
Abstract
A rare subset of aggressive SMARCA4-deficient uterine sarcomas has been recently proposed, with only a limited number of cases having been previously described. Here, we identify 16 additional cases of SMARCA4-deficient uterine sarcoma from the database of a large, CLIA-certified and CAP-accredited, reference molecular laboratory, and we expand on their clinicopathological and genomic features. Median patient's age was 49 years (range 32–70). Most tumors were aggressive with distant metastasis. SMARCA4-deficient uterine sarcoma demonstrated predominantly rhabdoid or large epithelioid cells with abundant cytoplasm, but also had varying degrees of small cell and spindle cell morphology. Tumors were microsatellite stable and exhibited no other or only few co-occurring genomic alterations by comprehensive genomic profiling. We discovered one patient, who developed SMARCA4-deficient uterine sarcoma at the age of 55, had a germline SMARCA4 mutation, whose daughter had previously died of small cell carcinoma of the ovary, hypercalcemic type, at the age of 32. Our data support the notion that SMARCA4 inactivation is the driver oncogenic event of a morphologically and molecularly distinct form of uterine sarcoma. Identification of SMARCA4-deficient uterine sarcomas may be clinically important due to their aggressive behavior, germline association, and emerging targeted therapies.
Similar content being viewed by others


Introduction
The genomic landscape of uterine sarcomas is an evolving field with recent characterization of molecularly defined subgroups such as BCOR-mutated and NTRK-rearranged sarcomas [1,2,3]. Inactivation of SMARCA4 (encoding the BRG1 protein) has recently been proposed to occur in a rare subset of undifferentiated uterine sarcomas with distinctive rhabdoid morphology and aggressive behavior [4]. Because of its unique clinicopathological, morphological, and molecular features, SMARCA4-deficient uterine sarcoma may be a new uterine sarcoma type [4]. However, to our knowledge, only five cases of SMARCA4-deficient uterine sarcomas have been reported in the literature to date. Affected patients were predominantly young and rapidly died of disease (median overall survival of 7 months). Morphologically, the tumors were characterized by poorly differentiated neoplasms with a diffuse growth pattern and epithelioid/rhabdoid cells [4], resembling the ‘large cell variant’ of small cell carcinoma of the ovary, hypercalcemic type [4, 5]. In addition, similarly to small cell carcinoma of the ovary, hypercalcemic type, the few previously reported SMARCA4-deficient uterine sarcomas cases exhibited inactivating SMARCA4 mutations [4, 5]; however, whether germline SMARCA4 alterations occurs in this provisional tumor entity is unknown.
Clinically, the five previously reported SMARCA4-deficient uterine sarcoma patients presented with vaginal bleeding or with uterine/cervical masses demonstrating highly metastatic behavior [4]. The tumors were found to be genetically distinct from uterine carcinomas with secondary SMARCA4 deficiency, which are often dedifferentiated, microsatellite unstable carcinomas with many co-occurring genomic alterations [6, 7]. In contrast, SMARCA4-deficient uterine sarcomas displayed a closer molecular and morphological resemblance to the ‘large cell variant’ of small cell carcinoma of the ovary, hypercalcemic type, wherein SMARCA4 inactivation appeared the main driver genetic alteration with few other co-occurring genomic alterations [4]. Because of these distinct clinicopathological, morphological, and molecular differences, the terms ‘SMARCA4-deficient uterine sarcomas’ or ‘malignant rhabdoid tumor of the uterus’ were proposed for this distinctive entity.
SMARCA4 is an ATPase and a member of the SWI/SNF chromatin remodeling complex [8], and its inactivation is the driver oncogenic event in a variety of tumor types with rhabdoid morphology such as small cell carcinoma of the ovary, hypercalcemic type, malignant rhabdoid tumors of central nervous system and extracranial sites and the newly described SMARCA4-deficient thoracic sarcomas, among others [9,10,11]. In addition, loss of function of SMARCA4 may be secondary oncogenic events in microsatellite unstable gynecological carcinomas leading to de-differentiated carcinomas [6, 7]. In the lung, SMARCA4-deficient pulmonary adenocarcinoma is also emerging as a distinctive type of aggressive lung nonsmall cell carcinoma that is CK7-positive, TTF1-negative, and with hepatoid (Hepar1-positive) phenotype [12, 13]. However, SMARCA4 germline mutations are not thought be associated with dedifferentiated gynecological carcinoma nor with either SMARCA4-deficient sarcoma or lung adenocarcinoma.
Identification of SMARCA4-deficient uterine sarcomas may have potential therapeutic implications. Recent preclinical data have demonstrated that both SMARCA4-deficient lung nonsmall cell lung cancer and small cell carcinoma of the ovary, hypercalcemic type, tumors are exquisitely sensitive to CDK4/6 inhibition [14, 15]. Phase 1 and Phase 2 clinical trials are investigating CDK4-6 inhibitors such as palbociclib and abemaciclib for the treatment of rhabdoid tumors, although predominantly in pediatric and young adult patients (e.g., NCT03526250, NCT03709680, and NCT02644460). Preclinical models have also demonstrated that inactivation of SMARCA4 may confer sensitivity to EZH2 inhibitors [16], such as tazemetostat, which are currently in Phase 2 clinical trials for SMARCA4-deficient tumors (e.g., NCT02601950). In this study, our main objectives were: (1) to validate the existence of SMARCA4-deficient uterine sarcoma as a distinctive cancer entity, (2) to determine the frequency of SMARCA4 inactivation as the main driver genomic alteration in advanced uterine sarcomas, and (3) to further expand on the spectrum of the clinicopathological, morphological and genomic features of these tumors, including any potential germline SMARCA4 mutation association.
Methods
SMARCA4-deficient uterine sarcoma cohort
Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol no. 20152817). A retrospective database search of a CLIA- and CAP-certified reference molecular pathology laboratory was performed for clinically advanced uterine sarcomas (excluding leiomyosarcomas) and uterine adenosarcomas with genomic profiles that contained inactivating SMARCA4 alterations. Of 301 uterine sarcoma cases (excluding leiomyosarcomas), 19 contained SMARCA4 alterations. Of the 19 cases, 2 cases were microsatellite unstable, which upon re-review were more compatible with undifferentiated carcinomas and excluded from the cohort. In addition, two cases contained PTEN, PIK3R1 alterations, and one of these two cases also contained CNNTB1 and ARID1A co-alterations, raising the possibility of an endometrioid-type tumor. For these reasons, these latter 2 cases were also excluded from the study. Another independent retrospective search of 64 advanced uterine adenosarcomas yielded 1 case with sarcomatous overgrowth and SMARCA4 inactivating mutation, which upon re-review was more compatible to SMARCA4-deficient uterine sarcoma, based on morphology and genomics (i.e., it lacked leaf-like, phyllodes-like architecture or peri-glandular stromal cuffing and contained inactivating SMARCA4 mutation as the sole driver alteration). Therefore, the cohort for this study was composed of 16 uterine sarcoma cases with inactivating SMARCA4 alterations and microsatellite stability and absence of alterations in the PI3K and ARID1A pathways. The cases were previously assayed with comprehensive genomic profiling (Foundation Medicine, Cambridge, MA) during the course of clinical care at other institutions. Clinicopathological data including patient age, tumor size and FIGO stage, and immunohistochemical profiles were extracted from the accompanying pathology report. The pathologic diagnosis of undifferentiated sarcoma and estimation of large cell, spindle cell and small cell components and other morphological features were evaluated on routine hematoxylin and eosin-stained slides of tissue sections submitted for genomic profiling by two board-certified gynecologic pathologists (DIL and JE).
Genomic profiling
Comprehensive next-generation sequencing-based genomic profiling was performed on hybridization-captured, adapter ligation-based libraries using DNA and RNA extracted from formalin-fixed paraffin-embedded tumor in a CLIA- and CAP-certified laboratory (Foundation Medicine, Inc). All samples forwarded for DNA and RNA extraction contained a minimum of 20% tumor cells. The samples were assayed using adapter-ligation and hybrid capture next-generation sequencing for all coding exons from up to 406 cancer-related genes, plus select introns from up to 31 genes frequently rearranged in cancer. Patient samples were fully sequenced and evaluated for genomic alterations including base substitutions, insertions, deletions, copy number alterations (amplifications and homozygous deletions), and select gene fusions/rearrangements, as previously described [17, 18]. The bioinformatics processes used in this study included Bayesian algorithms to detect base substitutions, local assembly algorithms to detect short insertions and deletions, a comparison with process-matched normal control samples to detect gene copy number alterations and an analysis of chimeric read pairs to identify gene fusions as previously described [19]. To help visualize the sequencing data results, an oncoprint plot was generated with online tools of cbio portal as described by Gao et al. [20] and Cerami et al. [21].
Calculation of tumor mutational burden and microsatellite instability
Tumor mutational burden was determined on 0.83–1.14 Mb of sequenced DNA using a mutation burden estimation algorithm that, based on the genomic alterations detected, extrapolates to the exome or the genome as a whole as previously described [22]. In this study, low tumor mutational burden score was defined ≤6 mut/Mb, intermediate tumor mutational burden as 6–19 mut/Mb and high-tumor mutational burden as >20 mut/Mb. Assessment of microsatellite instability was performed from DNA sequencing across 114 loci as previously described [22]. Each microsatellite locus had repeat length of 7–39 bp. The next-generation sequencing-based “microsatellite instability score” was translated into categorical microsatellite instability high, microsatellite instability ambiguous, or microsatellite stable by unsupervised clustering of specimens for which microsatellite instability status was previously assessed via gold standard methods [22].
Germline mutation algorithm and genetic testing
To assess whether a SMARCA4 alteration was germline, a validated somatic‐germline‐zygosity algorithm was applied to each genomic variant as previously described [23]. For each sample, variants were detected using the standard FoundationOne analysis pipeline, which aligns unique sequence reads and obtains candidate mutations with associated mutant allele frequencies [19]. Given the output of the copy number model, each variant’s measured allele frequency is compared to expectation at its local segment to determine whether a variant is predicted somatic, germline, or ambiguous. In addition, the somatic‐germline‐zygosity method classifies the tumor zygosity of the mutation (homozygous versus heterozygous) [23]. Confirmation of germline status of the single patient predicted to have a SMARCA4 germline alteration by the somatic‐germline‐zygosity algorithm was performed at the originating institution with a medical genetics counseling team, informed patient consent and germline genetic testing.
Immunohistochemistry
For the case exhibiting germline SMARCA4 mutation, immunohistochemistry was performed on 4-μm-thick sections from formalin-fixed paraffin-embedded tumor tissue using the DAKO linker 48 automated system. The following clones, dilutions, and vendors were used for each antibody: (1) cytokeratin cocktail, AE1/3 & Cam5.2, 1:50, DAKO, (2) epithelial membrane antigen E29, 1:800, DAKO, and (3) SMARCA4/BRG1, ERP3912, 1:50, Abcam. Appropriate positive and negative controls were examined for each immunohistochemical stain.
Results
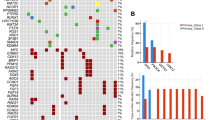
A retrospective search of 301 clinically advanced uterine sarcoma cases, excluding leiomyosarcomas, which had previously undergone comprehensive genomic profiling, from the archives of a large CLIA-certified and CAP-accredited reference molecular pathology laboratory (Foundation Medicine), yielded 19 cases (6%, 19 out of 301) with inactivating SMARCA4 alterations. Of the 19 cases, 2 cases were microsatellite unstable, and upon pathological re-review, they were more compatible with undifferentiated carcinomas and were excluded from the cohort. In addition, two cases contained PTEN, PIK3R1 alterations, and one of these latter two cases also contained CNNTB1 and ARID1A co-alterations, raising the possibility of an endometrioid-type tumor. For these reasons, these latter 2 cases were also excluded from the study. Another independent retrospective search of 64 advanced uterine adenosarcomas yielded 1 adenosarcoma case with sarcomatous overgrowth, which upon re-review was more compatible to SMARCA4-deficient uterine sarcoma, based on morphology and genomics. For instance, it lacked leaf-like (phyllodes-like) architecture or peri-glandular stromal condensation, and it contained inactivating SMARCA4 mutation as the sole driver alteration. Therefore, the SMARCA4-deficient uterine sarcoma cohort of this study was composed of 16 cases, of which 15 were previously submitted with the diagnosis of uterine sarcoma (5%, 15 of 301) plus 1 case with a submitted diagnosis of uterine adenosarcoma with sarcomatous overgrowth (2%, 1 of 64). All 16 cases contained inactivating SMARCA4 alterations, such as frameshift, nonsense, splice-site, missense, homozygous deletion (Fig. 1), low tumor mutational burden, and microsatellite stability. In this cohort of 16 cases, patients' age ranged from 32 to 70 years with a median age of 49 years (Table 1). Tumor size ranged from 4 to 17.7 cm with a median size of 6.9 cm. Most tumors were high stage and aggressive with spread of tumor beyond the uterus. Specifically, 13% of cases were stage I, 6% stage II, 31% stage III, and 50% stage IV (Table 1). In addition, most tumors exhibited lymphovascular invasion, and if a lymphadenectomy was performed, 31% of tumors-demonstrated lymph node metastasis (Table 1).

Mutational landscape of SMARCA4 across our SMARCA4-deficient uterine sarcoma cohort. a. The positions and types of alteration in SMARCA4 (NM_003072) identified in our cases. The SMARCA4/BRG1 protein domains include SNF2_N (SNF2 family N-terminal domain), HELICc (helicase superfamily C-terminal domain), Bromo (bromodomain), BRK (brahma and kismet domain), QLQ (Gln, Leu, Gln motif) and HSA (helicase/ SANT-associated domain). b. Copy number plot of single SMARCA4-deficient uterine sarcoma case demonstrating homozygous deletion of the SMARCA4 locus (arrow)
Whole slide images of H&E slides of tumor sections submitted for genomic profiling were rereviewed by two board-certified gynecological pathologists to confirm the diagnosis of undifferentiated uterine sarcoma and to assess morphological features. In this central pathology review, on low magnification, tumors exhibited a predominantly diffuse growth pattern (Fig. 2), often with identifiable necrosis, lymphovascular, and myometrial invasion (Fig. 2). In contrast to small cell carcinoma of the ovary, hypercalcemic type, follicle-like spaces were invariably absent. Morphological features are summarized in Table 2. On higher magnification, the vast majority of tumors (94%, 15 of 16) contained varying degrees of rhabdoid, large or epithelioid cells with abundant cytoplasm. However, a subset of tumors in our series also contained a small cell component with a high nuclear to cytoplasmic ratio (in 75% of cases, 12 of 16) and/or a more spindle cell morphology (in 50% of cases, 8 of 16) (Figs. 3 and 4). One tumor (6%, 1 of 16) exhibited no rhabdoid or large cell morphology (Table 1 and Fig. 3a). All tumors exhibited moderate to high grade nuclear atypia and brisk mitoses (Figs. 3 and 4). Of note, a subset of cases infiltrated the native endometrial stromal compartment, underneath superficial endometrial epithelium, adjacent to or around benign endometrial glands (Fig. 4), suggesting a cell origin from the endometrial stromal compartment and mimicking adenosarcoma. However, typical Mullerian adenosarcoma glandular and stromal morphological features, such as leaf-like (phyllodes-like) architecture or peri-glandular stromal condensation, were absent. Some tumors demonstrated a vaguely nested architectural pattern with occasional focal myxoid stroma and necrosis (Fig. 4). No malignant glands or epithelium and no areas of conventional Mullerian-type, endometroid, serous or clear cell carcinoma were identified in any case to suggest dedifferentiated carcinoma or carcinosarcoma.

SMARCA4-deficient uterine sarcoma with diffuse growth pattern involving endomyometrium, invading into deeper myometrium (a) and with peripheral lymphatic vascular invasion within outer uterine wall (b), corresponding to case #11. Higher magnification of large, epithelioid (c) and small cell (d) components

SMARCA4-deficient uterine sarcoma with predominant diffuse, solid growth pattern and different cell morphologies. Case #1 with spindle cell (a) and round nuclear (b) morphology and no associated rhabdoid morphology. Case #2 with large (c) and small cell (d) morphology. Case #3 (e) and case #12 (f) with rhabdoid, discohesive morphology

Morphologic spectrum of SMARCA4-deficient uterine sarcoma. Case #6 with both large (a) and small cell (b) components. (c) Case #7 with both spindle and large cell components. Case #4 with small and large cell component infiltrating endometrial stromal compartment adjacent to and around benign endometrial glands (d), small cell and vaguely nested component (e) and myxoid stromal change (f)
Comprehensive genomic profiling, via targeted DNA and RNA-based next-generation sequencing of up to 406 genes involved in tumorigenesis, revealed SMARCA4 mutations in the form of SMARCA4 substitutions and deletions resulting in predominantly frameshift and nonsense functional effects, rare splice site alterations and homozygous gene deletion, which were predicted to inactivate SMARCA4 function (Fig. 5 and Table 3). Inactivating mutations of SMARCA4 occurred throughout the gene, consistent with loss of function mutations of a tumor suppressor gene (Table 3 and Figs. 1 and 5). In addition, tumors had a low mutation burden (mean 1.7 mut/Mb) and were microsatellite stable (Table 3). In 13 of 16 cases (81%), inactivating SMARCA4 alterations was the only pathogenic/oncogenic driver mutation with no additional oncogenic alterations. In contrast, a minority of cases (3 of 16, 19%,) exhibited pathogenic alterations in genes other than SMARCA4, such as TP53 (n = 2), RB1 (n = 1), and CTNNB1 (n = 1) (Fig. 5 and Table 3). Variants of unknown significance were excluded from the analysis. RNA-based targeted next-generation sequencing revealed no gene fusions or rearrangements of any gene tested, including for JAZF1, PHF1, BCOR, or YWHAE-FAM22 that are frequently found in uterine sarcoma, suggesting that inactivation of SMARCA4 was the major oncogenic driver event in these tumors.

Oncoprint of the SMARCA4-deficient uterine sarcoma cohort demonstrating genomic profiles with inactivating oncogenic SMARCA4 mutations and no additional or only few other co-occurring alterations. Blue rectangle: homozygous gene deletion; black square: oncogenic truncating mutation; green square: oncogenic missense mutation, red rectangle: gene amplification. Variants of unknown significance were excluded
Using a validated computational approach to distinguish somatic versus germline origin of genomic alterations from deep sequencing of cancer specimens [23], SMARCA4 mutation was predicted to be germline in 1 of 16 (6%), ambiguous/indeterminate for germline in 8 of 16 (50%), somatic in 4 of 16 (25%), and unknown in 3 of 16 (19%) patients. Importantly, the patient predicted to have a germline SMARCA4 mutation, corresponding to case #14, underwent formal germline genetic testing via medical genetics counseling at the original institution. Upon formal germline testing, she was confirmed to have a germline SMARCA4 deleterious oncogenic nonsense mutation, 1831C>T (SMARCA4 transcript NM_003072), Q611*, which is predicted to truncate SMARCA4/BRG1. By immunohistochemistry, this patient’s tumor exhibited no expression of pancytokeratin or epithelial membrane antigen, consistent with the diagnosis of a sarcoma (Fig. 6). In addition, immunohistochemistry revealed that tumor cells lost expression of BRG1 (protein encoded by the SMARCA4 gene) with appropriate intact expression in benign endometrial glands and benign stromal cells (Fig. 6). Loss of BRG1 expression by immunohistochemistry suggests that the second truncating SMARCA4 mutation in this patient, K1602fs*8, occurred in trans resulting in inactivation of both SMARCA4 alleles. Of note, this patient was diagnosed with uterine sarcoma at the age of 55, and her family history was significant for a daughter who was previously diagnosed and died of small cell carcinoma of the ovary, hypercalcemic type, at the age of 32. Before her daughter died of small cell carcinoma of the ovary, the daughter had also been previously identified as carrying the same deleterious SMARCA4 mutation as her mother. However, her mother (SMARCA4-deficient uterine sarcoma patient) had not been previously tested. Follow-up and confirmatory germline genetic testing results are not available for the remaining patients who were predicted to have either somatic or ambiguous SMARCA4 mutation via the somatic‐germline‐zygosity computational algorithm in this study.

Case #14 with germline SMARCA4 mutation. a. Next-generation sequencing identified a 1831C>T (NM_003072), Q611*, nonsense tumor mutation that was further confirmed to be germline by genetic testing. b. H&E of patient’s tumor infiltrating endometrial stromal compartment underneath benign endometrial epithelium. Immunohistochemistry of the patient’s tumor revealed (c) no expression of cytokeratin cocktail AE1/3 Cam5.2, and (d) loss of BRG1 (protein encoded by SMARCA4) in tumor cells with appropriate labeling of benign endometrial epithelium and benign stromal cells (with BRG1 only) as internal control
Discussion
In this study, we demonstrate that inactivation of SMARCA4 is the main oncogenic driver event of a subset of aggressive undifferentiated uterine sarcomas predominantly with rhabdoid, but also less frequently with small cell and spindle cell morphology. These tumors contained no other or only few co-occurring genomic alterations as well as low tumor mutational burden and microsatellite stability. This work validates the proposal of SMARCA4-deficient uterine sarcomas (malignant rhabdoid tumor of the uterus) as a new distinct tumor entity [4] and further expands on the clinicopathological, morphological, and genomic features of these tumors, including identification a patient with germline, oncogenic SMARCA4 mutation.
Our cohort of 16 cases had several similarities to the findings of the five original cases described by Kolin et al.: (1) they were aggressive, (2) with lymphovascular invasion, lymph node metastasis and distant spread, (3) most contained rhabdoid/large cell morphology, (4) infiltrated benign endometrial glands, and (5) and contained few co-occurring genomic alterations. Morphologically, similar to Kolin et al., most tumors in our cohort were characterized by diffuse growth pattern with rhabdoid or large cell, epithelioid morphology with high-grade nuclear atypia in the form of vesicular chromatin and prominent nucleoli. However, in contrast to the few previously published cases, we have also identified cases with small cell and spindle cell morphology, necrosis, vaguely nested pattern, and focal myxoid changes.
In contrast to the prior proposed SMARCA4-deficient uterine sarcoma patients, in which these sarcomas occurred in younger women (median of 33 and mean 36 years), our cohort of patients in our larger series were older (median 49 years). The age distribution of our cohort was more comparable to the newly described SMARCA4-deficient thoracic sarcomas, which also occurred in older patients (median 48 years) [9, 11]. Our findings indicate SMARCA4-deficient uterine sarcoma should be considered in the differential diagnosis of tumors with rhabdoid/large, small and spindle cell morphologic features in patients of all ages, as in our study, this entity was not limited to young patients.
The median survival in the original study was 7 months. In our retrospective study, we did not have long term follow-up data to calculate survival. However, most tumors were high stage with lymphovascular invasion and extra uterine spread and metastasis, supporting the notion that SMARCA4-deficient uterine sarcomas exhibit aggressive behavior. Although we did not have enough tissue to confirm immunohistochemical findings of referring institutions, the tumors reportedly showed a nonspecific immunophenotype with all cases being essentially negative for cytokeratins and muscle markers and few cases showing focal staining for CD10 and focal weak staining for epithelial membrane antigen (Table 4).
Using a validated computational algorithm to distinguish somatic versus germline origin of genomic alterations from deep sequencing of cancer specimens, e.g., the somatic-germline-zygosity mutation algorithm [23], 1 of 16 (6%) patients (case #14, age 55), in our cohort was predicted to have a germline SMARCA4 mutation, which was confirmed via formal germline genetic testing and medical genetics counseling. This patient’s age of 55 is older than the typical age of 20–30s of patients with germline SMARCA4 mutations who develop small cell carcinoma of the ovary, hypercalcemic type [5]. In our cohort, SMARCA4-deficient uterine sarcomas may be associated with a germline SMARCA4 alteration in at least 6% of cases. However, this rate may be an underestimation since confirmatory direct germline genetic testing results are not available for the remaining patients that were called ambiguous with the somatic-germline-zygosity germline prediction algorithm.
Heterozygous germline mutations in SMARCA4 underlie rhabdoid tumor predisposition syndrome 2 (RTPS2; OMIM 613325) [24,25,26]. Patients with this syndrome may present with rhabdoid tumors in the central nervous system, or malignant rhabdoid tumors in extracranial sites such as small cell carcinoma, hypercalcemic type, in the ovary. Our study implicates SMARCA4 germline mutations in a subset of SMARCA4-deficient uterine sarcomas and potentially adds them to tumors that are predisposed in RTPS2. However, our findings and germline mutation rate frequency need to be validated with additional patients via direct prospective medical genetics counseling and germline testing in patients with newly diagnosed SMARCA4-deficient uterine sarcoma in order to determine the frequency of germline SMARCA4 alterations in these tumors.
Other tumors to be considered in the differential diagnosis of SMARCA4-deficient uterine sarcomas include undifferentiated/dedifferentiated carcinoma, small cell carcinoma of the ovary hypercalcemic type, high grade endometrial stromal sarcoma, adenosarcoma, among others. Morphology, tumor burden, and judicial use of immunohistochemistry (i.e., WT1, cytokeratins, epithelial membrane antigen, claudin-4, CD10, BCOR, cyclin D1, MMR, SMARCA4/BRG1) [27,28,29,30] and cytogenetics or molecular analysis (i.e., BCOR, YWHAE, JAZF1, PHF1) may be helpful in identifying SMARCA4-deficient uterine sarcoma. Negative cytokeratin, WT1 and epithelial membrane antigen immunostaining, location of tumor bulk and absence of a well to moderately differentiated, microsatellite unstable endometrioid adenocarcinoma, and the absence of PTEN, PI3K and ARID1A alterations in cases involving the uterus may be more compatible with SMARCA4-deficient uterine sarcoma.
While we did not have enough remaining tissue material to perform immunohistochemical studies in 15 of 16 cases, we extracted immunohistochemistry results that were performed at outside institutions, if they were available, from the submitting pathology reports. All tested tumors were reportedly negative for a wide spectrum of cytokeratins, and were either negative or focally and weakly positive for epithelial membrane antigen (Table 4). In contrast, majority of tumors were at least focally positive for CD10, which is traditionally viewed as a marker of endometrial stromal differentiation. They were also negative for muscle markers, desmin, smooth muscle actin, caldesmon, MyoD, as well as for CKIT, S100, and predominantly negative for Estrogen and Progesterone receptors (Table 4). These results support the original submitting diagnoses and our central pathology re-review diagnoses of uterine sarcoma. Reported expression of CD10 and infiltration of native endometrial stromal compartment suggests an endometrial stromal cell of origin for these tumors. Lack of leaf-like (phyllodes-like) architecture and peri-glandular stromal condensation argues against a diagnosis of Mullerian adenosarcoma.
Importantly, identification of SMARCA4-deficient uterine sarcoma may have therapy implications. Both in vitro and in vivo preclinical models have been demonstrated that selective EZH2 inhibitors, such as tazemetostat (currently in phase II clinical trials) [31], induce antiproliferative and antitumor effects in SMARCA4-deficient and rhabdoid tumor cell lines and mouse xenografts [16, 32]. Clinically, two patients with small cell carcinoma of the ovary, hypercalcemic type, who were treated with tazemetostat, have experienced preliminary clinical benefit (one partial response and one stable disease) [33, 34]. Given morphological and molecular similarities to small cell carcinoma of the ovary, hypercalcemic type, there may be similar potential benefit of EZH2 inhibitors for patients with SMARCA4-deficient uterine sarcoma. In addition, despite a low tumor mutational burden, immunotherapy has recently been shown to be effective in patients with small cell carcinoma of the ovary, hypercalcemic type [35], suggesting a potential similar benefit for SMARCA4-deficient uterine sarcoma patients.
More recently, preclinical models have demonstrated that patient-derived xenografts and cell lines from SMARCA4-deficient nonsmall cell lung carcinoma and small cell carcinoma of the ovary, hypercalcemic type, are highly sensitive to CDK4/6 inhibition through a synthetic lethal mechanism of reduced downstream cyclin D1 expression [14, 15]. Notably, drug sensitivity was detected in SMARCA4-deficient lung and ovarian tumors, suggesting that SMARCA4-deficient tumors are likely to be sensitive to CDK4/6 inhibition regardless of tissue of origin [14, 15]. The CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib are currently approved by the FDA for the treatment of hormone receptor-positive and Her2-negative breast cancer [36, 37], and Phase 1 and Phase 2 clinical trials of palbociclib and abemaciclib for the treatment of pediatric rhabdoid tumors are ongoing. Given the molecular and morphological similarities between small cell carcinoma of the ovary, hypercalcemic type, and SMARCA4-deficient uterine sarcoma, we hypothesize that CDK4/6 inhibitors may also suppress the growth of SMARCA4-deficient uterine sarcoma cells. However, additional clinical data are needed to validate the potential of emerging targeted therapies, such as Palbociclib and tazemetostat, for SMARCA4-defficient tumors.
In conclusion, here we identify and report the clinicopathological, morphological, and molecular features of the largest group to date of SMARCA4-deficient uterine sarcoma. Given the overall distinct morphological features and unique molecular profile of the tumors, with SMARCA4 mutation as the major oncogenic driver, we believe that our cohort validates the existence of a SMARCA4-deficient uterine sarcoma and further expands on the characteristics of these tumors. Our data demonstrate the usefulness of comprehensive genomic profiling in identifying this new group of uterine sarcomas, as correct identification of these group of tumors may have important prognostic, germline association and potential therapeutic implications.
References
Juckett LT, Lin DI, Madison R, Ross JS, Schrock AB, Ali S. A pan-cancer landscape analysis reveals a subset of endometrial stromal and pediatric tumors defined by internal tandem duplications of BCOR. Oncology. 2018;96:1–9.
Chiang S, Cotzia P, Hyman DM, Drilon A, Tap WD, Zhang L, et al. NTRK fusions define a novel uterine sarcoma subtype with features of fibrosarcoma. Am J Surg Pathol. 2018;42:791–8.
Ferreira J, Félix A, Lennerz JK, Oliva E. Recent advances in the histological and molecular classification of endometrial stromal neoplasms. Virchows Arch. 2018;473:665–78.
Kolin DL, Dong F, Baltay M, Lindeman N, MacConaill L, Nucci MR, et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod Pathol. 2018;31:1442–56.
Lin DI, Chudnovsky Y, Duggan B, Zajchowski D, Greenbowe J, Ross JS, et al. Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol Oncol. 2017;147:626–33.
Karnezis AN, Wang Y, Ramos P, Hendricks WP, Oliva E, D'Angelo E, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcemic type. J Pathol. 2016;238:389–400.
Köbel M, Hoang LN, Tessier-Cloutier B, Meng B, Soslow RA, Stewart CJR, et al. Undifferentiated endometrial carcinomas show frequent loss of core switch/sucrose nonfermentable complex proteins. Am J Surg Pathol. 2018;42:76–83.
Savas S, Skardasi G. The SWI/SNF complex subunit genes: their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol. 2018;123:114–31.
Le Loarer F, Watson S, Pierron G, de Montpreville VT, Ballet S, Firmin N, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet. 2015;47:1200–5.
Sauter JL, Graham RP, Larsen BT, Jenkins SM, Roden AC, Boland JM. SMARCA4-deficient thoracic sarcoma: a distinctive clinicopathological entity with undifferentiated rhabdoid morphology and aggressive behavior. Mod Pathol. 2017;30:1422–32.
Perret R, Chalabreysse L, Watson S, Serre I, Garcia S, Forest F, et al. SMARCA4-deficient thoracic sarcomas. Am J Surg Pathol. 2019;43:455–65.
Agaimy A, Fuchs F, Moskalev EA, Sirbu H, Hartmann A, Haller F. SMARCA4-deficient pulmonary adenocarcinoma: clinicopathological, immunohistochemical, and molecular characteristics of a novel aggressive neoplasm with a consistent TTF1neg/CK7pos/HepPar-1pos immunophenotype. Virchows Arch. 2017;471:599–609.
Haninger DM, Kloecker GH, Bousamra M II, Nowacki MR, Slone SP. Hepatoid adenocarcinoma of the lung: report of five cases and review of the literature. Mod Pathol. 2014;27:535–42.
Xue Y, Meehan B, Macdonald E, Venneti S, Wang XQD, Witkowski L, et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat Commun. 2019;10:558.
Xue Y, Meehan B, Fu Z, Wang XQD, Fiset PO, Rieker R, et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat Commun. 2019;10:557.
Chan-Penebre E, Armstrong K, Drew A, Grassian AR, Feldman I, Knutson SK, et al. Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: In vitro and in vivo preclinical models. Mol Cancer Ther. 2017;16:850–60.
Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4.
He J, Abdel-Wahab O, Nahas MK, Wang K, Rampal RK, Intlekofer AM, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–14.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput Biol. 2018;14:e1005965.
Schneppenheim R, Frühwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–84.
Witkowski L, Lalonde E, Zhang J, Albrecht S, Hamel N, Cavallone L, et al. Familial rhabdoid tumour’avant la lettre’–from pathology review to exome sequencing and back again. J Pathol. 2013;231:35–43.
Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35:933–5.
McCluggage WG, Oliva E, Connolly LE, McBride HA, Young RH. An immunohistochemical analysis of ovarian small cell carcinoma of hypercalcemic type. Int J Gynecol Pathol. 2004;23:330–6.
Schaefer I-M, Agaimy A, Fletcher CD, Hornick JL. Claudin-4 expression distinguishes SWI/SNF complex-deficient undifferentiated carcinomas from sarcomas. Mod Pathol. 2017;30:539–48.
Tessier-Cloutier B, Soslow RA, Stewart CJR, Köbel M, Lee CH. Frequent loss of claudin-4 expression in dedifferentiated and undifferentiated endometrial carcinomas. Histopathology. 2018;73:299–305.
Hoang LN, Lee Y-S, Karnezis AN, Tessier-Cloutier B, Almandani N, Coatham M, et al. Immunophenotypic features of dedifferentiated endometrial carcinoma—insights from BRG1/INI1-deficient tumours. Histopathology. 2016;69:560–9.
Italiano A, Soria J-C, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–59.
Kurmasheva RT, Sammons M, Favours E, Wu J, Kurmashev D, Cosmopoulos K, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pedia Blood Cancer. 2017;64:e26218.
Knutson SK, Drew AE, Plescia C, Copeland RA, Smith JJ, Heike K, et al. Abstract C87: EZH2 inhibition leads to decreased proliferation in SMARCA4-deleted ovarian cancer cell lines. Mol Cancer Ther. 2015;14:C87–C87.
Italiano A, Keilhack H, Toulmonde M, Coindre JMMJ, Massard C, Ottesen L, et al. 302 A phase 1 study of EPZ-6438 (E7438), an Enhancer of Zeste-Homolog 2 (EZH2) inhibitor: preliminary activity in INI1-negative tumors. Eur J Cancer. 2015;51:S54.
Jelinic P, Ricca J, Van Oudenhove E, Olvera N, Merghoub T, Levine DA, et al. Immune-active microenvironment in small cell carcinoma of the ovary, hypercalcemic type: rationale for immune checkpoint blockade. J Natl Cancer Inst. 2018;110:787–90.
Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209–19.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379:1926–36.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All study authors, except for JLH, are full-time employees of Foundation Medicine, Inc., which is a wholly owned subsidiary of Roche.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lin, D.I., Allen, J.M., Hecht, J.L. et al. SMARCA4 inactivation defines a subset of undifferentiated uterine sarcomas with rhabdoid and small cell features and germline mutation association. Mod Pathol 32, 1675–1687 (2019). https://doi.org/10.1038/s41379-019-0303-z
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41379-019-0303-z
This article is cited by
-
Pathological characteristics and immune microenvironment of SMARCA4-deficient undifferentiated uterine sarcoma
Diagnostic Pathology (2023)
-
SMARCA4-associated schwannomatosis
Acta Neuropathologica (2023)
-
Primary cutaneous SMARCA4-deficient undifferentiated malignant neoplasm: first two cases with clinicopathologic and molecular comparison to eight visceral counterparts
Modern Pathology (2022)
-
SMARCA4-deficient rectal carcinoma with a sarcomatoid component: a case report
Clinical Journal of Gastroenterology (2022)
-
The first case of SMARCA4-deficient sarcoma of stomach
Clinical Journal of Gastroenterology (2022)
