
Abstract
The DUX4 gene, located within repetitive subtelomeric arrays on chromosomes 4 and 10, plays a critical role in early embryogenesis and has been implicated in several human diseases, including facioscapulohumeral muscular dystrophy (FSHD) and cancer. In B-cell acute lymphoblastic leukemia (B-ALL), DUX4 rearrangements (DUX4-r) define a distinct genomic subtype affecting 5–10% of cases, which is more frequent among older children and teenagers. These rearrangements produce truncated DUX4 proteins with neomorphic transcriptional activity, resulting in aberrant gene expression programs and alternative splicing that disrupt normal B-cell precursor development. Patients with DUX4-r B-ALL often present with poor initial treatment responses, though they typically achieve excellent long-term survival rates with intensive chemotherapy regimens. The cryptic nature of DUX4 rearrangements has historically posed significant challenges to accurate detection, but recent advancements in next-generation sequencing technologies, including RNA and long-read sequencing, and improved immunophenotyping strategies—such as the use of CD371 as a surrogate marker—are enhancing diagnostic accuracy. This review explores the genetic and biological features of DUX4 and its rearrangements, shedding light on their role in leukemogenesis and associated clinical outcomes. Additionally, we highlight emerging technologies that enable the detection of DUX4-r and discuss their implications for clinical use and research. An improved understanding of DUX4 biology and its oncogenic potential may pave the way for novel treatment strategies, ultimately improving outcomes for patients with DUX4-r B-ALL.
Similar content being viewed by others
Introduction
The DUX4 gene has been implicated in human disease development, including leukemogenesis [1]. In 2016, genetic abnormalities of DUX4 were first discovered within a specific subtype of childhood acute lymphoblastic leukaemia (ALL), termed DUX4-r ALL [2]. DUX4-r ALL comprises 5–10% of B-cell precursor cases and tends to occur most commonly in older children and teenagers, a group of patients usually associated with poorer treatment response when compared to younger children [3]. In the majority of instances, the driving mutation is the translocation of DUX4 to the IGH locus in immature B-cells, leading to the expression of a truncated DUX4 protein [2]. The location of DUX4 within highly repetitive sequence on the subtelomeres of chomosomes 4 and 10 has made such rearrangements extremely challenging to genetically detect and biologically characterise, complicated further by variant isoform transcription and recombination between alleles of homologous sequence [4]. As a result, the mechanism by which DUX4-r induces leukemogenesis is unclear. In this review, we discuss what is known about the genetic composition of the DUX4 loci and the emerging genomic and biological features of wild-type (wt) and aberrant DUX4 expression in normal development and disease, focusing on leukemogenesis. DUX4-r exhibits a unique gene expression profile among ALL subtypes, driven by neomorphic transcriptional activity distinct from that of wt DUX4 protein [5]. Recent studies have made significant strides in characterising IGH::DUX4 cofactors and downstream pathway activation, creating promising new avenues of research for therapeutic invervention.
A metadata analysis of clinical features shows that DUX4-r patients have a specific demographic profile and excellent survival rates (>95% in children, >80% in adults), despite over half of patients responding poorly to initial induction therapy [6, 7]. There is uncertainty over whether the excellent survival rates are driven by the more intensive treatment received due to the association of measurable residual disease (MRD) at day 28 [8]. The accurate detection of these patients at diagnosis is currently a conundrum for most genetic testing laboratories. This review also summarises the technologies and laboratory approaches that have been assessed and used for identifying DUX4-r patients, and we discuss their broader utility for prospective screening. Recent studies have begun to unravel the role of DUX4-r in leukaemia development and treatment response, but future efforts must rely on accurate detection in prospective clinical trials and international collaboration to determine the origin, behaviour and outcomes of DUX4-r in leukaemia.
DUX4 genetics and wildtype function
Genetics
The double homebox (DUX) genes encode a family of transcription factors (TFs) that play key roles in the early embryonic development of eutherian mammals [9]. In humans, these consist of the three paralogues DUXA, DUXB and DUX4 [10], of which only DUX4 has been extensively characterised due to its roles in facioscapulohumeral muscular dystrophy (FSHD) [11] and multiple cancers [1, 12, 13].
DUX4 is expressed at two independent loci, 4q35 (also known as D4Z4) and 10q26, where it is nested within each 3.3 kb repeat unit of a macrosatellite array [14]. These arrays exhibit >99% sequence homology and recombination events between them are frequent [15]. The only consistent difference is the deletion of the 6-mer sequence “CGCCTC” in 10q26 repeats compared to D4Z4 [16]. Both arrays typically consist of 11–100 repeat units and only the final DUX4 copy is transcribed [11].
Each macrosatellite repeat unit includes a 1.4 kb intronless open reading frame (ORF) comprising the entire DUX4 protein coding sequence, while at the distal end of each ORF is a single non-coding exon. At both DUX4 loci, there are two major alleles that are equally frequent in the general population: A and B [17]. Downstream of the final ORF, A alleles exhibit five non-coding exons that contribute to tissue-specific splice isoforms (Fig. 1) [17, 18]. These exons are missing on both B alleles and it is not currently known how B-allele transcripts are stabilised [18].

Each contains the DUX4 open reading frame (ORF), of which only the distal unit is transcribed. The seven non-coding exons identified in A alleles are shown, along with polyadenylation signals (red lines) and known transcript isoforms. Created in BioRender. Ryan, S. (2025) https://BioRender.com/3zn0iuh.
All full-length DUX4 transcripts produce a protein at whose N-terminus are two homeoboxes that serve as its DNA binding domain [19], while at its C-terminus is a transactivation domain (TAD) which recruits histone acetyltransferases (HATs) for gene activation [20]. The amino acids spanning from the second homeobox to the TAD are intrinsically disordered, such that targeted deletion of this sequence does not affect DUX4 expression levels or function [21]. A single short-length transcript has been identified, arising from the A allele of the D4Z4 locus, which gives rise to a DUX4 protein without a TAD [18].
DUX4 in healthy tissue
DUX4 expression primarily occurs in the pre-implantation embryo, where it is tightly regulated by telomeric remodelling during embryo cleavage [22], acting as a pioneer factor in the first main wave of zygotic genome activation [23]. Hundreds of genes are directly activated by DUX4 [24], which collectively ensure genome stability, clearance of maternal transcripts and proteins, and the establishment of totipotentiality [25, 26]. Full-length chromosome 4 and 10 transcripts have also been detected in equal amounts in the germline cells of the testes [18] and in T cells of the thymus [27], though their function in these tissues is unclear.
DUX4 in non-leukaemic disease
DUX4 in FSHD
FSHD was the first disorder to be associated with aberrant expression of DUX4. It is the third most common type of muscular dystrophy, with a worldwide prevalence of 1 in 8000 to 20,000 individuals, depending on ancestry [28]. Symptoms tend to onset before the age of twenty and are characterised by a progressive deterioration of skeletal muscles in the face, upper body and lower extremities [29]. No effective treatment options currently exist; however, life expectancy is not shortened compared to the general population [30].
FSHD has only been observed in carriers of the D4Z4 4qA allele, present in around half of the European population [17]. This allele contains a non-canonical polyadenylation signal (PAS) at exon 3, which serves to efficiently stabilise transcripts, leading to a cytotoxic build-up of DUX4 protein and its targets [11, 17].
Cases can be categorised into two genetic subtypes: FSHD1 and FSHD2 (Fig. 2). The pathogenic variant in FSHD1 is the contraction of D4Z4 to 1-10 repeat units and is evident in 95% of cases [31]. D4Z4 contraction causes remodelling of chromatin, resulting in significant loss of repressive H3K9me3 methylation in the distal repeat unit and DUX4 gain-of-function, specifically in skeletal myocytes [31].

While FSHD1 involves contraction of the D4Z4 array to 1–10 repeat units, mutations of D4Z4 chromatin modifier genes are implicated in FSHD2. Both require the 4qA allele and lead to aberrant expression of DUX4 in skeletal muscle due to hypomethlyation of the DUX4 open reading frame (ORF) from the final repeat unit. Created in BioRender. Ryan, S. (2025) https://BioRender.com/ylvekuu.
The disease mechanism of FSDH2 is haploinsufficiency of genes associated with D4Z4 methylation [32]. Unlike FSHD1 wherein only the distal repeat unit is hypomethylated, FSHD2 is associated with global hypomethylation that impacts the whole D4Z4 array, though it does not result in increased levels of DUX4 expression or a more severe phenotype compared with FSHD1 [33]. However, these mutations can act as disease modifiers of D4Z4 contraction, resulting in worse symptoms compared to cases that exhibit only the latter [34].
Despite the ability of DUX4 to facilitate cancer progression, cancer risk in FSHD is not elevated above that of the general population [35], though recent findings suggest that gastrointestinal cancers in particular may be more prevalent among FSHD adults (>40 years) than in non-affected adults [36].
DUX4 in non-leukaemic cancers
DUX4 expression in advanced-stage solid cancers correlates with reduced expression of chemokines and MHC class 1 genes, and inhibition of T cell recruitment to tumour sites [13, 37, 38]. These findings indicate that DUX4 facilitates a tumour microenvironment that is less conducive to effective immune responses, promoting cancer progression and potential resistance to immunotherapy. It is not known why DUX4 is preferentially expressed in advanced-stage cancers, though transient expression of DUX4 transcriptional regulators DPPA2 and DPPA4 has been detected in cases, along with loss-of-function mutations in likely DUX4 repressors [13, 37], and it is possible that increased telomeric shortening derepresses DUX4 in a manner similar to that observed in cleavage-stage embryos [39].
DUX4 gene fusions have been identified as driver mutations in sarcomas, a rare group of malignancies that develop in connective tissue and bone [40]. There are more than 150 known subtypes of sarcoma, the majority (87%) of which occur in soft tissue of the extremities, trunk, head and neck [40]. They account for <1% of malignant cancers in adults but are significantly more common in children and adolescents, comprising 15-20% of paediatric malignancies [40].
DUX4 rearrangements have been implicated in ~1% of round cell sarcomas, a high-grade subset characterised by a round, small and undifferentiated cellular morphology [12], where its fusion partner is the Capicua gene CIC [41]. CIC-rearranged sarcomas typically affect children and young adults (median age 25–35 years), and are notable for their aggressive course and poor outcomes [42]. Capicua is a transcriptional repressor that plays a role in neuronal differentiation, immune cell development and embryogenesis [43]. It also acts a tumour suppressor, in part by silencing the polyomavirus enhancer activator 3 (PEA3) family of oncogenic transcription factors [44]. CIC::DUX4 produces a chimeric protein in which the C-terminal end of Capicua is fused with the DUX4 C-terminal TAD [41] (Fig. 3A), the addition of which results in the acetylation of Capicua targets by p300/CBP, dramatically increasing their expression [41].

Breakpoints of CIC-rearranged sarcoma (A) and DUX4 rearrangements identified in DUX4-r B-ALL (B). Translocation of DUX4 sequence is represented by red arrows in both diagrams. In B-ALL, breakpoints at DUX4 loci occur in the TAD, giving rise to proteins in which the TAD is truncated. Enhancer (Eμ at IGH) or promotor (likely P3 at ERG) regions are ‘hijacked’ to induce aberrant DUX4 expression in B cell precursors. In CIC-rearranged sarcoma, only the DUX4 TAD is translocated, fusing with the final exons of CIC or ATXN1, resulting in a chimeric protein that activates typically repressed CIC target genes in soft tissue of the central nervous system. Base pair coordinates were extracted from human genome build hg38. Created in BioRender. Ryan, S. (2025) https://BioRender.com/1pqisgg.
Two cases of round cell sarcomas have been reported in the CNS that share the gene expression profile of the CIC-rearranged subtype, and yet were negative for CIC alterations [45, 46]. In both, the DUX4 TAD was fused in-frame with the final exon of ATXN1, producing a protein that, similar to CIC::DUX4, contains almost the entire Ataxin 1 sequence flanked by the DUX4 TAD (Fig. 3A). Ataxin 1 interacts directly with Capicua to stabilise its repressor complex [47], suggesting that the addition of the TAD may activate Capicua targets in the same manner as CIC::DUX4.
Finally, a DUX4 fusion event has also been implicated in embryonal rhabdomyosarcoma (ERMS), another soft tissue sarcoma in which the affected cells morphologically resemble the developing skeletal myocytes of the embryo [48]. Fusion events are very rare in ERMS, for which driver events typically involve chromosomal gains [48]. In the single reported case, the patient showed a normal karyotype (46, XX) but carried a translocation of DUX4 to the EWSR1 gene locus at 22q12 [49]. Interestingly, some undifferentiated round cell sarcomas and RMS blasts have been found to originate from a common mesenchymal stem cell [50, 51]. The occurrence of DUX4 rearrangements among these sarcoma subtypes may indicate that DUX4 could have an oncogenic potential that is specific to this stem cell type.
DUX4-rearranged B-ALL
Description of DUX4 rearrangements
In DUX4-r B-ALL, repeat units from the D4Z4 or 10q26 loci are typically inserted out-of-frame into the immunoglobulin heavy locus (IGH) or – more rarely - the ETS-related gene (ERG) [3, 52], though a wide range of other gene partners have been identified in around 4.6% of DUX4-r cases (Fig. 4). The rearrangement involves insertion of a partial repeat copy, or one complete and one partial copy, of DUX4 in either strand orientation [52]. Reciprocal events, where IGH sequences are inserted into the DUX4 locus, can occur in some cases [53]. ‘Triple fusions’ have been detected in five cases, whereby DUX4 is fused with sequence from a third gene partner at the IGH locus, and it is conceivable that some non-recurrent DUX4 gene partners occur in triple fusion events wherein the IGH component was not detected [1].

Lines between genes represent translocation, with the colour of lines corresponding to destination chromosomes. In the case of triple fusions (e,g. IGH::QSOX1::DUX4), both DUX4 and the second gene partner fuse at the IGH locus, represented by two lines, first from DUX4 to the second gene parter, then to IGH. Among 695 DUX4-r cases for which a gene partner has been identified in the literature, 663 (95%) are IGH::DUX4 (excluding triple fusions), 7 (1%) are ERG::DUX4 and 3 (0.4%) are ETV6::DUX4. All others are single cases. Gene coordinates are derived from the human genome build hg38.
IGH breakpoints for IGH::DUX4 rearrangements mostly occur in a 3kb sequence overlapping the diversity (IGH-D) and joining (IGH-J) segments of IGH, near to the Eμ transcriptional enhancer (Fig. 3B) [2]. These segments undergo somatic rearrangement in B-cell precursors, wherein RAG endonuclease complexes induce double-stranded breaks at recombination signal sequences (RSS) [54]. DNA repair enzymes add random nucleotides and the processed ends are ligated [54]. RSS-like sequences have been detected at the breakpoints of translocation events in the IGH locus [55], suggesting the possibility that substrate-selection errors of RAG complexes are implicated in the insertion of DUX4 sequence.
DUX4 breakpoints are enriched in the 5’ region upstream of the DUX4 ORF and in the 3’ end of the ORF itself [2, 56]. Truncation of the DUX4 C-terminus is therefore observed in almost all IGH::DUX4 transcripts, leading to the replacement of up to twenty amino acids with IGH sequence. In the NALM6 cell line, for instance, an endogenous insertion of a full and partial repeat unit from the 10q26 array into IGH leads to a transcript involving only the partial unit, in which the final sixteen amino acids of wt DUX4 are replaced by those of the IGH-D sequence, including a PAS [53].
Phased RNA-seq data from NALM6 cells also showed allelic imbalance from the IGH locus, with the wt IGH allele being more active than the IGH::DUX4 rearrangement, indicating that the translocation took place on the silenced IGH allele [53]. During B-cell development, IGH allelic exclusion ensures that each B-cell produces a single type of antibody by first silencing both alleles through hypermethylation, then selectively activating one during RAG-mediated somatic rearrangement. While it is unknown whether the silenced IGH allele is affected in all DUX4-r cases, it is possible that excess expression of DUX4 hinders leukemogenesis due to cytotoxicity, though there is no evidence that overexpression of IGH::DUX4 is pro-apoptotic [5].
In ERG::DUX4, D4Z4/10q26 repeat units are inserted into ERG intron 3 (Fig. 3B) [52, 57]. Interestingly, this intron has been found to be a hotspot for double-strand breaks induced by aberrant activity of the androgen receptor complex [58], suggesting that DUX4 insertions at ERG take place through an alternate mechanism to that at the IGH locus. Similar to IGH::DUX4, however, transcripts typically result in a truncated C-terminal DUX4 protein and have the same gene expression profile [52], indicating that downstream leukaemic activity is identical.
Secondary genomic alterations
Intragenic ERG deletions (ERGdel) are a critical secondary event in DUX4-r B-ALL and occur almost exclusively in this subtype [59], with multiple studies showing that >95% of B-ALL cases with ERGdel harbour a DUX4 rearrangement [2, 60, 61]. Indeed, ERGdel was detected and proposed as a subgroup of BCP-ALL before DUX4 rearrangement events were discovered [62]. Typically, ERG deletions are focal, most commonly involving exons 3–7 or 3–9, and are thought to be secondary events arising from the binding of the DUX4 rearranged protein, but not wt DUX4, to an alternative transcription initiation site within intron 6 of ERG [2, 60, 61].
DUX4 rearranged proteins can also induce transcription of alternative ERG transcripts, ERGalt, originating from a non-canonical exon. The most common isoform results from splicing that joins the novel exon to exon 7, producing a truncated protein that has reduced transactivation ability and acts as a competitive inhibitor of wildtype ERG [2].
ERGalt transcripts contain exons that are not present in the deleted ERG allele, and are detectable in cases that do not carry the ERGdel [63], indicating that it arises from the unaffected allele. Moreover, ERGalt transcripts are more abundant in ERGdel cases, suggesting that activity of the DUX4 rearranged protein is positively correlated with the likelihood of ERGdel [63].
Interestingly, mouse models have revealed a role for isoform-specific ERG in oncogenesis. While animals expressing full-length ERG in bone marrow succumb to an aggressive erythromegakaryoblastic leukaemia, those expressing ERGalt develop lymphoid leukaemias with longer latency [2], suggesting that ERGalt may itself contribute to leukemogenesis in DUX4-r cases.
Mutational analyses of DUX4-r cases showed a mean of 17.5 non-silent somatic sequence mutations per case (range 2–42) [2], and a median of 0.25 SNVs per Mb [64]. A minority of DUX4-r patients show enrichment for the mutational signature associated with reactive oxygen species [64]. Across all currently published studies that have investigated the incidence of non-silent mutations in DUX4-r, 44.7% of cases carry mutations affecting genes involved in the RAS/MAPK pathway, while 32.3% carry mutations in epigenetic pathway genes, and 21.3% of cases carry mutations affecting transcription factor genes (Fig. 5).

Findings were meta-analyzed using data from seventeen studies in which non-silent mutations were investigated [1, 3, 52, 57, 59, 63, 64, 81, 86, 87, 98,99,100,101,102,103,104,105]. Gene sets were created by combining those used in Zhang et al. [2] and Brady et al. [64], which assessed mutations across all B-ALL subtypes.
DUX4-r B-ALL is characterized by a low incidence of other chromosomal alterations compared to other B-ALL subtypes [65], though DUX4-r carriers of ERGdel have been shown to harbour significantly more intragenic structural alterations than non-carriers [63]. Across published studies that have analysed intragenic structural variants in DUX4-r, the most common events are ERG deletion (43.1% of cases), CDKN2A/B deletion (31.2%), IKZF1 deletion (18.5%), and PAX5 deletion (12.9%) (Fig. 6).

Transactivation and gene expression profile
While IGH::DUX4 binds 97% of the same targets as wt DUX4 in NALM6 [56, 66] ten-fold fewer genes are transcriptionally activated by IGH::DUX4 compared to the wild-type [66]. Therefore, it has been suggested that the truncation of the C-terminal attenuates the ability of DUX4 rearranged protein to recruit p300/CEBP for transactivation, behaving as a less efficient isoform of wt DUX4 [53]. However IGH::DUX4 can recruit cofactors that are not involved in wt DUX4 transactivation and initiate novel mechanisms of alternative splicing, giving rise to a distinct gene expression profile that is unique among B-ALL subtypes [61].
Despite their identical DNA-binding domains, ChIP-seq studies against IGH::DUX4 have shown that active target sites are more enriched in intronic and intergenic sites compared to wt DUX4, while comparisons of transcriptional activity have found minimal overlap between both upregulated and downregulated gene sets [5, 66]. Target genes of wt DUX4 that play key roles in embryo development, such as ZSCAN4, DUXA and RFPL4A, are not activated by IGH::DUX4, which instead upregulates genes enriched in cell adhesion, migration, and lymphocyte activation pathways, including ERG, AGAP1, STAP1, ITGA6, PTPRM, C6orf89, TCF12 and CLEC12A [5, 66,67,68,69]. Genes downregulated in IGH::DUX4 cells, on the other hand, are implicated in pre-B cell receptor signalling and immune-related pathways.
The transcriptional activity of IGH::DUX4 is highly similar to that of DUX4 protein in which the entire TAD has been knocked out (DUX4-del50) [5]. p300 selectively binds wt DUX4 but not IGH::DUX4, and induction of the rearranged protein does not increase H3K27Ac modifications or chromatin accessibility at its DNA binding sites [5]. These findings suggest that IGH::DUX4 protein is not a pioneer factor, though it is able to initiate transcription and alternative splicing despite harbouring an inactive TAD. Recent studies have highlighted transcriptional cofactors that could account for this neomorphic activity.
Zhang et al. [70] detected recombination signal sequence (RSS) like motifs in IGH::DUX4 targets ERG, CLEC12A, and C6orf89, to which RAG1/2 binding was confirmed by in vitro cleavage assay. Knockdown of RAG1/2 significantly reduced expression levels of alternative transcripts, including ERGalt. Li et al. [68] found that the alternative splicing activity of transcription factor TFC12 is also increased in DUX4-r cells, while induction of the fusion protein upregulates TCF12 expression, suggesting a positive-feedback mechanism. Both RAG1/2 and TCF12 interact with a positively-charged pocket created by the dimerization of two IGH::DUX4 proteins at their homeodomains, disruption of which ceased IGH::DUX4-driven alternative splicing activity. Reduction of IGH::DUX4 activity by TCF12 knockdown suggests that TCF12 is a RAG1/2 cofactor rather than a competitive inhibitor.
IGH::DUX4 does not activate its targets in human embryonic kidney or T-ALL Jurkat cells, leading Campulungo et al. [5] to hypothesize that it interacts with a transcription factor that is preferentially expressed in B-cell precursors. General transcription factor IIi (GTF2I) is the most abundant interactor of IGH::DUX4 in REH cells - an ALL cell line - but not in the other cells types, and CUT&Tag analysis showed selective binding of GTF2I to DUX4-r target genes in REH cells even in the absence of IGH::DUX4. Increased expression of IGH::DUX4 also upregulated GTF2I, indicating positive feedback similar to that of TCF12, while GTF2I knockdown significantly reduced activity of IGH::DUX4 but not wt DUX4.
Further studies are required to ascertain possible interactions of RAG1/2, TCF12 and GTF2I in DUX4-r cells, among other potential cofactors, though the presented evidence indicates that all are implicated in IGH::DUX4-induced transcription. Expression of RAG1/2 and TCF12 are positively correlated during T cell receptor (TCR) development [71], suggesting TCF12 cooperates with RAG1/2 during rearrangement of TCR genes, and it is possible that a similar mechanism is aberrantly triggered by IGH::DUX4 expression in B cell precursors. Differences between wt DUX4 and DUX4-r expression and downstream activity are summarised in Table 1.
Lineage infidelity and immunophenotype
Single-cell analysis has revealed a heterogenous cell maturation profile in DUX4-r B-ALL that is not typically observed among other subtypes. While cells with BCR::ABL1, ETV6::RUNX1 and high hyperdiploid subtypes mainly resemble normal pro-B cells, DUX4-r cells appear to evade this ‘maturation block’ and display a broader range of differentiation states, including profiles corresponding to mature B cells [72]. Expression of NFAT3 is more abundant among early-stage DUX4-r cells, while mature cells preferentially express CEBPA and FLT3 [64, 72]. The extent of cell maturation differs among DUX4-r cases, leading Brady et al. [64] to delineate DUX4-r into DUX4-a and DUX4-b according to their differential gene expression, with DUX4-b corresponding to samples containing mature cell types.
DUX4-r leukaemic blasts also demonstrated a high level of lineage infidelity compared to other subtypes, exhibiting characteristics of myeloid (e.g., CD371 expression) and T cells (e.g., CD2 and GATA3 expression), coupled with downregulation of B-cell markers [72, 73]. Expression of CD371 is almost unique to the DUX4-r B-ALL [74, 75] and CAR T cell immunotherapy targeting CD371 has been shown to have a potent anti-leukaemic effect on DUX4-r cell populations [72].
In a high proportion of DUX4-r cases, blasts also undergo monocytic switch during induction therapy, characterised by a decrease in expression of leukaemic markers CD10, CD34, and CD20, and an increase in monocytic markers CD14 and CD33 [73, 75]. Higher expression of CEBPA, FLT3 and TLR10 has also been reported in DUX4-r cases with monocytic switching compared to cases without [72, 73], suggesting that it is a feature of mature B cells, while CEBPA has been shown to inhibit several B cell regulator genes [76]. No association has been detected between secondary alterations in DUX4-r B-ALL and monocytic switching [73].
Clinical features and outcome of patients with ALL and DUX4-r
DUX4-r has an overall frequency of 5–10% of B-cell precursor cases [64]. The frequency of this subtype varies by age with a peak frequency in older children/teenagers [3]. The median age of patients with DUX4-r is in the range of 8-13 years, which is significantly higher than other BCP-ALL patients (Table 2), who have a median age of ~5 years old. There is no reported association with white blood cell count at diagnosis or sex. Despite these differences, most paediatric and young adult studies have reported a very good outcome for patients with DUX4-r with several of the larger and more recent studies reporting event-free and overall survival rates as high as 95% at 5–10 years (Table 2). Even though the outcome of adults with DUX4-r are not quite as high, they are nonetheless very encouraging given the overall poor outcome reported for adults with ALL [77].
These excellent survival rates contrast with the relatively poor initial treatment response that has been reported by numerous studies. It is very difficult to directly compare initial treatment response rates across studies due to different induction regimens and variability in the timing and method of assessing measurable residual disease (MRD). Lineage infidelity may also confound immnophenotype-based approaches to detecting MRD that only take into account lymphoid markers, leading to an under-reporting of MRD among DUX4-r cases.
Nonetheless, there is a clear trend towards patients with DUX4-r responding more slowly to therapy than other patients. For example, in UKALL2003, 55% of patients with DUX4-r were MRD positive (≥0.01% at the end of induction, as assessed by PCR) compared to just 21% of patients with ETV6::RUNX1 [6]. Yoshimura and colleagues recently reported differential drug sensitivity by both age and genetic subtype, which may explain this apparent paradox, finding that older paediatric cohorts and DUX4-r cases may respond better to therapy stages later than induction [78]. However, as DUX4-r patients are more likely to be high risk due to their age and have MRD at the end of induction, they are also more likely to have been treated on more intensive treatment arms. Once again, as each trial has different risk stratification criteria, it is difficult to compare studies directly. However, for most studies the proportion of DUX4-r treated as standard risk is no more than one-third of cases.
In UKALL2003, only 36% of patients with DUX4-r were treated in regimen A (the lowest intensity protocol) compared with 70% of patients with ETV6::RUNX1 [6]. Therefore, at the moment we do not know if the excellent outcomes reported for patients with DUX4-r are because it is an intrinsically chemosensitive subtype, like ETV6::RUNX1, or because they were more likely to have received intensive chemotherapy. Few studies have reported outcome by treatment arm, but it is noteworthy that in UKALL2003, only one of the four relapses came from the cohort treated on regimen A [6]. In addition, CD371+ patients treated as standard/medium risk had an EFS of 91% at 5 years compared with 85% for those treated as high risk [75]. It is also currently unknown whether improved outcomes are observed among DUX4-r cases treated with more recently developed therapies, such as the immunotherapeutic Blinatumomab [79].
Even though IKZF1 deletions have been widely reported as an adverse biomarker [80] several studies have now shown that they are not associated with an inferior outcome among DUX4-r patients [60,61,62, 74]. Although one study has reported an adverse effect of ‘IKZF1plus’ [74] – the co-occurrence of IKZF1 deletions with deletions in CDKN2A, CDKN2B or PAX5, or P2RY8::CRLF2 fusion [80] – Li et al. [61]. reported an improved MRD response for DUX4-r patients with ERGdel, but this did not translate into a difference in outcome as reported by Schinnerl et al. [59]. Two studies have reported a small number of DUX4-r cases with co-occurring TP53 mutations and relapse rate (two of four cases and four of five cases) [74, 81]. Brady et al. [64] identified two gene expression sub-clusters within DUX4-r (DUX4-a and -b) with differential outcomes. DUX4-a cases had a superior outcome and were enriched for ERG and TBL1XR1 deletions, whereas DUX4-b cases were enriched with NRAS mutations, IKZF1 deletions and KMT2D mutations [64]. Additional dedicated biomarker studies that integrate treatment information are required to resolve some of these discrepancies and determine which risk factors within DUX4-r are clinically relevant.
Detection
Cytogenetics, FISH and SNP array
DUX4 rearrangement events cannot be detected by conventional cytogenetic approaches. Due to their small size (<10 kb) and typical insertion into the telomeric IGH locus, the translocated DUX4 sequence is not visible by G-banded karyotyping or fluorescence in situ hybridisation (FISH) [52]. Moreover, the repetitive nature of D4Z4/10q26 arrays, in addition to the presence of multiple D4Z4-like sequences across the genome, greatly increases the likelihood of cross-hybridisation by FISH or SNP (single-nucleotide polymorphism) array probes, leading to ambiguous or false-negative findings.
RNA-seq
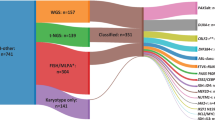
Transcriptomic analysis has proved to be successful for detecting DUX4-r cases, and is the approach utilised by 20/26 (77%) studies that have successfully identified them to date. Many have utilized alignment-based tools used to identify fusion breakpoints in RNA-seq data, such as STAR-fusion [82] and FusionCatcher [83]. The unique gene expression profile of DUX4-rearranged B-ALL also lends itself to accurate detection using differential gene expression (DEG) analysis. Studies utilising this approach typically employed statistical methods such as DESeq2 [84] to identify significantly upregulated/downregulated genes among cases using RNA-seq data. These are typically complemented by clustering and visualization techniques such as prediction of microarrays (PAM), hierarchical clustering, and t-distributed stochastic neighbour embedding (tSNE) to group samples according to their distinct transcriptional signatures. Machine learning algorithms ALLCatchR [69] and ALLsorts [85] build on these approaches to accurately assign B-ALL subtype, including DUX4-r, based on DEG. Both fusion analysis and DEG-based approaches consistently detect DUX4-r at a frequency of 5-10% in B-ALL cohorts [1, 2, 7, 52, 57, 59, 61, 63, 64, 74, 86, 87].
Short-read whole genome sequencing
Other approaches used to detect DUX4-r fusions at the genomic level include short-read whole-genome sequencing (WGS). Leongamornlert et al. [57]. found that the GRIDSS structural variant caller [88] could accurately identify DUX4-r fusions, as it employs a contig assembly approach to reconstruct sequences in WGS data around potential breakpoints from reads that show evidence of fusion events (such as splitting or discordant pair mapping), thereby mitigating the impact of off-target read alignments. This approach detected eight DUX4-r cases in a cohort of fifty-seven adult B-other ALL (14%).
The recently developed Pelops [89] tool is specifically designed to detect DUX4-r fusions. Pelops identifies read pairs that span the fusion junction between DUX4 and other genomic loci, such as IGH, in WGS data. These spanning read pairs are normalized using a spanning read pairs per billion total reads (SRPB) metric, which adjusts for differences in sequencing depth across samples to ensure accurate comparison. Pelops successfully identified all DUX4-r cases for which there was orthogonal evidence in a validation cohort [1], totalling 60/210 (28.6%) paediatric B-other cases.
Long-read sequencing
Long-read approaches have been used to successfully characterise the endogenous IGH::DUX4 fusion event in the NALM6 cell line [53]. By barcoding short reads that belong to the same high molecular weight DNA fragments using the 10x Genomics Chromium platform, 8 kb and 4.5 kb contigs could be assembled, which included the IGHM region, Eμ enhancer, translocated 10q26 DUX4 sequence, and other IGH sequences near the breakpoints.
PacBio and Oxford Nanopore Technology (ONT) long-read platforms can directly produce reads >10 kb in length, mitigating the potential for poor mapping at the IGH locus. Yasuda et al. [3] performed whole-genome sequencing on the NALM6 cell line using a PacBio single-molecule real-time (SMRT) platform, producing reads with a mean length of 13.3 kb [3]. Three reads successfully mapped the full and partial copy of chr10 DUX4 present at the IGH locus, confirming the IGH::DUX4 rearrangement reported in this cell line by previous studies. In the first published study that used ONT sequencing to subtype B-ALL cases, Kato et al. [90] used a targeted approach known as adaptive sampling to identify fusion breakpoints in tumour samples whose mean N50 was 11 kb. For detection of DUX4-r B-ALL, the DUX4, IGH and ERG loci were sequenced and subjected to structural variant (SV) detection using Nanomonsv [91], while de novo sequencing assembly was used for comprehensive breakpoint ascertainment. Three IGH::DUX4 cases were identified among thirteen B-ALL cases, all of which were found to carry ERGdel.
Indirect detection methods
As ERGdel is detectable by multiplex ligation-dependent probe amplification (MLPA), SNP array, and breakpoint-specific polymerase chain reaction (PCR) [92], it is often relied on as a surrogate marker for DUX4-r in B-ALL cases. Probe signal may be too weak to detect this alteration when it occurs subclonally, though qPCR and amplicon-sequencing have been shown to somewhat overcome this limitation [63]. However, ERGdel has been observed in a small number of cases that do not harbour a DUX4 rearrangement and is absent in around 40–60% of DUX4-r cases, limiting its reliability for diagnostic purposes [63]. ERGalt expression has also been suggested as a surrogate marker, but levels vary widely between cases and it has also been observed in low amounts among other B-ALL subtypes [2, 63].
Immunophenotyping has demonstrated much greater reliability in detecting DUX4-r cases, due to the non-lymphoid markers that are almost uniquely expressed in this subtype. In particular, flow cytometry of CD371 – an approach typically employed for the detection of myeloid malignancies – identified all but one DUX4-r in a cohort of 46 cases, 91.3% of which showed strong expression of this marker at diagnosis [74]. A subfraction of blasts showed weak CD371 expression in six instances of other subtypes, namely two high hyperdiploid, one near haploid, one BCR::ABL1 and two undefined B-other cases. Similarly, Buldini et al. [75]. found that 27/28 (96.4%) of B-ALL cases carrying DUX4-r fusions were CD371-positive at diagnosis, compared with 7/387 (1.8%) of cases of other subtypes, indicating very high sensitivity and specificity for CD371 detection as a marker for DUX4-r B-ALL.
Moreover, combined expression of CD371 and CD2 is thought to exclusively occur in DUX4-r cases [59, 74, 75], though, as CD2 is only expressed in 50-75% of samples, detection of both markers is not as sensitive as the detection of CD371 alone. In addition, Buldini et al. [75] found that CD371 had a sensitivity of 93% for predicting the myelomonocytic lineage infidelity among B-ALL cases at diagnosis, compared to 56% for CD2.
Immunohistochemistry targeting the N-terminus of the DUX4-r fusion protein has recently been suggested as a reliable approach for diagnosis. Among six B-ALL cases for which there was RNA-seq or immunohistological evidence of DUX4-r, Siegele et al. [93] reported strong nuclear staining with an N-terminus DUX4 antibody, while three additional cases that were known to be negative for IGH::DUX4 showed no evidence of staining. Further studies may be required to ascertain the specificity of this approach, given that low levels of DUX4 expression have been observed in DUX4-r negative B-ALL cases [63]. However, it is unclear whether this indicates the presence of translatable DUX4 mRNA or is due to technical artifacts such as cross-sample contamination.
Comparing DUX4-r detection methods
Despite technical differences between direct detection approaches, the reported frequency of DUX4-r cases remains consistent across studies (5-10%), with exceptions observed only among smaller studies. Given the relative lack of studies that have utilised DNA-seq data compared to RNA-seq, a robust comparison of the sensitivities of the corresponding detection methods is not yet feasible. However, on the basis of current literature, there is no evidence to suggest that optimised short-read WGS approaches (e.g., GRIDSS, Pelops) have greater sensitivity than RNA-seq and vice versa, while the additional utility of long-read technologies currently seems to be limited to more accurate resolution of breakpoints, particularly at partner gene loci.
A disadvantage of DEG analysis is that is it only applicable in the context of B-ALL, as it depends on comparison of the DUX4-r gene expression profile (GEP) with that of other B-ALL cases. Similarly, CD371 can only be used as a surrogate marker for DUX4-r relative to its expression among cases of other B-ALL subtypes. By contrast, gene fusion analysis (using both short-read/long-read WGS and RNA-seq) can be used to detect DUX4-r B-ALL even where diagnostic information is unavailable, and can also detect DUX4 rearrangements occurring in non-leukaemic cancers. For example, the Pelops tool has successfully identified DUX4 fusion events in other cancer types across Genomics England cohorts, many of which were not previously known to be associated with DUX4 rearrangements (Alona Solinsky, personal communication, January 2025).
On the other hand, clinically relevant differences in GEP associated with cell maturation profiles are not detectable by gene fusion analysis and there is no evidence that different DUX4 partner genes in B-ALL contribute to differential gene expression among DUX4-r cases. Moreover, while CD371+ is associated with good long-term outcomes in B-ALL, almost all DUX4-r cases show strong expression of this antigen, so it cannot be used to assess outcomes between DUX4-r cases. DEG analysis may therefore be more informative for the evaluation of outcomes within this subtype than gene fusion analysis and CD371 flow cytometry.
The costs associated with each detection method are an important consideration in both clinical and research settings. High-throughput RNA-seq is more affordable than both short-read and long-read WGS. Given the potential to differentiate between the two DUX4-r groups (DUX4-a and DUX4-b) from RNA data but not WGS, it is the most cost-effective option for both DUX4-r detection and downstream clinical analyses. CD371 flow cytometry is relatively cheap, highly accessible and faster than sequencing [94], which, combined with its high sensitivity and specificity for DUX4-r detection, makes it the best option in clinical settings where access to more expensive sequencing equipment is limited.
Long-read WGS is currently one of the most costly options, with PacBio platforms being more expensive than ONT, which are capable of producing longer read lengths [95]. ONT platforms have a higher sequencing error rate, which may impact variant calling; however, these have largely been mitigated by improved basecalling algorithms [96]. In addition, ONT platforms generate data in real-time during runs, leading to improved efficiency over PacBio and short-read platforms. Further optimisations of ONT platforms are likely to reduce the per-sample cost of long-read WGS below that of short-read alternatives [97]. Long-read platforms can also generate RNA-seq data, though the utility of this in the context of B-ALL is yet to be investigated.
Conclusions
The unique biology of the DUX4 transcription factor and its rearrangements underpins its roles in both normal developmental processes and disease. In B-ALL, DUX4-rearranged cases occur at a frequency of 5-10%, and exhibit a distinct transcriptional profile driven by a truncated DUX4 protein, which recruits alternate cofactors for gene activation and splicing. The downstream consequences of its unusual activity are demonstrated by aberrant expression of non-lymphoid markers (e.g., CD371), unique structural variation (ERGdel), a broad cell maturation profile, and a higher incidence of MRD at the end of induction therapy. Despite these features, the overall prognosis for DUX4-rearranged B-ALL patients remains favourable, with event-free and overall survival exceeding 80% in most paediatric cases. Advancements in sequencing technologies have the potential to both markedly improve and simplify detection of this notoriously cryptic subtype, leading a more comprehensive genomic profiling and a robust understanding of the mechanisms behind DUX4 rearrangement.
Future work
-
Understand the mechanisms by which DUX4 rearrangements occur, and the genomic, transcriptional and biological features shared among its many identified gene partners.
-
Understand how DUX4-r activity - and in particular its capacity for alternative splicing - leads to B-cell lineage development, infidelity and the evasion of maturation blocks typically not observed in B-ALL.
-
Comparative studies of DUX4 rearrangements among other cancer types, given preliminary findings that DUX4 rearrangements can occur in several other malignancies beyond B-ALL and CIC-r sarcoma, which may reveal shared oncogenic pathways.
-
Reach an international consensus on the best approaches to DUX4-r detection at diagnosis, with a focus on those that can be adopted in clinical settings without access to cutting-edge sequencing technology.
-
Improved risk stratification and prognosis using chemotherapy and, more recently, developed therapies such as Blinatumomab.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Ryan SL, Peden JF, Kingsbury Z, Schwab CJ, James T, Polonen P, et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia. 2023;37:518–28.
Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48:1481–9.
Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569–74.
van Geel M, Dickson MC, Beck AF, Bolland DJ, Frants RR, van der Maarel SM, et al. Genomic Analysis of Human Chromosome 10q and 4q Telomeres Suggests a Common Origin. Genomics. 2002;79:210–7.
Campolungo D, Salomé M, Biferali B, Tascini AS, Gabellini D. DUX4-r exerts a neomorphic activity that depends on GTF2I in acute lymphoblastic leukemia. Sci Adv. 2023;9:eadi3771.
Schwab C, Cranston RE, Ryan SL, Butler E, Winterman E, Hawking Z, et al. Integrative genomic analysis of childhood acute lymphoblastic leukaemia lacking a genetic biomarker in the UKALL2003 clinical trial. Leukemia. 2023;37:529–38.
Paietta E, Roberts KG, Wang V, Gu Z, Buck GAN, Pei D, et al. Molecular classification improves risk assessment in adult BCR-ABL1-negative B-ALL. Blood. 2021;138:948–58.
Yu CH, Jou ST, Su YH, Coustan-Smith E, Wu G, Cheng CN, et al. Clinical impact of minimal residual disease and genetic subtypes on the prognosis of childhood acute lymphoblastic leukemia. Cancer. 2023;129:790–802.
Leidenroth A, Hewitt JE. A family history of DUX4: phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol Biol. 2010;10:364.
Bosnakovski D, Toso EA, Ener ET, Gearhart MD, Yin L, Lüttmann FF, et al. Antagonism among DUX family members evolved from an ancestral toxic single homeodomain protein. iScience. 2023;26:107823.
Mocciaro E, Runfola V, Ghezzi P, Pannese M, Gabellini D. DUX4 Role in Normal Physiology and in FSHD Muscular Dystrophy. Cells. 2021;10:3322.
Yoshimoto T, Tanaka M, Homme M, Yamazaki Y, Takazawa Y, Antonescu CR, et al. CIC-DUX4 induces small round cell Sarcomas distinct from Ewing Sarcoma. Cancer Res. 2017;77:2927–37.
Smith AA, Nip Y, Bennett SR, Hamm DC, Lemmers RJLF, van der Vliet PJ, et al. DUX4 expression in cancer induces a metastable early embryonic totipotent program. Cell Rep. 2023;42:113114.
Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci. 2007;104:18157–62.
Dai Y, Li P, Wang Z, Liang F, Yang F, Fang L, et al. Single-molecule optical mapping enables quantitative measurement of D4Z4 repeats in facioscapulohumeral muscular dystrophy (FSHD). J Med Genet. 2020;57:109–20.
Wang T, Antonacci-Fulton L, Howe K, Lawson HA, Lucas JK, Phillippy AM, et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature. 2022;604:437–46.
Lemmers RJLF, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science (1979). 2010;329:1650–3.
Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, et al. Facioscapulohumeral Dystrophy: Incomplete Suppression of a Retrotransposed Gene. PLoS Genet. 2010;6:e1001181.
Lee JK, Bosnakovski D, Toso EA, Dinh T, Banerjee S, Bohl TE, et al. Crystal structure of the Double Homeodomain of DUX4 in complex with DNA. Cell Rep. 2018;25:2955–2962.e3.
Choi SH, Gearhart MD, Cui Z, Bosnakovski D, Kim M, Schennum N, et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016;44:5161–73.
Mitsuhashi H, Ishimaru S, Homma S, Yu B, Honma Y, Beermann ML, et al. Functional domains of the FSHD-associated DUX4 protein. Biol Open. 2018;7:bio033977.
Zhang X, Zhang C, Zhou D, Zhang T, Chen X, Ren J, et al. Telomeres cooperate in zygotic genome activation by affecting DUX4/Dux transcription. iScience. 2023;26:106158.
Hendrickson PG, Doráis JA, Grow EJ, Whiddon JL, Lim JW, Wike CL, et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet. 2017;49:925–34.
Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, et al. DUX4 Activates Germline Genes, Retroelements, and Immune Mediators: Implications for Facioscapulohumeral Dystrophy. Dev Cell. 2012;22:38–51.
Grow EJ, Weaver BD, Smith CM, Guo J, Stein P, Shadle SC, et al. p53 convergently activates Dux/DUX4 in embryonic stem cells and in facioscapulohumeral muscular dystrophy cell models. Nat Genet. 2021;53:1207–20.
Hamm DC, Paatela EM, Bennett SR, Wong CJ, Campbell AE, Wladyka CL, et al. The transcription factor DUX4 orchestrates translational reprogramming by broadly suppressing translation efficiency and promoting expression of DUX4-induced mRNAs. PLoS Biol. 2023;21:e3002317.
Das S, Chadwick BP. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS One. 2016;11:e0160022.
Deenen JCW, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJGM, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–9.
Hamel J, Johnson N, Tawil R, Martens WB, Dilek N, McDermott MP, et al. Patient-Reported Symptoms in Facioscapulohumeral Muscular Dystrophy (PRISM-FSHD). Neurology. 2019;93:1180–92.
Hamel J, Tawil R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics. 2018;15:863–71.
Jones TI, King OD, Himeda CL, Homma S, Chen JCJ, Beermann ML, et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin Epigenetics. 2015;7:37.
Hamanaka K, Šikrová D, Mitsuhashi S, Masuda H, Sekiguchi Y, Sugiyama A, et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology. 2020;94:2441–47.
Jia FF, Drew AP, Nicholson GA, Corbett A, Kumar KR. Facioscapulohumeral muscular dystrophy type 2: an update on the clinical, genetic, and molecular findings. Neuromuscul Disord. 2021;31:1101–12.
Sacconi S, Briand-Suleau A, Gros M, Baudoin C, Lemmers RJLF, Rondeau S, et al. FSHD1 and FSHD2 form a disease continuum. Neurology. 2019;92:2273–85.
Dmitriev P, Kairov U, Robert T, Barat A, Lazar V, Carnac G, et al. Cancer-related genes in the transcription signature of facioscapulohumeral dystrophy myoblasts and myotubes. J Cell Mol Med. 2014;18:208–17.
Kurashige T, Morino H, Ueno H, Murao T, Watanabe T, Hinoi T, et al. Gastrointestinal cancer occurs as extramuscular manifestation in FSHD1 patients. J Hum Genet. 2023;68:91–5.
Chew GL, Campbell AE, De Neef E, Sutliff NA, Shadle SC, Tapscott SJ, et al. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev Cell. 2019;50:658–671.e7.
Pineda JMB, Bradley RK. DUX4 is a common driver of immune evasion and immunotherapy failure in metastatic cancers. eLife. 2024;12:RP89017.
Okamoto K, Seimiya H. Revisiting Telomere Shortening in Cancer. Cells. 2019;8:107.
Burningham Z, Hashibe M, Spector L, Schiffman JD. The Epidemiology of Sarcoma. Clin Sarcoma Res. 2012;2:14.
Mancarella C, Carrabotta M, Toracchio L, Scotlandi K. CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers. Cancers (Basel). 2022;14:5411.
Sparber-Sauer M, Corradini N, Affinita MC, Milano GM, Pierron G, Carton M, et al. Clinical characteristics and outcomes for children, adolescents and young adults with “CIC-fused” or “BCOR-rearranged” soft tissue sarcomas: A multi-institutional European retrospective analysis. Cancer Med. 2023;12:14346–59.
Lee Y. Regulation and function of capicua in mammals. Exp Mol Med. 2020;52:531–7.
Weissmann S, Cloos PA, Sidoli S, Jensen ON, Pollard S, Helin K. The Tumor Suppressor CIC Directly Regulates MAPK Pathway Genes via Histone Deacetylation. Cancer Res. 2018;78:4114–25.
Pratt D, Kumar-Sinha C, Cieślik M, Mehra R, Xiao H, Shao L, et al. A novel ATXN1-DUX4 fusion expands the spectrum of ‘CIC-rearranged sarcoma’ of the CNS to include non-CIC alterations. Acta Neuropathol. 2021;141:619–22.
Satomi K, Ohno M, Kubo T, Honda-Kitahara M, Matsushita Y, Ichimura K, et al. Central nervous system sarcoma with ATXN1::DUX4 fusion expands the concept of CIC-rearranged sarcoma. Genes Chromosomes Cancer. 2022;61:683–8.
Lee Y, Fryer JD, Kang H, Crespo-Barreto J, Bowman AB, Gao Y, et al. ATXN1 Protein Family and CIC Regulate Extracellular Matrix Remodeling and Lung Alveolarization. Dev Cell. 2011;21:746–57.
Agaram NP. Evolving classification of rhabdomyosarcoma. Histopathology. 2022;80:98–108.
Sirvent N, Trassard M, Ebran N, Attias R, Pedeutour F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from t(4;22)(q35;q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet Cytogenet. 2009;195:12–8.
Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007;11:421–9.
Blum JM, Añó L, Li Z, Van Mater D, Bennett BD, Sachdeva M, et al. Distinct and Overlapping Sarcoma Subtypes Initiated from Muscle Stem and Progenitor Cells. Cell Rep. 2013;5:933–40.
Lilljebjorn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, von Palffy S, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790.
Tian L, Shao Y, Nance S, Dang J, Xu B, Ma X, et al. Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Nat Commun. 2019;10:2789.
Roth DB, Gellert M, Craig N. V(D)J Recombination: Mechanism, Errors, and Fidelity. Microbiol Spectr. 2014;2;MDNA3-0041-2014.
Heinäniemi M, Vuorenmaa T, Teppo S, Kaikkonen MU, Bouvy-Liivrand M, Mehtonen J, et al. Transcription-coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. eLife. 2016;5:e13087.
Dong X, Zhang W, Wu H, Huang J, Zhang M, Wang P, et al. Structural basis of DUX4/IGH-driven transactivation. Leukemia. 2018;32:1466–76.
Leongamornlert D, Gutierrez-Abril J, Lee S, Barretta E, Creasey T, Gundem G, et al. Diagnostic utility of whole genome sequencing in adults with B-other acute lymphoblastic leukemia. Blood Adv. 2023;7:3862–73.
Weier C, Haffner MC, Mosbruger T, Esopi DM, Hicks J, Zheng Q, et al. Nucleotide resolution analysis of TMPRSS2 and ERG rearrangements in prostate cancer. J Pathol. 2013;230:174–83.
Schinnerl D, Riebler M, Schumich A, Haslinger S, Bramböck A, Inthal A, et al. Risk factors in DUX4-positive childhood and adolescent B-cell acute lymphoblastic leukemia. Blood Cancer J. 2024;14:119.
Zaliova M, Zimmermannova O, Dorge P, Eckert C, Moricke A, Zimmermann M, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014;28:182–5.
Li Z, Lee SHR, Chin WHN, Lu Y, Jiang N, Lim EH, et al. Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. 2021;5:5226–38.
Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2013;28:70–7.
Zaliova M, Potuckova E, Hovorkova L, Musilova A, Winkowska L, Fiser K, et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica. 2019;104:1407–16.
Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54:1376–89.
Vendramini E, Giordan M, Giarin E, Michielotto B, Fazio G, Cazzaniga G, et al. High expression of miR-125b-2 and SNORD116 noncoding RNA clusters characterize ERG-related B cell precursor acute lymphoblastic leukemia. Oncotarget. 2017;8:42398–413.
Tanaka Y, Kawazu M, Yasuda T, Tamura M, Hayakawa F, Kojima S, et al. Transcriptional activities of DUX4 fusions in B-cell acute lymphoblastic leukemia. Haematologica. 2018;103:e522–6.
Steeghs EMP, Bakker M, Hoogkamer AQ, Boer JM, Hartman QJ, Stalpers F, et al. High STAP1 expression in DUX4-rearranged cases is not suitable as therapeutic target in pediatric B-cell precursor acute lymphoblastic leukemia. Sci Rep. 2018;8:693.
Li Z, Jiang M, Wang J, Zhuo Z, Zhang S, Tan Y, et al. Transcription factor 12-mediated self-feedback regulatory mechanism is required in DUX4 fusion leukaemia. Clin Transl Med. 2023;13:e1514.
Beder T, Hansen BT, Hartmann AM, Zimmermann J, Amelunxen E, Wolgast N, et al. The Gene Expression Classifier ALLCatchR Identifies B-cell Precursor ALL Subtypes and Underlying Developmental Trajectories Across Age. Hemasphere. 2023;7:e939.
Zhang H, Cheng N, Li Z, Bai L, Fang C, Li Y, et al. DNA crosslinking and recombination-activating genes 1/2 (RAG1/2) are required for oncogenic splicing in acute lymphoblastic leukemia. Cancer Commun. 2021;41:1116–36.
Roels J, Van Hulle J, Lavaert M, Kuchmiy A, Strubbe S, Putteman T, et al. Transcriptional dynamics and epigenetic regulation of E and ID protein encoding genes during human T cell development. Front Immunol. 2022;13:960918.
Thorsson H, Henningsson R, Puente-Moncada N, Peña-Martínez P, Sjöström L, Ågerstam H, et al. Single-cell genomics details the maturation block in BCP-ALL and identifies therapeutic vulnerabilities in DUX4-r cases. Blood. 2024;144:1399–411.
Novakova M, Zaliova M, Fiser K, Vakrmanova B, Slamova L, Musilova A, et al. DUX4r; ZNF384r, and PAX5-P80R mutated B-cell precursor acute lymphoblastic leukemia frequently undergo monocytic switch. Haematologica. 2020;106:2066–75.
Schinnerl D, Mejstrikova E, Schumich A, Zaliova M, Fortschegger K, Nebral K, et al. CD371 cell surface expression: a unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica. 2019;104:e352–5.
Buldini B, Varotto E, Maurer-Granofszky M, Gaipa G, Schumich A, Brüggemann M, et al. CD371-positive pediatric B-cell acute lymphoblastic leukemia: propensity to lineage switch and slow early response to treatment. Blood. 2024;143:1738–51.
Collombet S, van Oevelen C, Sardina Ortega JL, Abou-Jaoudé W, Di Stefano B, Thomas-Chollier M, et al. Logical modeling of lymphoid and myeloid cell specification and transdifferentiation. Proc Natl Acad Sci. 2017;114:5792–9.
Samra B, Jabbour E, Ravandi F, Kantarjian H, Short NJ. Evolving therapy of adult acute lymphoblastic leukemia: state-of-the-art treatment and future directions. J Hematol Oncol. 2020;13:70.
Yoshimura S, Li ZH, Gocho Y, Yang WJ, Crews KR, Lee SHR, et al. Impact of Age on Pharmacogenomics and Treatment Outcomes of B-Cell Acute Lymphoblastic Leukemia. Journal of Clinical Oncology. 2024;42:3478–90.
Hodder A, Mishra AK, Enshaei A, Baird S, Bhuller K, Elbeshlawi I, et al. Blinatumomab for First-Line Treatment of Children and Young Persons With B-ALL. J Clin Oncol. 2024;42:907–14.
Stanulla M, Cave H, Moorman AV. IKZF1 deletions in pediatric acute lymphoblastic leukemia: still a poor prognostic marker?. Blood. 2020;135:252–60.
Ueno H, Yoshida K, Shiozawa Y, Nannya Y, Iijima-Yamashita Y, Kiyokawa N, et al. Landscape of driver mutations and their clinical impacts in pediatric B-cell precursor acute lymphoblastic leukemia. Blood Adv. 2020;4:5165–73.
Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20:213.
Nicorici D, Şatalan M, Edgren H, Kangaspeska S, Murumägi A, Kallioniemi O, et al. FusionCatcher – a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv [Preprint]. 2014.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Schmidt B, Brown LM, Ryland GL, Lonsdale A, Kosasih HJ, Ludlow LE, et al. ALLSorts: an RNA-Seq subtype classifier for B-cell acute lymphoblastic leukemia. Blood Adv. 2022;6:4093–7.
Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine. 2016;8:173–83.
Rehn J, Mayoh C, Heatley SL, McClure BJ, Eadie LN, Schutz C, et al. RaScALL: Rapid (Ra) screening (Sc) of RNA-seq data for prognostically significant genomic alterations in acute lymphoblastic leukaemia (ALL). PLoS Genet. 2022;18:e1010300.
Cameron DL, Schröder J, Penington JS, Do H, Molania R, Dobrovic A, et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017;27:2050–60.
Grobecker P, Berri S, Peden JF, Chow KJ, Fielding C, Armogida I, et al. A dedicated caller for DUX4 rearrangements from whole-genome sequencing data. BMC Med Genomics. 2025;18:24.
Kato S, Sato-Otsubo A, Nakamura W, Sugawa M, Okada A, Chiba K, et al. Genome profiling with targeted adaptive sampling long-read sequencing for pediatric leukemia. Blood Cancer J. 2024;14:145.
Shiraishi Y, Koya J, Chiba K, Okada A, Arai Y, Saito Y, et al. Precise characterization of somatic complex structural variations from tumor/control paired long-read sequencing data with nanomonsv. Nucleic Acids Res. 2023;51:e74–e74.
Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28:70–7.
Siegele BJ, Stemmer-Rachamimov AO, Lilljebjorn H, Fioretos T, Winters AC, Dal Cin P, et al. N-terminus DUX4-immunohistochemistry is a reliable methodology for the diagnosis of DUX4-fused B-lymphoblastic leukemia/lymphoma. Genes Chromosomes Cancer. 2022;61:449–58.
Couillez G, Morel P, Clichet V, Fourdrain L, Delette C, Harrivel V, et al. Flow cytometry as a fast, cost-effective tool to assess IGHV mutational status in CLL. Blood Adv. 2023;7:4701–4.
Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: An overview. Hum Immunol. 2021;82:801–11.
Sereika M, Kirkegaard RH, Karst SM, Michaelsen TY, Sørensen EA, Wollenberg RD, et al. Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat Methods. 2022;19:823–6.
Geyer J, Opoku KB, Lin J, Ramkissoon L, Mullighan C, Bhakta N, et al. Real-time genomic characterization of pediatric acute leukemia using adaptive sampling. Leukemia. 2025;39:1069–77.
Li JF, Dai YT, Lilljebjorn H, Shen SH, Cui BW, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci USA. 2018;115:E11711–20.
Jeha S, Choi J, Roberts KG, Pei D, Coustan-Smith E, Inaba H, et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021;2:326–37.
Marincevic-Zuniga Y, Dahlberg J, Nilsson S, Raine A, Nystedt S, Lindqvist CM, et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J Hematol Oncol. 2017;10:148.
Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307.
Tran TH, Langlois S, Meloche C, Caron M, Saint-Onge P, Rouette A, et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: a report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022;6:1329–41.
Yu CH, Wu G, Chang CC, Jou ST, Lu MY, Lin KH, et al. Sequential Approach to Improve the Molecular Classification of Childhood Acute Lymphoblastic Leukemia. J Mol Diagn. 2022;24:1195–206.
Migita NA, Jotta PY, Nascimento NPD, Vasconcelos VS, Centoducatte GL, Massirer KB, et al. Classification and genetics of pediatric B-other acute lymphoblastic leukemia by targeted RNA sequencing. Blood Adv. 2023;7:2957–71.
Hu Z, Kovach AE, Yellapantula V, Ostrow D, Doan A, Ji J, et al. Transcriptome Sequencing Allows Comprehensive Genomic Characterization of Pediatric B-Acute Lymphoblastic Leukemia in an Academic Clinical Laboratory. J Mol Diagn. 2024;26:49–60.
zur Stadt U, Alawi M, Adao M, Indenbirken D, Escherich G, Horstmann MA. Characterization of novel, recurrent genomic rearrangements as sensitive MRD targets in childhood B-cell precursor ALL. Blood Cancer J. 2019;9:96.
Acknowledgements
The authors would like to thank Alona Sosinsky (Scientific Director for Cancer, Genomics England) for the personal communication. S.L.R. is a Career Development Fellow supported by Cancer Research UK (CRUK) (grant reference C60802/A27193), and J.B. is supported by Blood Cancer UK (BCUK) (grant reference 23016).
Author information
Authors and Affiliations
Contributions
JB, AVM and SLR contributed to writing of the manuscript, read, and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Ethics approval and consent to participate
This review of the published literature was performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bakewell, J., Moorman, A.V. & Ryan, S.L. DUX4-rearranged B-ALL: deciphering a biological and clinical conundrum. Leukemia 39, 2835–2847 (2025). https://doi.org/10.1038/s41375-025-02758-5
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-025-02758-5
