
Abstract
Spasmolytic polypeptide-expressing metaplasia (SPEM) and pyloric gland adenoma (PGA) in the stomach are metaplastic and neoplastic lesions, respectively, in which gastric body glands are replaced by pyloric glands. The aim of this study was to evaluate the genomic profile of SPEM and compare it with intestinal-type gastric cancer (GC) and PGA. Thirteen gastrectomies showing PGA with or without dysplasia, GC and SPEM were retrospectively selected. MUC5AC, MUC6, gastrin, and TFF2 IHC were performed. Lesions were subjected to laser capture microdissection followed by DNA extraction. Forty-three DNA samples were extracted from PGA without cytological dysplasia, PGA with low-grade and high-grade dysplasia and pyloric gland adenocarcinoma, GC, SPEM, and adjacent normal tissue from the body of the stomach and were subjected to exome sequencing for 49 genes that are commonly dysregulated in GC. Sanger sequencing was performed for confirmation. Twenty nonsynonymous mutations were identified in SPEM, and none of these were frameshifts or indels. PGA with or without cytological dysplasia showed a significantly higher number of mutations compared with SPEM. As cytological dysplasia increased from no dysplasia to dysplasia in PGA, the percentage of frameshift mutations, indels, and missense variations increased. Further missense or frameshift mutations were observed in the KRAS, APC, TP53, and CTNNB1 genes in the PGA group. In GC, mutations were observed in the TP53 gene (p.Arg248Gln). Missense mutations in the MUC5AC, KRAS, BRAF, and EZH2 genes were common between SPEM and GC. SPEM showed fewer genomic variations than GC and PGA, and was genomically distinct from the pyloric epithelium in PGA. Stepwise progression of PGA from PGA without dysplasia to PGA with dysplasia/adenocarcinoma was associated an increase in mutations. SPEM appears to be more genomically similar to GC than PGA.
Similar content being viewed by others


Introduction
Spasmolytic polypeptide-expressing metaplasia (SPEM), also known as pseudopyloric metaplasia, is a metaplastic lesion observed in the body of the stomach, wherein the normal native glands (specialized gastric glands) are replaced by gastric pyloric glands [1]. Wang (in a mouse model) and later Schimdt (in humans) observed SPEM adjacent to intestinal-type gastric cancer (GC) in humans [2]. While SPEM has been shown to progress to intestinal metaplasia (IM) and GC in mouse models, its role in human gastric carcinogenesis is still unclear. Extensive research performed on mouse models and Mongolian gerbils has shown that due to chronic Helicobacter pylori infection, oxyntic gland atrophy occurs, causing decreased acid secretion followed by the simultaneous loss/differentiation/reprogramming of mature chief cells into metaplastic mucous-secreting cells, which are not native to the deeper regions of the corpus of the stomach [3,4,5]. Autoimmune gastritis (AIG) is one of the causes of chronic gastritis, in which autoimmune antibodies specifically target parietal cells in the stomach, leading to their loss or atrophy [6,7,8]. AIG leads to a spectrum of morphological changes in the gastric mucosa, including diffuse chronic lymphoplasmacytic infiltration, oxyntic atrophy, pseudopyloric metaplasia, neuroendocrine hyperplasia and tumors, pyloric gland adenoma (PGA), and gastric adenocarcinoma [6,7,8,9,10]. PGA, similar to SPEM, is a recently recognized entity that can occur in the entire gastrointestinal tract (predominantly in the stomach, gall bladder, and duodenum), wherein the gastric pyloric glands replace the normal native glands, subsequently proliferate as a neoplasm and can even undergo malignant transformation. PGAs were first described by Elster in 1976 as not just any benign or hyperplastic growth but as neoplasms showing malignant potential [11]. In the stomach, PGAs are most commonly observed in the corpus.
Based on our experience, PGA and SPEM share overlapping etiologies, as both of these lesions are observed exclusively in the gastric corpus and are associated with AIG. Histologically, these lesions are characterized by the proliferation of pyloric-type glands in PGA and focal pseudopyloric metaplastic glands in SPEM. Therefore, based on the morphological similarity between SPEM and PGA, it was hypothesized that SPEM could also be related to PGA at the genomic level. Moreover, in the model of intestinal-type gastric carcinogenesis, it is speculated that SPEM could be one of the precursor lesions to IM and, hence, to GC. Therefore, it is possible that SPEM could show some genomic similarity to intestinal-type GC as well. Although high GNAS and KRAS mutation rates have been reported previously in PGA [12,13,14], little is known about the genomic alterations in human SPEM. Therefore, the aims of this study were first to understand the genomic spectrum of SPEM using formalin-fixed paraffin-embedded (FFPE) samples, and second to explore and evaluate the genomic similarities between SPEM, IM, and intestinal-type GC and between SPEM and PGA, including pyloric-type adenocarcinoma.
Materials and methods
Sample selection
A retrospective analysis of 13 patients who had undergone gastrectomy at the Department of Surgery, NUHS, Singapore (2010–2016) was performed. These surgical resections were performed in patients with PGA and/or GC. Among the 13 gastrectomies, 8 received a pathological diagnosis of PGA with or without cytological dysplasia, and 5 presented with intestinal-type GC. Our aim was to identify PGA cases located only in the body of the stomach (and not in other sites such as the gall bladder and duodenum). Furthermore, the selected PGAs showed a transition from areas of no cytological dysplasia to low-grade and/or high-grade dysplasias. Within the GC group, we aimed to select GC cases only from the body of the stomach. We selected the case for this study only when we could concurrently observe SPEM, IM, and intestinal-type GC. All tissue specimens were fixed in 10% formalin, embedded in paraffin blocks as FFPE blocks, and routinely processed. Serial sections of 4–6 microns were obtained and subjected to hematoxylin and eosin (H&E) and immunohistochemistry (IHC) for diagnosis (Supplementary Methods).
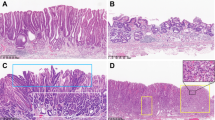
From these 13 cases, 43 representative areas from different lesions were selected. From each case of GC, representative areas of SPEM, IM, GC, and normal tissue from the body of the stomach (as controls) (n = 20 areas) were selected, microdissected, and DNA was extracted (n = 20 DNA samples). Similarly, from the eight PGA eases, foci of PGA without cytological dysplasia, PGA with LGD, PGA with HGD and/or pyloric gland adenocarcinoma, and matched normal tissue from the body of the stomach (as controls) (n = 23 areas) were selected, microdissected, and DNA was extracted (n = 23 DNA samples) (Supplementary Table 1) (Fig. 1). The study was approved by the National University Hospital Singapore, the Domain-Specific Review Board Committee, and the Department of the Pathology Research Committee.

Representative H&E of PGN, PGA with LGD, PGA with HGD, pyloric gland adenocarcinoma, SPEM (black circle), IM (red circle), and intestinal-type GC.
Laser capture microdissection (LCM) and DNA sequencing (targeted exome sequencing)
Six-micron sections were obtained on special membrane slides (Thermo Fisher Scientific, USA) for LCM. Different lesions of interest were identified using the reference H&E slide and microdissected by using the Arcturus XTM microdissection system (Thermo Fisher Scientific, USA). The microdissected cells of interest were collected in the CapSure® LCM caps, which were placed over the target area. Genomic DNA (gDNA) was extracted using the QIAmp DNA microkit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Selection of the gene panel was performed after a comprehensive review of the literature, identifying candidate genes that were frequently found to be dysregulated in intestinal-type gastric adenocarcinoma and PGA. The selection of the gene panel was also partially based on our recent publication of genomic changes in gastric IM [15]. Forty-nine genes were selected, which comprised tumor suppressor genes, oncogenes, and other key tumorigenic functions in GC and PGA. The coding exomes of the 49 genes (Supplementary Table 2) were sequenced across 43 samples in this study.
Quality control of the gDNA samples was performed by the Qubit and gel electrophoresis method. A minimum of 40 ng DNA was required for exome sequencing, which was performed using the QIAseq DNA panel kit according to the manufacturer’s protocol. NGS was performed by using an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA, USA). The raw sequence data were aligned to the human reference genome (hs37d5), and the smCounter algorithm was applied to detect somatic single nucleotide variations (SNVs) and insertions and deletions [16]. Forward and reverse sequence reads were checked to ensure that there were no false reads. For variant annotation, the SnpEff (SNP effect) tool (a variant annotation and effect prediction tool) was used [17], which categorized the variants into low, moderate, and high annotation impacts. Only high- and moderate-impact mutations were considered for evaluation between the different groups.
In addition to the default output from the smCounter program, several filters were applied. For Sanger sequencing, only variants with reads showing an at least 10% variant allele frequency (VAF), and five variant-supporting reads were included for further confirmatory analysis to reduce false positive predictions. To identify likely pathogenic variants, the filtered variants were compared with reported somatic cancer mutations in the COSMIC database and the single nucleotide polymorphism database (dbSNP).
Sanger sequencing
Validation of the results obtained by targeted exome sequencing was carried out by Sanger sequencing whenever DNA was available (n = 7 DNA samples). Fifty nanograms of extracted genomic DNA was PCR amplified using Taq master mix (Promega, Wisconsin, USA). Forward and reverse primer sequences are provided in Supplementary Table 3. The optimized PCR conditions were as follows: 98 °C for 2 min, followed by 40 cycles of 98 °C for 30 s, 60 °C for 30 s, 72 °C for 60 s, and a final extension at 72 °C for 5 min. Sequencing was performed using an ABI BigDye® Terminator v3.1 Cycle Sequencing Kit according to the manufacturer’s protocol. The samples were then cleaned up by using the magnetic beads method and analyzed in a 3730 DNA Analyzer (3730s, Life technologies).
Statistical analysis
IBM SPSS statistical package version 23 was used in this study to analyze and compare the genomic profile across the different groups of patients. Nonparametric tests (Mann–Whitney and Kruskal–Wallis tests) were used to identify the differences in SNVs in the different groups. A p value < 0.05 was considered significant.
Results
A total of 13 patients were included in the study, among whom 7 were males, and 6 were females. The first group comprised patients with PGAs with matched normal body tissues (total samples n = 23 from eight patients), and the second group consisted of five patients with SPEM, IM, intestinal-type GC, and matched normal body tissues (total samples n = 20 from five patients).
Diagnosis of PGA
All the cases were subjected to H&E staining to assess their respective histological diagnoses. All eight PGAs were located in the gastric body and were labeled PG1-PG9 (PG4 presented suboptimal DNA and hence was removed from the study). Each PGA could show the features of PGA without cytological dysplasia with or without LGD, HGD or pyloric gland adenocarcinoma within the same polyp. Histologically, PGA appeared as a lesion with tightly packed pyloric glands lined by cuboidal or columnar epithelium [18]. The cytoplasm was abundant and appeared eosinophilic, with round to oval bland nuclei. Dysplasia was categorized as low grade or high grade according to a previously reported system [19]. The presence of LGD was characterized by irregularly shaped glands, elongated and hyperchromatic nuclei, eosinophilic cytoplasm, and some stratification. PGA with HGD appeared as a lesion with irregular and occasional cribriform glands, vesicular nuclei with prominent nucleoli and few mitoses. The cytoplasm was eosinophilic in quality. Pyloric gland adenocarcinoma was diagnosed when there were invasive irregular complex glands with a high nucleus/cytoplasmic ratio, vesicular nuclei, irregular nuclear membrane, prominent nucleoli, numerous mitoses, and the loss of polarity of cells. All the pyloric gland samples showed superficial cytoplasmic expression of MUC5AC and strong cytoplasmic expression of MUC6 (Fig. 2). In addition, adjacent normal areas from the body of the stomach comprising specialized glands were obtained as the corresponding control in all eight cases of PGA.

Positive expression of MUC6 and MUC5AC in PGN; negative expression of gastrin and positive expression of TFF2 in SPEM.
Diagnosis of SPEM, IM, and GC
All five intestinal-type gastric adenocarcinomas were labeled GC1–GC5, and the location of the carcinoma in each case was the gastric corpus. Each GC case additionally showed features of IM (IM1–IM5) as well as SPEM (SP1–SP5) in the adjacent gastric mucosae. SPEM morphologically resembled the mucous/pyloric glands in the deeper part of the body of the stomach. IM was characterized by metaplasia of the native stomach glands with intestinal-type mucosa replete with a brush border and goblet cells. GC was categorized as intestinal-type GC according to the WHO classification of gastric tumors [20] and was characterized by complex, irregular glandular proliferation invading the submucosa, cells with a high N/C ratio, and vesicular and pleomorphic nuclei with prominent nucleoli and multiple mitoses lining the glands. All five GC cases were evaluated for TFF2 and gastrin by IHC to confirm the presence of SPEM. SPEM was diagnosed by negative gastrin expression and positive TFF2 expression. In addition, adjacent normal body mucosa tissue comprising specialized glands was obtained as a control in each of five GC cases. Upon confirmation, the area of interest was microdissected by LCM, and the genomic DNA was extracted and subjected to exome sequencing (Fig. 2).
Histological comparison between PGA and SPEM
While PGA shows the proliferation of pyloric-type glands, SPEM shows pseudopyloric metaplastic glands. According to the IHC results, both lesions were negative for gastrin and positive for MUC6 and/or TFF2. The two lesion types can be distinguished by the fact that PGA occurs as a polypoid mass secondary to the proliferation of such glands, while SPEM occurs as a focal lesion that is not detected grossly.
Genomic alterations in the study cohort
A total of 129 (SPEM), 9596 (IM), 6030 (GC), 15,576 (PGN), 11,248 (PGL), and 6,012 (PGH/PGCa) synonymous, and nonsynonymous point mutations were observed in the different lesions (Table 1). All the synonymous point mutations and those showing low annotation were filtered from the total number of point mutations observed in the DNA samples. PGN, PGL, and PGH/PGCa showed a significantly greater number of nonsynonymous mutations than SPEM (Mann–Whitney test, U = 0.000, p < 0.05).
Genomic alteration in SPEM
Five areas of GC showing features of SPEM (positive histology, gastrin negativity, and TFF2 positivity) were subjected to targeted exome sequencing and were labeled SP1–SP5. Twenty distinct point mutations (nonsynonymous and high impact) were identified in SPEM, none of which were frameshifts or indels (Table 2). Some of the mutations were observed in more than one sample. In sample SP2, KRAS showed a point mutation in the splice-donor region that has not been reported previously (chr12: 25380167, C>T). An exhaustive search in the COSMIC and dbSNP databases showed only two known point mutations in the MUC5AC and MYC genes (missense variants) in SPEM samples. While the mutation in MUC5AC has not been reported to be associated with any pathology, the mutation in the MYC gene is associated with carcinoma of the esophagus and other solid organ carcinomas [21]. To confirm this finding, IHC for c-myc was performed in the corresponding sample, and nuclear expression in the pseudopyloric glands was observed in the tissue section (Fig. 3). In addition, missense point mutations were observed in several other genes, as mentioned in Table 2. Approximately 55.6% of the SNPs in the SPEM samples showed C>T transitions and G>A transversions. These transitions and transversions are commonly observed in GC [22].

IHC for c-myc in SP2 sample showing no expression of c-myc in SPEM glands and a positive nuclear expression in metaplastic glands in SP4 sample.
Germline mutations in SPEM
In addition to the genomic mutations mentioned above, four germline mutations were observed in the SPEM DNA samples. These were also observed in the matched normal body tissue DNA samples. Three of these mutations were observed in the ALK gene, at chr2: 29416481 (c.4472A>G, p.Lys1491Arg), chr2: 29416366 (c.458C>G, p.Asp1529Glu), and chr2: 29416572 (c.4381T>C, pIle1461Val). One germline mutation was observed in APC at chr5: 112176756 (c.5465T>A, p.Val1822.Gly). These variants were confirmed by Sanger sequencing in the SP4 DNA sample (Fig. 4a).

Confirmatory Sanger sequencing in SPEM and PGA samples. a PCR gel image for KRAS, APC, and ALK for sample SP4 followed by sequencing of APC Chr5:112176756 (C>T) and ALK Chr2: 29416572 (T>C); b PCR gel image for KRAS for PG2L, PG2H, PG6A, and PG6L. Left image showing the sequencing for PG2H sample for point mutation located at chr12: 25398284 (C>T).
Genomic alterations in PGA
In the DNA samples of PGN, PGL, and PGH/PGCa, approximately 6012, 4892, and 2630 mutations were observed, respectively. Furthermore, as cytological dysplasia increased (i.e., from PGN to PGL to PGH/PGCa), the percentage of frameshift mutations, indels, and missense variations increased gradually (Table 3). Since thousands of point mutations were observed in the three groups, variants with at least a 10% VAF and 5′ supporting reads were filtered. After filtration, mutations were observed in the KRAS, APC, TP53, and CTNNB1 genes, which were missense, frameshift, or protein–protein types of variations. The COSMIC and dbSNP databases revealed the corresponding mutations, all of which have been reported to be pathogenic and have been implicated in adenocarcinomas of solid organs (Table 4). The results of targeted exome sequencing were further confirmed by Sanger sequencing in five DNA samples. The verified mutations were as follows: chr12: 25398284 (c.35G>T, p.Gly12Val) for PG2H, PG2L, PG6A, and PG8L samples and chr12: 25398282 (C>A, p.Gly13Cys) for PG6L. A representative PCR gel image and Sanger sequencing image are provided in Fig. 4b.
Next, an evaluation was performed within the PGA groups to identify any stepwise genomic differences between PGN, PGL, and PGH/PGCa within the same case. Only significant frameshift mutations and insertion–deletions were considered in this assessment. In the PG1 and PG2 cases, PGH/PGCa exhibited more frameshifts/indels than PGN (PG1) or PGL (PG2). Similarly, in PG5, the PGL exhibited more frameshifts/indels than the matched PGN. Finally, in PG6, PG8, and PG9, there was no difference in frameshifts/indels between the matched corresponding lesions (Supplementary Table 4).
Genomic profile of IM and GC (intestinal type)
A total of 3656 and 2111 distinct nonsynonymous, high-impact mutations with 98.5% C>T transitions and G>A transversions were found in the IM and GC groups, respectively. Variants with reads exhibiting an at least 10% VAF and 5 variant-supporting reads were filtered, which showed that predominant mutations were in the TP53 gene in 60% (n = 3) of GC cases (Table 5). Apart from TP53, mutations were also observed in the PIK3CA, ERBB4, and ATM genes. The PIK3CA gene is known to be actively mutated in EBV-positive tumors, and the missense mutation observed in this gene in our study has been reported to be mutated in adenocarcinoma of the stomach [23, 24]. None of the variants in IM samples were able to pass the filter. For further confirmation, Sanger sequencing was performed in GC5 only for TP53 at chr17: 7577538 (p.Arg248Gln), as the DNA was sufficient to perform Sanger sequencing only for the GC5 sample (Fig. 5). In addition, IHC was performed to verify the sequencing results. The GC5 FFPE sample exhibited nuclear p53 expression in malignant gastric glands, while the nonmutated GC2 sample presented no p53 expression in malignant glands (Fig. 5b).

a PCR gel image for TP53 and corresponding point mutation at chr17: 7577738 (C>T) in GC5 sample. b IHC for p53 in GC5 sample showing nuclear expression in malignant gland versus no expression of P53 in GC2 nonmutated sample.
SPEM vs IM
Since SPEM has been considered a precursor of IM, at least in mouse models, the genomic profile of SPEM was compared with that of IM. KRAS, BRAF, and ERBB4 were common genomic mutations between the two lesions.
PGH/Adca vs GC
We compared the common point mutations between the PGH/Adca and GC groups. Since thousands of point mutations were observed in the three groups, variants with at least a 10% VAF and 5′ supporting reads were filtered between the PGH/Adca and GC groups according to the standardized method. After filtration, it was observed that none of the mutations were shared between the two groups. However, when unfiltered mutations were compared, it was observed that 20 point mutations were shared between the PGA with high-grade dysplasia and/or pyloric gland adenocarcinoma and GC groups. Thirteen of the 20 SNPs were reported previously in various solid carcinomas, while the remaining SNPs were of benign or uncertain significance (Supplementary Table 5).
SPEM vs PGH/Adca and GC
Finally, the genomic profile of SPEM was compared with PGH/Adca and intestinal-type GC. Although common point mutations in the ALK, EGFR, BRAF, FGFR3, and ABL1 genes were observed, there were no mutations in key genes such as KRAS and APC (involved in PGA carcinogenesis) in the SPEM samples. On the other hand, both the SPEM and GC samples showed point (missense) mutations in the MUC5AC, KRAS, BRAF, and EZH2 genes, which are commonly implicated in gastric carcinogenesis.
Discussion
Previous proteomic and microarray profiling of SPEM FFPE samples has shed light on the dysregulated proteins between the normal mucosa, SPEM, and GC [25,26,27]. However, there remains a lack of reports comparing the targeted genomic profile of commonly dysregulated genes in PGA and GC with that of human SPEM. While whole-genome and whole-exome sequencing can shed light on genomic mutations and identify new driver mutations, targeted sequencing is more cost effective than sequencing the entire genome to a depth sufficient to find variants that could affect phenotypic expression [28]. Here, we attempted to evaluate 43 FFPE samples from 13 patients with different pathological diagnoses. A panel of 49 candidate genes that are commonly dysregulated in GC were selected for customized exome sequencing using QIAseq™ targeted DNA panels and were further sequenced using an Illumina HiSeq 2500 sequencer. The results of this study show that overall SPEM is characterized much less genomic variation than PGA and GC. SPEM is genomically distinct from PGA. Finally, SPEM may bear some resemblance to intestinal-type GC, as it shares some of the genomic mutations involved in gastric carcinogenesis.
We observed that overall, the total number of nonsynonymous point mutations in SPEM was significantly lower than that in other lesions in this study. This could indicate that although SPEM may be a premalignant condition, additional mutations and/or changes may be necessary for it to progress to gastric carcinoma. Very few point mutations were observed in SPEM. We found point mutations in MUC5AC (rs35783651) and MYC (rs750664148), which have also been reported previously in carcinoma of the esophagus and other solid organ carcinomas [21]. In addition, a mutation in codon 290 in KRAS (chr12: 25380167, C>T) was also observed. This mutation has not been reported previously in any lesion; however, the role of KRAS in SPEM development has been previously elucidated. The expression of activated KRAS in the chief cells of Mist1-KRAS mice has been shown to induce the development of SPEM [29]. Germline missense mutations were also observed in the APC and ALK genes, which are known to be involved in FAP syndrome, hereditary colorectal carcinomas, neuroblastomas, and hereditary cancer-predisposing syndromes [30]. These results shed important light on the genomic mutations in human SPEM, about which not much is known.
Another important finding of this study was that the genomic profile of SPEM differs from that of PGA, with or without dysplasias. None of the key regions in the genes that are known to be involved in the pathogenesis of PGA (e.g., KRAS and APC) were observed in SPEM. First, there was a significant difference between the mean nonsynonymous point mutations between PGA with or without cytological dysplasia and SPEM (Mann–Whitney test U = 0.000, p < 0.05). This difference existed despite the fact that the pyloric epithelium observed in PGA morphologically and phenotypically resembles that of SPEM, and SPEM and PGA share a common etiology and occur predominantly in the gastric corpus. Second, although point mutations in the ALK, EGFR, ABL1, BRAF, and FGFR3 genes were common among these groups, none of these genes have been reported to be critical for the neoplastic transformation of PGA. Thus, at the genomic level, SPEM and PGA are distinct from each other, which may be significant, as the pyloric epithelia seen in PGA show a propensity toward stepwise progression to malignancy, while SPEM may not.
On the other hand, when the genomic profile of SPEM was compared with that of intestinal-type GC, point mutations in the MUC5AC, KRAS, BRAF, and EZH2 genes were found to be common [23]. MUC5AC is located on chromosome 11p.15.5, a site that is commonly involved in the loss of heterozygosity in GC [31, 32]. Decreased MUC5AC protein expression is also associated with a poor prognosis in GC [33]. On the other hand, not only amplification and/or point mutations in KRAS have been reported in GC, but KRAS activation can lead to the development of SPEM in mouse models [29, 34,35,36]. High protein expression of EZH2, an enhancer of zeste homolog 2, is also associated with a poor prognosis in GC [37]. Based on these findings, it seems that SPEM resembles the intestinal type of GC genomically and could be a precursor condition in human GC as well.
In the PGA group, 9 out of 15 samples (60%) showed a KRAS mutation in exon 2 (c.35G>A and amino acid change, p.G12D) that has been previously reported in PGA [13, 14]. In two samples (i.e., PG6L and PG6N), KRAS mutations were observed in exon 2 but at the c.37G>T position (amino acid change p.G13C), which is implicated in NSCLC and colorectal cancer [38]. In addition, in 7 out of 15 PGA samples (46%), a frameshift deletion in the APC gene at c.4385_4386delAG (amino acid change p.S1465fs*3), which has been previously reported in colorectal cancer [35]. In addition to KRAS and APC, GNAS is reported to be involved in the pathogenesis of PGA; however, in this study, no significant genomic variations were observed in GNAS. Point mutations in GNAS were observed, but they were not pathogenic and were of little clinical importance.
Another interesting result of this study was that within the same pyloric gland, there can be morphological differences ranging from no cytological dysplasia to low- and high-grade dysplasia and even adenocarcinoma. Our study seems to suggest that this morphological change to a higher-grade dysplasia/malignancy is associated with genomic changes that have been shown to be involved in other gastrointestinal malignancies. To this end, PGA without cytological dysplasia (PGN), PGA with LGD and PGA with HGD and/or adenocarcinoma were compared with each other within the same case. Only the highly annotated, nonsynonymous, frameshift mutations were compared with their counterparts. In PG1, both HGD and PGN samples showed CTNNB1 mutations, but in addition to PG1H, there were multiple genes that showed frameshift mutations that were absent in PG1N. Similarly, in PG2, both HGD and LGD showed KRAS mutation, but PG2H also showed a frameshift deletion in the APC gene that was absent in PG2L. In PG5 cases, areas without dysplasia as well as LGD presented frameshift mutations in the APC gene, but the LGD areas also showed frameshift mutations in the IDH2 gene. The difference in mutations in higher-grade lesions could be due to the subpopulation of cells that had further progressed along the path of malignant transformation. At a genomic level, we considered this to be a manifestation of the stepwise progression of the pyloric glandular epithelium within a PGA toward pyloric gland adenocarcinoma. However, in PG6, PG8, and PG9 cases, the PGN, PGL, and PGH/PGCa samples from within the same case showed similar genomic profiles in the context of frameshift mutations. This may have occurred because only frameshift mutations were compared within the same case, even though there may have been other genes that were upregulated or differentially expressed in the groups within the same case that could not be evaluated in this study.
In conclusion, this study shows the presence of few genomic variations in SPEM compared with GC and PGA. The pseudopyloric epithelium appears to be distinctly different from the pyloric epithelium in PGA even when it does not show any cytological dysplasia. Stepwise progression of PGA without cytological dysplasia to LGD and to HGD/Pyloric adenocarcinoma shows increased genetic mutations as dysplasia increases. Finally, there seem to be some genomic similarities between SPEM and intestinal-type gastric adenocarcinoma based on the common genomic mutations observed in genes commonly implicated in gastric carcinogenesis.
References
Wang TC, Goldenring JR, Dangler C, Ito S, Mueller A, Jeon WK, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114:675–89.
Schmidt PH, Lee JR, Joshi V, Playford RJ, Poulsom R, Wright NA, et al. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Investig. 1999;79:639–46.
Fox JG, Li X, Cahill RJ, Andrutis K, Rustgi AK, Odze R, et al. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology. 1996;110:155–66.
Weis VG, Petersen CP, Mills JC, Tuma PL, Whitehead RH, Goldenring JR. Establishment of novel in vitro mouse chief cell and SPEM cultures identifies MAL2 as a marker of metaplasia in the stomach. AJP Gastrointest Liver Physiol. 2014;307:G777–92.
Goldenring JR, Nam KT, Wang TC, Mills JC, Nicholas A. Origins of gastric cancer. 2013;138:2207–10.
Petersen CP, Mills JC, Goldenring JR. Murine models of gastric corpus preneoplasia. Cell Mol Gastroenterol Hepatol. 2017;3:11–26.
Nookaew I, Thorell K, Worah K, Wang S, Hibberd ML, Sjövall H, et al. Transcriptome signatures in Helicobacter pylori-infected mucosa identifies acidic mammalian chitinase loss as a corpus atrophy marker. BMC Med Genom. 2013;6:41.
Jeong S, Choi E, Petersen CP, Roland JT, Federico A, Ippolito R, et al. Distinct metaplastic and inflammatory phenotypes in autoimmune and adenocarcinoma-associated chronic atrophic gastritis. United Eur Gastroenterol J. 2017;5:37–44.
Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis-pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10:529–41.
Pittman ME, Voltaggio L, Bhaijee F, Robertson SA, Montgomery EA. Autoimmune metaplastic atrophic gastritis: recognizing precursor lesions for appropriate patient evaluation. Am J Surg Pathol. 2015;39:1611–20.
Elster K. Histologic classification of gastric polyps. Berlin Heidelberg: Springer; 1976. p. 77–93.
Kushima R, Vieth M, Borchard F, Stolte M, Mukaisho KI, Hattori T. Gastric-type well-differentiated adenocarcinoma and pyloric gland adenoma of the stomach. Gastric Cancer. 2006;9:177–84.
Matsubara A, Sekine S, Kushima R, Ogawa R, Taniguchi H, Tsuda H, et al. Frequent GNAS and KRAS mutations in pyloric gland adenoma of the stomach and duodenum. J Pathol. 2013;229:579–87.
Hashimoto T, Ogawa R, Matsubara A, Taniguchi H, Sugano K, Ushiama M, et al. Familial adenomatous polyposis-associated and sporadic pyloric gland adenomas of the upper gastrointestinal tract share common genetic features. Histopathology. 2015;67:689–98.
Huang KK, Ramnarayanan K, Zhu F, Srivastava S, Xu C, Tan ALK, et al. Genomic and epigenomic profiling of high-risk intestinal metaplasia reveals molecular determinants of progression to gastric cancer. Cancer Cell. 2018;33:137–50.e5.
Xu C, Nezami Ranjbar MR, Wu Z, DiCarlo J, Wang Y. Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller. BMC Genom. 2017;18:5.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 2012;6:80–92.
Vieth M, Montgomery EA. Some observations on pyloric gland adenoma: an uncommon and long ignored entity! J Clin Pathol. 2014;67:883–90.
Stolte M. The new Vienna classification of epithelial neoplasia of the gastrointestinal tract: advantages and disadvantages. Virchows Arch 2003;442:99–106.
Hamilton SR, Aaltonen LA pathology and genetics of tumours of the digestive system. 2000;2:59–104.
Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34:155–63.
Esser D, Holze N, Haag J, Schreiber S, Krüger S, Warneke V, et al. Interpreting whole genome and exome sequencing data of individual gastric cancer samples. BMC Genomics. 2017;18:517.
Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9.
List of variants in gene PIK3CA reported as likely pathogenic for adenocarcinoma of stomach—ClinVar Miner. https://clinvarminer.genetics.utah.edu/variants-by-gene/PIK3CA/condition/Adenocarcinoma%20of%20stomach/likely%20pathogenic.
Sousa JF, Ham AJL, Whitwell C, Nam KT, Lee HJ, Yang HK, et al. Proteomic profiling of paraffin-embedded samples identifies metaplasia-specific and early-stage gastric cancer biomarkers. Am J Pathol. 2012;181:1560–72.
Lee JR, Baxter TM, Yamaguchi H, Wang TC, Goldenring JR, Anderson MG. Differential protein analysis of spasomolytic polypeptide expressing metaplasia using laser capture microdissection and two-dimensional difference gel electrophoresis. Appl Immunohistochem Mol Morphol. 2003;11:188–93.
LaFleur BJ, Yang H, Aburatani H, Kim WH, Kim MA, Nam KT, et al. Gene expression profiling of metaplastic lineages identifies CDH17 as a prognostic marker in early stage gastric cancer. Gastroenterology. 2010;139:213–25.e3.
Oetting WS. Exome and genome analysis as a tool for disease identification and treatment: the 2011 human genome variation society scientific meeting. Hum Mutat. 2012;33:586–90.
Choi E, Hendley AM, Bailey JM, Leach SD, Goldenring JR. Expression of activated ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology. 2016;150:918–30.e13.
Picelli S, Zajac P, Zhou X-L, Edler D, Lenander C, Dalén J, et al. Common variants in human CRC genes as low-risk alleles. Eur J Cancer. 2010;46:1041–8.
Pigny P, Guyonnet-Duperat V, Hill AS, Pratt WS, Galiegue-Zouitina S, d’Hooge MC, et al. Human mucin genes assigned to 11p15.5: identification and organization of a cluster of genes. Genomics. 1996;38:340–52.
Moskaluk CA, Rumpel CA. Allelic deletion in 11p15 is a common occurrence in esophageal and gastric adenocarcinoma. Cancer.1998;83:232–9.
Kim SM, Kwon CH, Shin N, Park DY, Moon HJ, Kim GH, et al. Decreased Muc5AC expression is associated with poor prognosis in gastric cancer. Int J Cancer. 2014;134:114–24.
Polom K, Das K, Marrelli D, Roviello G, Pascale V, Voglino C, et al. KRAS mutation in gastric cancer and prognostication associated with microsatellite instability status. Pathol Oncol Res. 2019;25:333–40.
Lee SH, Jung SH, Kim T-M, Rhee J-K, Park H-C, Kim MS, et al. Whole-exome sequencing identified mutational profiles of high-grade colon adenomas. Oncotarget. 2017;8:6579–88.
Rahman R, Asombang AW, Ibdah JA, Yamada N, Kitamoto S, Yokoyama S, et al. NIH public access. J Clin Pathol. 2015;67:529–36.
Matsukawa Y, Semba S, Kato H, Ito A, Yanagihara K, Yokozaki H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006;97:484–91.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Funding
This work was supported by a CISSP grant, Department of Pathology, Singapore.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Srivastava, S., Huang, K.K., Rebbani, K. et al. An LCM-based genomic analysis of SPEM, Gastric Cancer and Pyloric Gland Adenoma in an Asian cohort. Mod Pathol 33, 2075–2086 (2020). https://doi.org/10.1038/s41379-020-0520-5
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41379-020-0520-5
This article is cited by
-
Revolutionizing Gastric Cancer Prevention: Novel Insights on Gastric Mucosal Inflammation-Cancer Transformation and Chinese Medicine
Chinese Journal of Integrative Medicine (2025)
