
Abstract
Positive program death-ligand 1 (PD-L1) immunohistochemistry (IHC) is an approved companion diagnostic guiding the use of immune checkpoint inhibitors in uterine cervical carcinoma (CXC). The clinical and genomic features of PD-L1-positive (PD-L1positive) CXC have not been previously described. We reviewed the clinicopathologic and molecular features of 647 CXC cases that were tested using DAKO 22C3 PD-L1 IHC and comprehensive genomic profiling during the course of clinical care. PD-L1positive cases were defined via a combined positive score of ≥ 1. No differences were found in age, genetic ancestry, and HPV status of the PD-L1positive (n = 548) and PD-L1negative disease subset. The PD-L1 positivity rate varied by histologic subtype of CXC with squamous cell carcinoma (SCC) having a PD-L1 positivity rate of 91% (397/437) and usual-type adenocarcinoma’s PD-L1 positivity rate being 60% (35/58). In addition, the PD-L1 positivity rate varied depending on site of the specimen with 89.1% (261/293) positivity rate observed in cervix specimens compared to 25% (2/8) in brain metastases specimens. No significant difference in tumor mutational burden (TMB), microsatellite instability, and CD274 (encoding PD-L1) amplification was observed between PD-L1positive and PD-L1negative CXC subsets. By combining TMB with PD-L1, an additional 17 patients are eligible for pembrolizumab when compared to PD-L1 testing alone. TERT promoter alterations and APOBEC mutational signature were enriched in the PD-L1positive CXC SCC (p = 0.011, and p = 0.004, respectively). Our study reveals important prevalence data on PD-L1 positivity in CXC non-SCC and suggests that further studies in these histologic subtypes are warranted. In addition, we also provide a key framework to guide both specimen selection and future investigations of predictors of immunotherapy response in cervical cancer patients. Lastly, TERT promoter alterations and APOBEC mutational signature may be a biologically unique subset of PD-L1positive CXC SCC.
Similar content being viewed by others

Introduction
The most frequent biomarker associated with uterine cervical carcinoma (CXC) is human papillomavirus (HPV), in particular HPV types 16 and 18 [1]. Even though the rate of CXC deaths has declined with the long-standing use of cervical screening cytology and the more-recent use of HPV detection, cervical cancer remains a significant disease with an estimated 13,800 new cases of invasive cervical cancer and 4290 deaths in the United States during 2020 [2]. Histologically, CXC are most often squamous cell carcinomas (SCC), but can also be usual-type adenocarcinomas or adenosquamous carcinomas, all of which are HPV driven. Rare HPV-independent subtypes also exist, such as, gastric-type adenocarcinoma, adenoma malignum/minimal deviation carcinoma, mesonephric carcinoma, clear cell carcinoma, and serous carcinoma. Currently, the standard of care therapy includes a combination of surgery, chemotherapy, radiation therapy, and/or targeted therapies such as bevacizumab [3]. Recently, new treatment options utilizing anti-program death-ligand 1/programmed cell death protein 1 (PD-L1/PD-1) immunotherapies have been approved for CXC.
In June 2018, pembrolizumab was approved for recurrent or metastatic CXC in patients whose tumors express PD-L1 using the DAKO 22C3 companion diagnostic (CDx) immunohistochemistry (IHC) assay based on the KEYNOTE-158 clinical trial [4]. The specific cutoff used in this approval was a combined positive score (CPS) ≥ 1 being considered as eligible for pembrolizumab. In KEYNOTE-158 trial, 83.7% (82/98) of the patients had a CPS ≥ 1 with an objective response rate (ORR) of 14.6% and with no responses in patients whose tumors had PD-L1 CPS < 1 [5]. PD-L1 IHC is a recognized CDx for pembrolizumab in CXC. In addition, patients with a tumor mutational burden (TMB) ≥ 10 mutations/Mb with FoundationOne®CDx are eligible for pembrolizumab based on a pan-tumor CDx approval in June 2020 that followed the KEYNOTE-158 trial [6]. And finally, as a third FDA-approved pan-tumor option, cervical cancer patients with microsatellite instability-high (MSI-High) tumors are also eligible for pembrolizumab [7]. Additional predictive biomarkers include CD274 (PD-L1) gene amplification that has shown some evidence as a predictor to PD-L1 therapy response in solid tumors [8]. Recently, potential resistance and hyper-progression biomarkers for patients treated with immune checkpoint inhibitors (ICPI) have emerged in several tumor types. STK11 and KEAP1 alterations have been shown to be predictors of immunotherapy resistance in non-small cell lung cancer (NSCLC), and patients with solid tumors featuring MDM2/4 amplification have been linked to tumor hyper-progression when ICPI are used for their treatment [9,10,11,12].
Currently, there is expanding interest in uncovering new ways of defining disease subsets in CXC that are based on PD-L1 IHC status. In this study, we characterize the clinicopathologic and genomic features of the CXC patients whose tumors are PD-L1 positive using comprehensive genomic profiling (CGP).
Materials and methods
Patient cohort selection
Approval for this study was obtained from the Western Institutional Review Board Protocol No. 20152817. We re-reviewed all CXC cases that were tested with PD-L1 DAKO 22C3 CDx IHC assay between September 2018 and March 2020. These cases were sent to Foundation Medicine for CGP and PD-L1 IHC during routine clinical care. Age of patient and specimen site of sample were extracted from accompanying pathology reports.
PD-L1 IHC
All PD-L1 IHC testing was performed using the DAKO 22C3 PharmDx assay per manufacturer’s instructions in a Clinical Laboratory Improvement Amendments (CLIA)-certified and College of American Pathologists (CAP)-accredited reference laboratory (Foundation Medicine, Morrisville, NC). In brief, the DAKO PD-L1 22C3 PharmDx assay consists of a monoclonal mouse anti-PD-L1, the 22C3 clone, with the envision flex visualization system on the autostainer link 48 using the staining protocol provided by the package insert and interpreted with the guidelines of the DAKO interpretation guide [13, 14]. All cases have accompanying controls, hematoxylin and eosin (H&E)-stained patient slide, negative reagent control stained patient slide, and a DAKO PD-L1 22C3-stained patient slide. PD-L1 IHC slides were interpreted by board-certified pathologists using the CPS, which is the number of PD-L1 staining cells (tumor cells, lymphocytes, macrophages) divided by the total number of viable tumor cells, multiplied by 100 [14]. As per DAKO’s interpretation guide, any convincing partial or complete linear membrane staining (≥1+) of viable tumor cells that is perceived as distinct from cytoplasmic staining and any convincing membrane and/or cytoplasmic staining (≥1+) of lymphocytes and macrophages (mononuclear inflammatory cells) within tumor nests and/or immediately adjacent supporting stroma were counted for scoring. A CPS ≥ 1 is considered positive for PD-L1 in CXC patient per the CDx claim [4].
Comprehensive genomic profiling
CGP was performed using the FDA-approved FoundationOne®CDx assay (Foundation Medicine, Cambridge, MA) using previously described methods in a CLIA-certified and CAP-accredited laboratory [15]. FoundationOne®CDx uses a hybrid capture methodology and detects base substitutions, insertions/deletions, and copy number alterations in 324 genes and select gene rearrangements in 36 genes, as well as TMB and MSI. A board-certified pathologist reviewed a H&E slide from each sample under light microscopy to determine tumor adequacy (at least 20% tumor nuclei present) and to confirm these cases were cervical cancer before sequencing the case. TMB was determined on 0.79 Mb of sequenced DNA and assessment of MSI was performed from DNA sequencing across 95 loci as previously described [16, 17]. TMB ≥ 10 mutations/Mb was considered High (TMB-High) based on the CDx approval in pan-solid tumors for pembrolizumab. As research use only: genetic ancestry, APOBEC mutational signature, and HPV were determined based on CGP results. Genomic ancestry of patients was determined using a principal component analysis of genomic single nucleotide polymorphisms trained on data from the 1000 Genomes Project and each patient was classified as belonging to one of the following super populations: African, Central and South American, East Asian, European, and South Asian [18, 19]. APOBEC mutational signatures were called as described by Zehir et al. [20]. De novo assembly of non-human sequencing reads and BLASTn comparison against all viral nucleotide sequences in the NCBI RefSeq database was used to detect the presence of HPV genome sequences.
Analysis methodology
We analyzed patient characteristic differences in age, predominant genetic ancestry, metastatic vs. primary site specimen, APOBEC mutational signature, and presence of HPV between the PD-L1positive and PD-L1negative disease subsets. In addition, the immunotherapy biomarkers of TMB, MSI, and CD274 amplification were also examined with respect to PD-L1positive and PD-L1negative disease subsets. Statistical analysis was performed using ANOVA, χ2 contingency test, or Fisher’s exact test.
To examine the genomic biomarker landscape of our patient cohort, we extracted the top 25 genes that have genomic alterations (GA) and compared these same genes between the CXC PD-L1positive and PD-L1negative disease subsets with Fisher’s exact test. P value was adjusted for multiple comparisons using the Bonferroni method and p < 0.05 was considered significant [21]. As a secondary analysis, we compared the same top 25 genes with GA and compared these genes in the CXC SCC PD-L1positive and PD-L1negative disease subsets; and also the CXC non-SCC PD-L1positive and PD-L1negative disease subsets, both with Fisher’s exact test.
Results
Clinicopathologic characteristics
A total of 647 CXC specimens were tested with both PD-L1 IHC and CGP at Foundation Medicine between September 2018 and March 2020. The mean and median age of the cohort were both 53 years old with no difference in the age of the patients in the PD-L1positive and PD-L1negative disease subset (ANOVA, p = 0.589) (Table 1). Most patients were of predominant European genetic ancestry with no significant differences in predominant genetic ancestry between the PD-L1positive and PD-L1negative disease subset (χ2 contingency, p = 0.597) (Table 1). Slightly more than half of the specimens were obtained from metastatic sites 54.7% (354/647). There was a significantly higher PD-L1 positivity rate in the specimens from primary sites (89.1%, 261/293) when compared to the specimens from metastatic sites (81.1%, 287/354) (Fisher’s exact test, p = 0.006). A difference in PD-L1 positivity based on the location of the metastatic specimen was observed with multiple sites having 100% positivity and metastatic specimens from the brain having only 25% (2/8) PD-L1 positivity (Table 2). When we examined the histologic subtypes of brain metastatic specimens, we found that 33.4% (1/3) of SCC histology were positive and 20% (1/5) non-SCC histology were positive for PD-L1. In addition, we saw a significantly higher rate of APOBEC mutational signature in the PD-L1positive disease subset (Fisher’s exact test, p < 0.001) (Table 1). When we examined the CXC SCC histology subtype, we also saw a significant enrichment of APOBEC mutational signature in the PD-L1positive (19.1%, 76/397) vs the PD-L1negative (2.5%, 1/40) disease subset (Fisher’s exact test, p = 0.004). In CXC non-SCC histologic subtype, while we did see a higher rate in APOBEC mutational signature in the PD-L1positive (7.3%, 11/151) vs PD-L1negative (1.7%, 1/59) disease subset, the results were not significant (Fisher’s exact test, p = 0.190). Lastly, we saw no difference in the HPV positivity rate in the PD-L1positive and PD-L1negative disease subsets (Fisher’s exact test, p = 0.122).
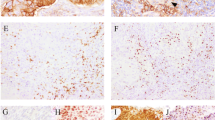
Overall, 84.7% (548/647) CXC patients were positive for PD-L1 based on the CDx CPS score cutoff of ≥ 1. When examining the PD-L1 prevalence rate in different CXC subtypes, we saw differences in PD-L1 positivity rate (Fig. 1). For example, SCC histologic subtype has a PD-L1 positivity rate of 91% (397/437) and usual-type adenocarcinoma has a PD-L1 positivity rate of 60% (35/58). In addition, the signet-ring cell type adenocarcinoma has the lowest PD-L1 positivity rate at 50.0% (2/4) and glassy cell carcinoma, mesonephric adenocarcinoma, and invasive stratified mucin-producing carcinoma, all had 100% PD-L1 positivity rates, although the small sample size for these CXC subtypes restricts the ability to achieve definitive conclusions (Table 2).

Histologic subtypes: A squamous cell carcinoma; B squamous cell carcinoma; C mesonephric adenocarcinoma; D gastric-type adenocarcinoma; E serous-type adenocarcinoma; F glassy cell carcinoma; G invasive stratified mucin-producing carcinoma; H clear cell adenocarcinoma; I endometrioid-type adenocarcinoma; and J usual-type adenocarcinoma. All digital images are at 200× magnification.
Immunotherapy biomarkers
When we examined the PD-L1 CPS scores of the CXC patients, we saw a clustering of patients with SCC histology with high PD-L1 expression with a CPS score of around 100 and in non-SCC histology with a CPS score around 50 (Fig. 2A). Mean and median (mutations/Mb) TMB in the overall CXC cohort is low (7.74, 5.00). There is no significant difference in TMB between the PD-L1positive CXC subset (7.68, 5.00) and in the PD-L1negative CXC subset (7.34, 5.00) (ANOVA, p = 0.592). When using the TMB CDx cutoff of 10 mutations/Mb, the prevalence of TMB positivity was 25% (161/647) in the overall cohort (Table 1). There was also no significant difference in the TMB positivity rate (using a 10 mutations/Mb TMB as the cutoff) between the PD-L1positive (26.3%, 144/548) and PD-L1negative (17.2%, 17/99) CXC subsets (Fisher’s exact test, p = 0.058) (Table 1). MSI was high (H) in 1.9% (12/647) of the overall cohort, all of which were PD-L1positive, although no significance was found (Fisher’s exact test, p = 0.230). CD274 (PD-L1 gene) amplification was higher in the PD-L1positive CXC subset (2.4%, 13/549) when compared to the PD-L1negative cohort (0%, 0/99), although no significance was found (Fisher’s exact test, p = 0.236).

A Violin plot of combined positive score (CPS) values in uterine cervical carcinoma squamous cell carcinoma (SCC) and non-SCC histologic subtype. In the SCC histologic subtype, we saw a clustering of high PD-L1 expression at a CPS score of around 100; and in non-SCC histologic subtype with a clustering around a CPS score of 50. B Figure of relationship between PD-L1 positivity (CPS ≥ 1), TMB-High (≥ 10 mutations/Mb), and MSI-High. pos positive, neg negative.
At a TMB cutoff of 10 mutations/Mb, 26.3% (144/548) of PD-L1positive CXC patients were also positive for TMB-High (Fig. 2B). On the other hand, 89.4% 144/161 of TMB-High patients were also PD-L1positive. All 12 MSI-High CXC were PD-L1positive and were TMB-High (except one CXC that was PD-L1positive, but the TMB status was indeterminate). These 12 MSI-H patients had relatively high PD-L1 expression (CPS ≥ 1) and TMB levels (median = 32.5 mutations/Mb; mean = 35.6 mutations/Mb).
The frequencies of GA in genes that have been associated with immunotherapy resistance or hyper-progression in other tumor types such as NSCLC including STK11/KEAP1 alterations, MDM2/MDM4 amplification were 11.7% (64/548), 1.6% (9/548), 0.7% (4/548), and 0.2% (1/548) in PD-L1positive; and 13.1% (13/99), 1.0% (1/99), 4.0% (4/99), and 0% (0/99) in PD-L1negative CXC subsets, respectively.
Genomic landscape of PD-L1positive and PD-L1negative disease subsets
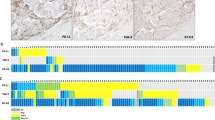
Co-mutation plots of the top 25 genes with GA in PD-L1positive and PD-L1negative CXC disease groups are shown in Fig. 3A, B. GA in PIK3CA was the most common in both the PD-L1positive and PD-L1negative disease subsets. However, the next four most common GA in the PD-L1positive disease subset were MLL2 (17.7%), TP53 (13.1%), FBXW7 (12.6%), and TERT (12.6%) which contrasted with the PD-L1negative disease subset (KRAS (21.2%), ERBB2 (14.1%), ARID1A (14.1%), and TP53 (14.1%)).

A Co-mutation plots of the top 25 genes with genomic alterations (GA) in the PD-L1positive uterine cervical carcinoma (CXC) disease subset. The top five genes with GA in the PD-L1positive disease subset are PIK3CA, MLL2, TP53, FBXW7, and TERT. B Co-mutation plots of the top 25 genes for the PD-L1negative CXC disease subset. The top five genes with GA in the PD-L1negative CXC were PIK3CA, KRAS, ERBB2, ARID1A, and TP53.
Of the top 25 genes with GA in the overall CXC cohort, two genes had significantly different frequencies between the PD-L1positive and PD-L1negative disease subset (Supplementary Table 1). The frequency of GA in TERT promoter was enriched in the PD-L1positive disease subset and KRAS was enriched in the PD-L1negative disease subset (Fisher’s exact test with adjusted p value, p = 0.003 and p = 0.011, respectively).
In our secondary analysis of the CXC SCC histologic subtype, we saw that the frequency of GA in TERT promoter was still enriched in the PD-L1positive disease subset and CTNNB1 was enriched in the PD-L1negative disease subset (Fisher’s exact test, p = 0.011 and p = 0.031, respectively) (Supplementary Table 2). When we examined the CXC non-SCC histologic subtypes, we did not see any significant difference in the prevalence of GA between the PD-L1positive and PD-L1negative subset.
Discussion
Investigators have previously examined the prevalence rates of PD-L1 positivity by IHC in two large pan-tumor study and had also examined the clinicopathologic and genomic differences in PD-L1positive and PD-L1negative disease subsets in tumor types such as triple-negative breast carcinoma [22,23,24]. However, to the best of our knowledge, this is the first study that examined the clinicopathologic and genomic differences between the PD-L1positive and PD-L1negative CXC patients in a large cohort of clinical samples.
Depending on the status of the PD-L1 DAKO 22C3 CDx assay, a CXC patient can be potentially eligible for pembrolizumab-based ICPI treatment. With our large cohort of PD-L1positive and PD-L1negative cases, we examined if there were differences in PD-L1 status based on CXC histologic subtype and found that the rate of PD-L1 positivity varied amongst CXC subtypes with signet-ring cell type adenocarcinoma having the lowest rate of PD-L1 positivity at 50%; and glassy cell carcinoma, mesonephric adenocarcinoma, and invasive stratified mucin-producing carcinoma having 100% PD-L1 positivity rates, although the sample size is small in these specific subtypes. Of note, most of the cases in this CXC cohort have SCC histology and the PD-L1 positivity rate for these tumors was 91% (397/437). In the KEYNOTE-158 trial, only 6.1% (6/98) of the patients were of non-SCC histology and all of them were PD-L1 positive [5]. This differed from our data of non-SCC histology where many of the cases were not PD-L1 positive. For example, in CXC usual-type adenocarcinoma the positivity rate was 60% (35/58) and interestingly, the CXC adenosquamous carcinoma PD-L1 positivity rate was more closely aligned with the usual-type adenocarcinoma subtype then the SCC subtype. The PD-L1 positivity prevalence data in different CXC histology subtype suggests that further trials needs to be performed to assess the cutoff and clinical utility of PD-L1 IHC in CXC non-SCC histologic subtypes.
In addition, in SCC histologic subtype, we saw a clustering of high PD-L1 expression at a CPS score of around 100; and in non-SCC histologic subtype with a clustering around a CPS score of 50. We hypothesize that this represented a small group of patients who could be hyper-responders to ICPI and should be studied for ICPI use in an earlier line of therapy. In addition, we saw a significantly higher PD-L1 positivity rate in specimens from a primary site when compared to metastatic sites and differences in PD-L1 positivity rate based on site of metastasis. Notably, there was a low PD-L1 positivity rates in brain metastases. While initial examination of the histologic makeup in these samples revealed a higher percentage of non-SCC histology (62.5%, 5/8), histology does not seem to entirely account for the difference in PD-L1 prevalence. Whether this lower prevalence is due to pre-analytic processing of brain specimens or biologic reasons remains elusive. Regardless, these results could be helpful when determining which samples to select when multiple samples are available.
From the KEYNOTE-158 trail, it was shown that the negative predictive value of PD-L1 IHC in predicting the non-response to ICPI is high with no responses in the PD-L1 negative group; however, the positive predictive value is not as high given that the ORR was only 14.6% (three complete and nine partial responses) in the PD-L1-positive group [5]. Given this relatively low ORR, additional biomarkers are needed to further identify CXC patients who are likely to respond to ICPI. While PD-L1 IHC is the only CDx specifically approved for ICPI in CXC, TMB-High and MSI-High are also approved for pembrolizumab eligibility based on FDA pan-tumor approvals. In this cohort, 22.3% (144/647) patients were both PD-L1 positive and TMB-High, and we hypothesize that this group of patients could potentially have a higher ORR as compared to patients with only PD-L1 positivity, illustrating the potential value of combining TMB with PD-L1 IHC for ICPI in CXC patients. MSI-High was identified in only a small number of patients in our CXC cohort (12/647), and all these CXC cases were also positive for PD-L1 IHC and TMB (except one CXC that was PD-L1positive, but the TMB status was indeterminate). It is interesting to note that the MSI-High patients had relatively high levels of PD-L1 and TMB, and we hypothesize that these triple-biomarker positive patients could represent a subset of patients that are hyper-responders to ICPI. These hypotheses should be validated in future clinical studies. In addition, we identified a subset of CXC patients with STK11 and/or KEAP1 alterations, and MDM2/4 amplifications, which have been described as potential resistance or hyper-progression biomarkers for ICPI. This further illustrates the value of combining CGP with PD-L1 IHC in predicting immunotherapy response.
When examining the genomic landscape of PD-L1positive and PD-L1negative CXC, we saw a similar overall genomic landscape, but also some key differences. In our PD-L1positive CXC and CXC SCC histologic subtype, we saw an increased association with TERT promoter alterations and APOBEC mutational signature. Mutations in TERT have been well described in CXC, and TERT promoter alterations has been investigated as a target for immunotherapy with TERT-based vaccines albeit with only limited responses to date [25, 26]. However, there has been a resurgence of interest in targeting TERT GA based on a personalized immunotherapy approach that combined TERT targeting with ICPI [27]. APOBEC mutational signature, while prevalent in multiple tumor types, has been shown to be enriched in certain tumor types such as CXC [28]. In various tumor types, APOBEC mutational signature has been shown to be highly correlated with PD-L1 expression and the APOBEC signature has been suggested as a predictive marker for immunotherapy response [29, 30]. In our cohort of CXC and CXC SCC histologic subtype, a significant correlation of APOBEC mutational signature was seen in the PD-L1positive disease subset, adding further evidence of the potential predictive value of APOBEC mutational signature in ICPI response across multiple tumor types. However, it is important to note that we did not see a significant difference in TERT promoter alterations and APOBEC mutational signature between the PD-L1positive and PD-L1negative non-SCC histologic subtype.
In conclusion, we characterize the clinicopathologic and genomic features PD-L1positive CXC. Our study presents important prevalence data on PD-L1 positivity in non-SCC histologic subtypes of CXC and suggests that further studies in these histologic subtypes are warranted. Given that 1% positivity for PD-L1 is a wide net to capture eligible CXC patients, who may have low ICPI responses, our results also provide a resource to study the criteria to potentially narrow the net for CXC high ICPI responders. In addition, these data provide the framework for future studies and a resource with regard to the influence of site of the specimen, and primary tumor versus metastasis in relation to PD-L1 positivity. Combining TMB and PD-L1 IHC further expands ICPI eligibility for a subset of CXC patients. Lastly, future studies are warranted to determine whether TERT promoter alterations and APOBEC mutational signature in the PD-L1positive CXC SCC histologic subtype can be further predictors of immunotherapy response in cervical SCC patients and whether they can further stratify PD-L1positive patients into high responders.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev. 2003;16:1–17.
American Cancer Society. Key statistics for cervical cancer; 2020.
Board PDQATE. Cervical cancer treatment (PDQ®): patient version. In: PDQ cancer information summaries. National Cancer Institute (US), Bethesda (MD); 2002.
FDA. FDA approves pembrolizumab for advanced cervical cancer with disease progression during or after chemotherapy; 2018.
Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2019;37:1470–8.
FDA. FDA approves pembrolizumab for adults and children with TMB-H solid tumors; 2020.
FDA. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication; 2017.
Goodman AM, Piccioni D, Kato S, Boichard A, Wang HY, Frampton G, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4:1237–44.
Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–50.
Fuentes-Antrás J, Provencio M, Díaz-Rubio E. Hyperprogression as a distinct outcome after immunotherapy. Cancer Treat Rev. 2018;70:16–21.
Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Disco. 2018;8:822–35.
Marinelli D, Mazzotta M, Scalera S, Terrenato I, Sperati F, D’Ambrosio L, et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann Oncol. 2020. https://doi.org/10.1016/j.annonc.2020.08.2105.
DAKO. DAKO PD-L1 IHC 22C3 pharmDx Package Insert. January 6.
DAKO. PD-L1 IHC 22C3 pharmDx Interpretation Manual—Cervical Cancer; 2018.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn. 2019;21:1053–66.
Newberg J, Connelly C, Frampton G. Abstract 1599: determining patient ancestry based on targeted tumor comprehensive genomic profiling. Cancer Res. 2019;79:1599–1599.
Carrot-Zhang J, Chambwe N, Damrauer JS, Knijnenburg TA, Robertson AG, Yau C, et al. Comprehensive analysis of genetic ancestry and its molecular correlates in cancer. Cancer Cell. 2020;37:639–54.e636.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Goeman JJ, Solari A. Multiple hypothesis testing in genomics. Stat Med. 2014;33:1946–78.
Huang RSP, Haberberger J, Severson E, Duncan DL, Hemmerich A, Edgerly C, et al. A pan-cancer analysis of PD-L1 immunohistochemistry and gene amplification, tumor mutation burden and microsatellite instability in 48,782 cases. Mod Pathol. 2020. https://doi.org/10.1038/s41379-020-00664-y.
Huang RSP, Li X, Haberberger J, Sokol E, Severson E, Duncan DL, et al. Biomarkers in breast cancer: an integrated analysis of comprehensive genomic profiling and PD-L1 immunohistochemistry biomarkers in 312 patients with breast cancer. Oncologist. 2020 https://doi.org/10.1634/theoncologist.2020-0449.
O’Malley DP, Yang Y, Boisot S, Sudarsanam S, Wang JF, Chizhevsky V, et al. Immunohistochemical detection of PD-L1 among diverse human neoplasms in a reference laboratory: observations based upon 62,896 cases. Mod Pathol. 2019;32:929–42.
Vinothkumar V, Arunkumar G, Revathidevi S, Arun K, Manikandan M, Rao AK, et al. TERT promoter hot spot mutations are frequent in Indian cervical and oral squamous cell carcinomas. Tumour Biol. 2016;37:7907–13.
Lin J, Li Y, Zou H, Zhang L, Xiao Y, Pan Q, et al. The alteration of TERT in 10,000 Chinese patients with solid tumors. J Clin Oncol. 2020;38:e13655.
Zanetti M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat Rev Clin Oncol. 2017;14:115–28.
Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–6.
Boichard A, Pham TV, Yeerna H, Goodman A, Tamayo P, Lippman S, et al. APOBEC-related mutagenesis and neo-peptide hydrophobicity: implications for response to immunotherapy. Oncoimmunology. 2018;8:1550341–1550341.
Wang S, Jia M, He Z, Liu XS. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene. 2018;37:3924–36.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
RSPH and DIL performed study concept and design. JH and ND performed analysis and acquired data. All authors interpreted data and provided the study materials; and all authors read, revised, and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
All authors of the manuscript are employees of Foundation Medicine, Inc., which is a wholly owned subsidiary of Roche and receives stock from Roche.
Ethics approval and consent to participate
Approval for this study was obtained from the Western Institutional Review Board Protocol No. 20152817.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Huang, R.S.P., Haberberger, J., Murugesan, K. et al. Clinicopathologic and genomic characterization of PD-L1-positive uterine cervical carcinoma. Mod Pathol 34, 1425–1433 (2021). https://doi.org/10.1038/s41379-021-00780-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41379-021-00780-3
This article is cited by
-
Real-world multicenter study of immune checkpoint inhibitors in advanced cervical cancer across HPV-associated and HPV-independent subtypes
International Journal of Clinical Oncology (2026)
-
Systemic immune-inflammatory index predict short-term outcome in recurrent/metastatic and locally advanced cervical cancer patients treated with PD-1 inhibitor
Scientific Reports (2024)
-
Metabolism-associated molecular classification of cervical cancer
BMC Women's Health (2023)
-
Comparison of PD-L1 expression and MMR status between primary and matched metastatic lesions in patients with cervical cancer
Journal of Cancer Research and Clinical Oncology (2023)
