX2, the tumors were minced into cells and the proteins were extracted for western blotting analysis. The results showed that ZHX2-siRNA@LNP effectively downregulated ZHX2 and SLC3A2 expression in SU-DHL-4 cells (Supplementary Fig. 12E). Notably, we confirmed that ZHX2-siRNA@LNP promoted ferroptosis by measuring the ROS (n = 3; relative MFI mean: 1.00 vs. 2.06, p = 0.0012; Student’s t-test) and GSH levels (n = 3; 24.66 vs. 10.32, p = 0.0035; Student’s t-test) (Supplementary Fig. 12F, G). Meanwhile, similar results were also validated in the U2932 xenograft mouse models (n = 4; tumor weight: PBS vs. ZHX2-siCtrl@LNP/ZHX2-siRNA@LNP: 0.6393 vs. 0.6588/0.1100, p = 0.9022/0.0027; Student’s t-test; tumor volume: p = 0.8676, p < 0.0001; Two-way ANOVA analysis) (n = 4; PBS vs. ZHX2-siCtrl@LNP/ZHX2-siRNA@LNP, HE, cell number/100*100 μm2, mean: 98, 97, 48; Ki-67 index: 55.1 vs. 53.8/22.54) (n = 3; ROS, relative MFI mean: 1.00 vs. 2.02, p = 0.0015; GSH, 29.04 vs. 15.38, p = 0.0117; all Student’s t-test) (Supplementary Fig. 13A–F). In conclusion, we demonstrated that ZHX2-siRNA@LNP downregulates ZHX2, thus repressing the expression of SLC3A2 and promoting ferroptosis in DLBCL.
Discussion
DLBCL is a malignant lymphoma with heterogeneous clinical manifestations, morphology, and biology [34, 35]. Despite new breakthroughs in various therapeutic options, patients with DLBCL still show refractory recurrence and a poor prognosis [36, 37]. Hence, it is critical to explore more effective therapeutic targets. Here, the overexpression of ZHX2 promoted DLBCL cells proliferation, suggesting that it could be a promising therapeutic target for DLBCL. Notably, we also designed ZHX2-siRNA@LNP to inhibit DLBCL by interfering with ZHX2 in vivo, which provides a new strategy for the treatment of patients with DLBCL. However, the mechanism underlying the high expression of ZHX2 in DLBCL remains unclear. Studies have demonstrated that copy number variations (CNVs) could lead to the overexpression of target genes, such as MYC [38] and KRAB [39]. Subsequently, we preliminarily analyzed the CNVs of ZHX2 in 5 patients with DLBCL but found no significant change compared to healthy individuals (data not shown). Considering that previous related studies have utilized sample sizes in the hundreds [38, 39], we speculated that the negative results observed may be due to the limited sample size in our study. Therefore, we intend to increase the number of patient samples to further investigate whether CNVs contribute to the increased expression of ZHX2 in DLBCL. Additionally, the expression of transcription factors may be driven by microRNA dysregulation [40, 41], abnormal DNA methylation [42, 43], and histone modifications [44], which also provides valuable insights into elucidating the overexpression of ZHX2 in DLBCL.
As a member of the ZHX family, ZHX2 has been reported to play a tumor-suppressive role in a variety of cancers, including hepatocellular carcinoma [14, 15, 45], thyroid cancer [46], hematological tumors Hodgkin lymphoma [16, 47], and multiple myeloma [17]. Conversely, ZHX2 is significantly amplified in ccRCC and breast cancer, and promotes tumor proliferation by binding downstream NF-κB and HIF1α, respectively [18,19,20,21]. Nevertheless, the specific functional mechanism of ZHX2 in DLBCL remains unclear. Here, we found that ZHX2 protects the development of DLBCL by inhibiting ferroptosis, and knockdown of ZHX2 strengthens the activation of the ferroptosis pathway. These studies highlight that the function of ZHX2 is closely related to the disease-type-specific signaling pathways.
LLPS is a dynamic and complex physiological process in which some specific molecular components spontaneously form aggregates that regulate key cellular processes, such as cell differentiation, cell division, and autophagy [48,49,50]. Recently, several studies have confirmed that proteins can modulate ferroptosis through LLPS and regulate tumorigenesis. A previous study showed that stimulating the phase separation of FSP1 promotes ferroptosis in cancer cells and then inhibits tumor growth [28]. Additionally, lncFASA binds to peroxiredoxin 1 and promotes ferroptosis by inducing LLPS to disrupt breast cancer proliferation [51]. Another study also proved that manganese promoted the LLPS of YAP/TAZ by mediating its nuclear translocation and activating ACSL4, thus promoting the ferroptosis of oral squamous cell carcinoma [52]. In this study, we found that ZHX2 undergoes LLPS to promote the proliferation of DLBCL cells by inhibiting ferroptosis, which provides new evidence for the role of LLPS in cancer. Mechanistically, ChIP-seq and RNA-seq data suggested that ZHX2 with LLPS could activate its downstream negative ferroptosis regulator SLC3A2 through phase separation mediated by the HD3 domain, which may be due to the LLPS of ZHX2 modifying the distribution and functional status of its bound chromosomes. Nevertheless, the detailed mechanism underlying this remains unclear and requires further investigation.
Although previous studies have indicated that ferroptosis is associated with the pathogenesis of DLBCL [7,8,9], the specific regulatory mechanism remains unclear. In the present study, our results clarified a critical ferroptosis inhibition pathway regulated by ZHX2-mediated in DLBCL. Specifically, we observed that inhibition of ZHX2 reduced the proliferation of DLBCL cells and induced ferroptosis of DLBCL. Mechanistically, ZHX2 with LLPS could bind to the promoter region of the ferroptosis negative regulator SLC3A2. Recent studies have emphasized that SLC3A2 could inhibit ferroptosis in various tumors [34, 35, 53], but its function in DLBCL is still unclear. Our results revealed that interference with SLC3A2 also inhibits cell proliferation by promoting ferroptosis in DLBCL, providing a new target for the treatment of DLBCL from the perspective of regulating ferroptosis.
In summary, we confirmed that ZHX2, as a transcription factor, protects DLBCL cells against ferroptosis through LLPS. Mechanistically, the depletion of HD3 domain abolished LLPS of ZHX2, further inhibiting the combination of ZHX2 and the SLC3A2 promoter, thus promoting ferroptosis. Strikingly, our designed ZHX2-siRNA@LNP could effectively promote the activation of ferroptosis, thus impeding the growth of DLBCL in vivo and providing a novel therapeutic strategy for DLBCL.
Data availability
The raw sequencing data of ChIP-Seq and RNA-Seq generated from this study have been deposited in the GEO database under the accession codes GSE292497, GSE292498, and GSE292652.
References
Silkenstedt E, Salles G, Campo E, Dreyling M. B-cell non-Hodgkin lymphomas. Lancet. 2024;403:1791–807.
Pfreundschuh M, Schubert J, Ziepert M, Schmits R, Mohren M, Lengfelder E, et al. Six versus eight cycles of bi-weekly CHOP-14 with or without rituximab in elderly patients with aggressive CD20+ B-cell lymphomas: a randomised controlled trial (RICOVER-60). Lancet Oncol. 2008;9:105–16.
Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37:551–68.e14.
Salles G, Duell J, González Barca E, Tournilhac O, Jurczak W, Liberati AM, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020;21:978–88.
Poletto S, Novo M, Paruzzo L, Frascione PMM, Vitolo U. Treatment strategies for patients with diffuse large B-cell lymphoma. Cancer Treat Rev. 2022;110:102443.
Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, Skouta R, Zaitsev Eleina M, Gleason Caroline E, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Yang Wan S, SriRamaratnam R, Welsch Matthew E, Shimada K, Skouta R, Viswanathan Vasanthi S, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Schmitt A, Grimm M, Kreienkamp N, Junge H, Labisch J, Schuhknecht L, et al. BRD4 inhibition sensitizes diffuse large B-cell lymphoma cells to ferroptosis. Blood. 2023;142:1143–55.
Cai Y, Lv L, Lu T, Ding M, Yu Z, Chen X, et al. α-KG inhibits tumor growth of diffuse large B-cell lymphoma by inducing ROS and TP53-mediated ferroptosis. Cell Death Discov. 2023;9:182.
Kawata H, Yamada K, Shou Z, Mizutani T, Yazawa T, Yoshino M, et al. Zinc-fingers and homeoboxes (ZHX) 2, a novel member of the ZHX family, functions as a transcriptional repressor. Biochem J. 2003;373:747–57.
Zhang Y, Fan Y, Hu H, Zhang X, Wang Z, Wu Z, et al. ZHX2 emerges as a negative regulator of mitochondrial oxidative phosphorylation during acute liver injury. Nat Commun. 2023;14:7527.
Tan S, Guo X, Li M, Wang T, Wang Z, Li C, et al. Transcription factor Zhx2 restricts NK cell maturation and suppresses their antitumor immunity. J Exp Med. 2021;218:e20210009.
Tan S, Wang Z, Li N, Guo X, Zhang Y, Ma H, et al. Transcription factor Zhx2 is a checkpoint that programs macrophage polarization and antitumor response. Cell Death Differ. 2023;30:2104–19.
Wu Z, Ma H, Wang L, Song X, Zhang J, Liu W, et al. Tumor suppressor ZHX2 inhibits NAFLD–HCC progression via blocking LPL-mediated lipid uptake. Cell Death Differ. 2019;27:1693–708.
Zhou S-J, Deng Y-L, Liang H-F, Jaoude JC, Liu F-Y. Hepatitis B virus X protein promotes CREB-mediated activation of miR-3188 and Notch signaling in hepatocellular carcinoma. Cell Death Differ. 2017;24:1577–87.
Nagel S, Schneider B, Meyer C, Kaufmann M, Drexler HG, MacLeod RAF. Transcriptional deregulation of homeobox gene ZHX2 in Hodgkin lymphoma. Leuk Res. 2012;36:646–55.
Armellini A, Sarasquete ME, García-Sanz R, Chillón MC, Balanzategui A, Alcoceba M, et al. Low expression of ZHX2, but not RCBTB2 or RAN, is associated with poor outcome in multiple myeloma. Br J Haematol. 2008;141:212–5.
Zhang J, Wu T, Simon J, Takada M, Saito R, Fan C, et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science. 2018;361:290–5.
Zhu L, Ding R, Yan H, Zhang J, Lin Z. ZHX2 drives cell growth and migration via activating MEK/ERK signal and induces Sunitinib resistance by regulating the autophagy in clear cell renal cell carcinoma. Cell Death Dis. 2020;11:399.
Fang W, Liao C, Shi R, Simon JM, Ptacek TS, Zurlo G, et al. ZHX2 promotes HIF1α oncogenic signaling in triple-negative breast cancer. eLife. 2021;10:e70412.
He Y, Zhang Q, Chen Y, Wu Y, Quan Y, Chen W, et al. ZHX2 deficiency enriches hybrid MET cells through regulating E-cadherin expression. Cell Death Dis. 2023;14:444.
de Haan K, Zhang Y, Zuckerman JE, Liu T, Sisk AE, Diaz MFP, et al. Deep learning-based transformation of H&E stained tissues into special stains. Nat Commun. 2021;12:4884.
Stamatiou K, Huguet F, Serapinas LV, Spanos C, Rappsilber J, Vagnarelli P. Ki-67 is necessary during DNA replication for fork protection and genome stability. Genome Biol. 2024;25:105.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Yang R, Gao W, Wang Z, Jian H, Peng L, Yu X, et al. Polyphyllin I induced ferroptosis to suppress the progression of hepatocellular carcinoma through activation of the mitochondrial dysfunction via Nrf2/HO-1/GPX4 axis. Phytomedicine. 2024;122:155135.
Yang Z, Zou S, Zhang Y, Zhang J, Zhang P, Xiao L, et al. ACTL6A protects gastric cancer cells against ferroptosis through induction of glutathione synthesis. Nat Commun. 2023;14:4193.
Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019;176:419–34.
Nakamura T, Hipp C, Santos Dias Mourão A, Borggräfe J, Aldrovandi M, Henkelmann B, et al. Phase separation of FSP1 promotes ferroptosis. Nature. 2023;619:371–7.
Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci. 2012;37:509–16.
Wu F, Xiong G, Chen Z, Lei C, Liu Q, Bai Y. SLC3A2 inhibits ferroptosis in laryngeal carcinoma via mTOR pathway. Hereditas. 2022;159:6.
Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y, et al. Targeting SLC3A2 subunit of system XC− is essential for m6A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021;168:25–43.
Kara G, Calin GA, Ozpolat B. RNAi-based therapeutics and tumor targeted delivery in cancer. Adv Drug Deliv Rev. 2022;182:114113.
Liu Q, Dai G, Wu Y, Zhang M, Yang M, Wang X, et al. iRGD-modified exosomes-delivered BCL6 siRNA inhibit the progression of diffuse large B-cell lymphoma. Front Oncol. 2022;12:822805.
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11.
Tirado CA, Chen W, García R, Kohlman KA, Rao N. Genomic profiling using array comparative genomic hybridization define distinct subtypes of diffuse large B-cell lymphoma: a review of the literature. J Hematol Oncol. 2012;5:54.
Lugtenburg PJ, de Nully Brown P, van der Holt B, D’Amore FA, Koene HR, de Jongh E, et al. Rituximab-CHOP with early rituximab intensification for diffuse large B-cell lymphoma: a randomized phase III tTrial of the HOVON and the Nordic Lymphoma Group (HOVON-84). J Clin Oncol. 2020;38:3377–87.
Vaidya R, Witzig TE. Prognostic factors for diffuse large B-cell lymphoma in the R(X)CHOP era. Ann Oncol. 2014;25:2124–33.
Stokes ME, Wenzl K, Huang CC, Ortiz M, Hsu C-C, Maurer MJ, et al. Transcriptomic classification of diffuse large B-cell lymphoma identifies a high-risk activated B-cell-like subpopulation with targetable MYC dysregulation. Nat Commun. 2024;15:6790.
Martins F, Rosspopoff O, Carlevaro-Fita J, Forey R, Offner S, Planet E, et al. A cluster of evolutionarily recent KRAB zinc finger proteins protects cancer cells from replicative stress–induced inflammation. Cancer Res. 2024;84:808–26.
Wei X, Yu S, Zhang T, Liu L, Wang X, Wang X, et al. MicroRNA-200 loaded lipid nanoparticles promote intestinal epithelium regeneration in canonical MicroRNA-deficient mice. ACS Nano. 2023;17:22901–15.
Wei F, Ren W, Zhang X, Wu P, Fan J. miR-425-5p is negatively associated with atrial fibrosis and promotes atrial remodeling by targeting CREB1 in atrial fibrillation. J Cardiol. 2022;79:202–10.
Liang W-W, Lu RJ-H, Jayasinghe RG, Foltz SM, Porta-Pardo E, Geffen Y, et al. Integrative multi-omic cancer profiling reveals DNA methylation patterns associated with therapeutic vulnerability and cell-of-origin. Cancer Cell. 2023;41:1567–85.e7.
Hájková H, Fritz MH, Haškovec C, Schwarz J, Šálek C, Marková J, et al. CBFB-MYH11 hypomethylation signature and PBX3 differential methylation revealed by targeted bisulfite sequencing in patients with acute myeloid leukemia. J Hematol Oncol. 2014;7:66.
Li F, Si W, Xia L, Yin D, Wei T, Tao M, et al. Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol Cancer. 2024;23:90.
Yue X, Zhang Z, Liang X, Gao L, Zhang X, Zhao D, et al. Zinc fingers and homeoboxes 2 inhibits hepatocellular carcinoma cell proliferation and represses expression of cyclins A and E. Gastroenterology. 2012;142:1559–70.e2.
Zhang Y, Sun M, Gao L, Liang X, Ma C, Lu J, et al. ZHX2 inhibits thyroid cancer metastasis through transcriptional inhibition of S100A14. Cancer Cell Int. 2022;22:76.
Nagel S, Schneider B, Rosenwald A, Meyer C, Kaufmann M, Drexler HG, et al. t (4;8) (q27; q24) in Hodgkin lymphoma cells targets phosphodiesterase PDE5A and homeobox gene ZHX2. Genes Chromosomes Cancer. 2011;50:996–1009.
Wang D, Sun T, Xia Y, Zhao Z, Sheng X, Li S, et al. Homodimer-mediated phosphorylation of C/EBPα-p42 S16 modulates acute myeloid leukaemia differentiation through liquid-liquid phase separation. Nat Commun. 2023;14:6907.
Ong JY, Torres JZ. Phase separation in cell division. Mol Cell. 2020;80:9–20.
Fujioka Y, Alam JM, Noshiro D, Mouri K, Ando T, Okada Y, et al. Phase separation organizes the site of autophagosome formation. Nature. 2020;578:301–5.
Fan X, Liu F, Wang X, Wang Y, Chen Y, Shi C, et al. LncFASA promotes cancer ferroptosis via modulating PRDX1 phase separation. Sci China Life Sci. 2023;67:488–503.
Chen H, Cui H, Liu W, Li BW, Tian Z, Zhao YY, et al. Manganese drives ferroptosis of cancer cells via YAP/TAZ phase separation activated ACSL4 in OSCC. Oral Dis. 2024;30:4898–908.
Ma H, Chen X, Mo S, Zhang Y, Mao X, Chen J, et al. Targeting N-glycosylation of 4F2hc mediated by glycosyltransferase B3GNT3 sensitizes ferroptosis of pancreatic ductal adenocarcinoma. Cell Death Differ. 2023;30:1988–2004.
Acknowledgements
This work was supported by grants from the Distinguished Taishan Scholars in Climbing Plan (tspd20210321), the Distinguished Taishan Scholars Plan (NO. tstp20230653), the National Natural Science Foundation of China (82170182, 82370165, 82470153, 82470180), the Natural Science Foundation of Shandong Province (ZR2024QH026), Qingdao Science and Technology for People’s Livelihood Demonstration Special Project (25-1-5-smjk-17-nsh), the ECCM Program of Clinical Research Center of Shandong University (2021SDUCRCB008), the Innovative Team of Outstanding Youth in Colleges and Universities of Shandong Province (2023KJ022) and the Young Elite Sponsorship Program of Shandong Provincial Medical Association (2023-LC-0336). Additionally, we thank the Translational Medicine Core Facility of Shandong University for consultation and instrument availability, which supported this work.
Author information
Authors and Affiliations
Contributions
JZ and DMW designed and performed the experiments, analyzed the data, and wrote the manuscript. MFL, XML, NNW, and XS performed the experiments. SYL and BYL contributed to obtaining the patient samples used in the study. TS and DXM wrote the manuscript. DMW, JJY, FL, and CYJ provided supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Wang, D., Luan, M. et al. Liquid-liquid phase separation of ZHX2 protects DLBCL cells against ferroptosis through induction of SLC3A2. Leukemia 39, 2442–2451 (2025). https://doi.org/10.1038/s41375-025-02718-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-025-02718-z
