
Abstract
Eosinophilic, solid and cystic (ESC) renal cell carcinoma (RCC) is characterized by a solid and cystic architecture with cells showing abundant eosinophilic cytoplasm with hobnail arrangement and a cytokeratin 7-negative/cytokeratin 20-positive immunophenotype. Recent studies have suggested that bi-allelic events affecting TSC genes might play an important role for such tumors. However, only indirect evidence of the clonal origin of TSC mutation has been gathered so far. Therefore, in this paper we aimed to perform multi-regional tumor sampling molecular analysis in four ESC RCC cases that had been completely embedded, three sporadic and one occurring in a patient with tuberous sclerosis complex (TSC). Histologically, the 4 cases showed cystic and solid architecture and cells with abundant eosinophilic cytoplasm with cytoplasmic stippling and round to oval nuclei. Immunohistochemistry showed at least focal expression of cytokeratin 20 in all tissue samples and negative cytokeratin 7, as well as diffuse positivity for S100A1 and at least focal expression of cathepsin K in three out of four cases. The sporadic cases showed the same somatic TSC1 mutations in all tissue samples analyzed, while the TSC-associated case showed the same TSC1 alteration in both normal tissue and all tumor samples analyzed, proving the germline nature of the alteration. In conclusion, our data demonstrate that clonal TSC loss is a key event in ESC RCC and support considering ESC RCC as an entity given its distinct morphologic, immunophenotypical and molecular characteristics.
Similar content being viewed by others

Introduction
Patients with tuberous sclerosis complex (TSC) are at high risk for developing different kinds of tumors involving multiple organs (subependymal giant cell tumors of the brain, lymphangioleiomyomatosis of the lung, angiomyolipoma of the kidney and liver, angiofibroma of the skin); eventually, up to 4% of them are diagnosed with renal cell carcinoma (RCC) [1]. In this setting, three morphological groups of RCC have been described: one characterized by tumors similar to RCC with smooth muscle stroma, another with chromophobe-like features and a third showing eosinophilic-macrocystic histology [2]. The latter group of tumors has been then reported in patients without TSC by Trpkov et al. and has been defined eosinophilic solid and cystic (ESC) RCC [3]. Such neoplasms show solid and cystic architecture with tumor cells characterized by voluminous eosinophilic cytoplasm with granular cytoplasmic “stippling” and usually demonstrate at least focal expression of cytokeratin 20 (CK20). The majority of tumors that have been described occurred in adult females, were monofocal and showed an indolent behavior [3, 4].
However, ESC RCC has also been reported in younger individuals (less than 35 years of age) and males; moreover, multifocal tumors and at least two metastatic cases have been recorded [5,6,7].
Recently, ESC RCC has been demonstrated to harbor somatic TSC1 or TSC2 mutations, which appear to be a key alteration in these tumors [6, 8, 9]. However, it has been noted that a subset of clear-cell RCCs from the Cancer Genome Atlas cohort harbor TSC1 or TSC2 mutations, although biallelic inactivation seems to be very uncommon [9, 10]. In addition, relatively few cases have been sequenced for TSC1 or TSC2 in both neoplastic and normal tissue, to confirm their sporadic nature. Finally, only indirect evidence suggests the clonal nature of TSC1/TSC2 mutations, as multi-regional analysis has never been performed so far in such tumors.
Therefore, the aim of this work was to prove the clonal nature of TSC mutations in ESC RCC by multi-regional tumor sampling next generation sequencing (NGS). In addition, this study aims to provide an expanded immunohistochemical analysis of ESC RCC.
Materials and methods
Cases search and immunohistochemistry
The archives of one of the authors’ institution (GM) were searched for unclassified RCCs with eosinophilic histology. All hematoxylin and eosin (HE) and immunohistochemistry (IHC) slides were reviewed to confirm the diagnosis according to reported ESC RCC criteria [3, 4].
IHC stains were performed on a Ventana BenchMark XT platform (Ventana Medical Systems, Tucson, Arizona, USA) with appropriate controls using the antibodies listed in Table 1.
IHC staining for Cytokeratin 20 was performed on multiple tumor samples for each case; the other reactions were performed on available unstained sections. IHC results were considered “negative” if <5% of cells were stained, “focal” if 5% to 25% of cells were stained, and “positive” if >25% of cells were reactive.
Fluorescence in situ hybridization (FISH)
Fluorescence in situ hybridization (FISH) was carried out on all cases using dual color break apart TFE3 and TFEB probes (Cytotest Inc, Rockville, MD 20850, USA) as previously described [11]. Briefly, 3 µm sections were cut from formalin-fixed paraffin-embedded tissue blocks and mounted on positively charged slides. The slides were dried for one hour at 60 °C then deparaffinized, rehydrated and fixed in methanol/acetic acid 3:1 for 5 min. Pretreatment was performed at 85 °C for 30 min with 0,1 citrate buffer (pH6) solution followed by pepsin (4 mg./ml in 0.9% NaCl, pH 1,5) treatment for 8 min at 37 °C. After washing and dehydration, 10 µl probe was applied on selected area and sealed with rubber cement. Denaturation was assessed by incubating the slides at 80 °C for 10 min in a humidified atmosphere (Thermobrite System) followed by hybridization overnight at 37 °C. The rubber cement and the cover slip were removed and the slides were washed in 2X SSC/0,3% NP40 for 15 min at room temperature and then at 72 °C for 2 min. Next, the tissue sections were counterstained with DAPI antifade (Prolong Gold Antifade Reagent Life Technologies) and examined under an X60- X100 oil immersion objective using an Olympus BX61 fluorescence microscope equipped with filters that visualize the different wavelengths of the fluorescent probe. Scoring was performed by an experienced pathologist (AC). At least 100 neoplastic non-overlapping nuclei were included in the scoring. To avoid false positive results due to nuclear truncation, cells with a single fluorescent signal were not evaluated.
DNA extraction
Sections (5 µm thick) were cut from multiple FFPE blocks from tumor samples and manually microdissected to isolate a high percentage of neoplastic cells (>50%). Normal renal parenchyma was manually microdissected from the edge of specimens. Sections were treated with xylene and 100% ethanol to remove paraffin and then DNA was isolated using the GeneRead DNA FFPE kit (Qiagen, Hilden, Germany, http://www.qiagen.com Cat. n. 180134). DNA amount and quality were identified using NanoVue Plus Spectrophotometer (BioChrom Ltd) following the manufacturer’s instructions.
Library preparation and deep amplicons sequencing
Deep sequencing of the whole coding region of 17 kidney-cancer-related genes was performed with a custom panel created using the Ampliseq Designer pipeline (Thermo Fisher Scientific). The genes included: TSC1, TSC2, MTOR, AKT1, PIK3CA, PTEN, SDHB, FH, VHL, SETD2, BAP1, PBRM1, MET, FLCN, SMARCA4, SMARCB1, TCEB1. Briefly, we used 30 ng (10 ng per pool) of genomic DNA to amplify the 689 amplicons included in the panel according to the manufacturer’s manual. Primer sequences were partially digested and ligated to adapters and barcodes. Libraries purification was performed using Agencourt® AMPure® XP (Beckman Coulter). Purified libraries were amplified for a total of five cycles. Libraries obtained were quantified and quality assessed using the Agilent D100 High Sensitivity Kit (Agilent Technologies) on the 4200 Tape Station instrument (Agilent Technologies) and then pooled to equimolar concentrations. Emulsion PCR and sample enrichment were performed using the Ion PGM Hi-Q View OT2 kit (Thermo Fisher Scientific) on an Ion One touch 2DX (Thermo Fisher Scientific) instrument according to the manufacturer’s instruction. Briefly, an input concentration of DNA library obtained with the first amplification step was added to the emulsion PCR master mix and the Ion sphere particles (ISPs) and a double phase (oil/water) PCR was performed. Then ISPs were recovered and template positive ISPs were enriched using Dynabeads MyOne Streptavidin C1 beads (Thermo Fisher Scientific). 318 Chips V2 (Thermo Fisher Scientific) were used to sequence samples on Ion torrent PGM DX (Thermo Fisher Scientific) using Ion PGM Hi-Q View Sequencing kit (Thermo Fisher Scientific) following the manufacturer’s instructions.
Variant calling
Data from the PGM sequencing were initially processed using the Ion Torrent platform-specific software (Torrent Suite AD 5.6.4) to generate sequence reads, alignment of the reads on the reference genome Hg19, trim adapter sequences, filter and remove poor signal-profile reads. The variant calling from the sequencing data was generated using the Variant Caller plugin.
To provide reliable somatic variant analysis were considered suitable only samples with more than 400,000 reads, an average coverage >500X and a coverage uniformity >95%. We also applied the following filters to the Variant Caller plugin: a minimum allele frequency value of 2% and a minimum phred quality score of 30. We also analyzed data uploading.bam files on the Ion Reporter 5.10 software (Thermo Fisher Scientific) in order to provide variants annotation and Copy Number Variations (CNVs) analysis. To provide a reliable CNVs analysis we built an amplicons baseline by analyzing with the same panel 15 healthy FFPE spleen samples processed in our laboratory during the past three years. Variants annotations was also made using the Ensembl Variant Effect Predictor pipeline of the Wellcome Trust Sanger institute [12] as second database check. Filtered variants were visually examinated using the Integrative Genomic Viewer (IGV) tool to test their level of quality and to confirm the variant presence on both “+” and “−“ strand. The clinical relevance (pathogenicity) of the annotated variants was assessed using ClinVar database (NCBI) and LOVD (IARC).
Results
Clinicopathological and morphological features
Four ESC RCC cases were retrieved, which had been completely embedded (4, 9, 1 and 15 blocks respectively). Clinicopathological features are summarized in Table 2.
Three patients did not have clinical stigmata suspicious for tuberous sclerosis. Medical history of the fourth patient revealed tuberous sclerosis diagnosed at the age of 14, Hashimoto thyroiditis, hypertension, dyslipidemia and uterine fibromatosis; the patient underwent multiple resections of bilateral renal angiomyolipomas (AMLs). The patient showed also a second small, separated nodule 1 cm in diameter and microscopic perilesional proliferations that resulted to be AML.
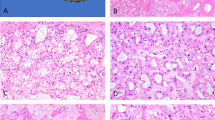
Tumors were exophytic and solitary (Fig. 1A), well circumscribed with a tan color and cystic and/or solid growth (Fig. 1B).

A MR image of ESC RCC1; B Gross features of ESC RCC1, showing brownish color and prevalently cystic aspect; C Histologically, ESC RCC1 recapitulated the macroscopic features with macrocysts and microcysts admixed with more solid foci; D ESC RCC2 showed overlapping histological features with ESC RCC1, with cystic areas showing septa lined by cells with voluminous eosinophilic cytoplasm with cytoplasmic “stippling”; E ESC RCC3 was characterized by a solid growth with medium-sized neoplastic cells with sparse histiocytes; F ESC RCC4 also showed cells with voluminous eosinophilic cytoplasm, often vacuolated, and sparse macrophages and lymphocytes.
Histologically, ESC RCC1 and ESC RCC2 appeared to be very similar and were characterized by microcysts and macrocysts (Fig. 1C) with variably thick septa lined by cells with hobnail arrangements and abundant eosinophilic, stippled and vacuolated cytoplasm and round to oval nuclei (Fig. 1D). Cystic spaces were frequently admixed with more solid foci showing a nested architecture with cells showing similar features. Macrophages and sparse lymphocytes were readily appreciable; multinucleated neoplastic cells were also present. ESC RCC3 was completely solid while ESC RCC4 showed a solid-microcystic architecture without macrocysts; however, the neoplastic cells were identical to those of the other two cases with abundant eosinophilic cytoplasm, often vacuolated, and round to oval nuclei (Fig. 1E, F). Follow up was available for three patients, who all were alive without evidence of disease after a median time of 34 months.
Immunophenotypical characteristics and FISH results
Immunohistochemistry results are shown in Table 3; representative immunostains are shown in Fig. 2. We found CK20 expressed at least focally in all tumor blocks tested (5–20%), while CK7 resulted always negative. S100A1 was positive, whereas CD117 and Parvalbumin were negative in all four cases. Interestingly, Cathepsin K expression was observed at least focally in 3 out of 4 cases. FH and SHDB showed maintained expression. Other positive stains included PAX8, CK8-18 and vimentin; negative stains included CAIX and melanocytic markers HMB45, SOX10, MART1/Melan A and S100.

Representative IHC stains for PAX8 (A), CK20 (B), CK8-18 (C), Vimentin (D), S100A1 (E) and Cathepsin K (F).
None of the tumors showed TFE3 or TFEB gene alterations (neither rearrangement nor amplification).
Molecular analysis
Multi-regional tumor sampling NGS of ESC RCC1, 2 and 3 showed deleterious frameshift mutations in the TSC1 gene in all tumor blocks (ESC RCC1: c.324delT, p.Gln109fs; ESC RCC2: c.1561delT, p.Ser521fs; ESC RCC3: c.2511_2512insAAAC, p.Ser838fs) (Table 4). As the TSC1 mutations were not present in the normal tissue analyzed we have assumed them as somatic events. Interestingly, we also found a somatic deleterious frameshift causing mutation in the MTOR gene in ESC RCC1 (c.6342_6346delGCTGC, p.Gln2114fs).
ESC RCC4 was associated with multiple capsular AMLs, consistent with TSC. As expected, a heterozygous deleterious mutation in an intronic splice-site of TSC1 gene (c.1997+1G>A) was found in the normal tissue, thus confirming the presence of a germline involvement, and in a capsular AML as well. The subsequent multi-regional tumor analysis showed the same mutation in all samples but with a significantly increased allele fraction, suggesting a second mutational event on the wild-type allele and subsequent loss of heterozygosity of the mutational spot. To further investigate on the presence of a bi-allelic event in the mutated TSC1 loci, we performed CNVs analysis using a custom-made baseline. As expected from the high allele fraction reported, in ESC RCC4 we observed bi-allelic event remarked by CNV analysis in terms of ploidy dropdown. The same information has been assessed for case 1 and 2, revealing also in those cases a bi-allelic event, consistent with the high variant allele fraction reported. No copy number alterations were observed in case 3, suggesting that there could be a second event that contributes to pathogenesis outside from the observed alterations of our NGS panel.
Discussion
In this study, we performed a multi-regional tumor sampling NGS study of ESC RCC and demonstrated the consistency of TSC1 mutation in all tumor samples analyzed, proving its clonal origin and providing further evidence that loss of function of TSC genes is a constant finding in ESC RCC. Also, we provide an expanded immunohistochemical analysis of ESC RCC confirming that CK20 is expressed at least focally in all tumor samples evaluated.
ESC RCC has been recently proposed as a novel tumor entity given its morphological, immunohistochemical (CK20+/CK7− immunoprofile) and molecular characteristics defined by bi-allelic mutations in TSC genes [13]. Most of the cases reported in the Literature are sporadic, although few tumors with the same characteristics have been reported in patients with TSC [3,4,5, 9, 14]. The great majority of the cases have been described in female patients with a wide age range distribution as solitary, low-stage tumors, even though rare cases with metastasis have been observed [3,4,5,6,7, 14]. Also in our series, all tumors occurred in female patients, of whom three did not show clinical stigmata of TSC while one was diagnosed with TSC at the age of 14. All tumors were solitary and low-stage, with well-defined borders and a solid and/or cystic macroscopic appearance. Microscopically, they showed the typical morphologic appearance described in previous works [3, 4], characterized by solid and/or cystic growths with cells with voluminous eosinophilic stippled cytoplasm and round to irregular nuclei, without necrosis or mitotic activity. Variable degree of cytoplasmic vacuolization was also present, as well as interspersed macrophages and small lymphoid elements.
One of the peculiar aspects of ESC RCC is the IHC reactivity for CK20 in about 85% of cases, generally associated with negative CK7 [3, 4]. Although negative CK20 has been reported in around 15% of cases, it might be possible that such result can be due to the fact that most of cases described in previous works are consultation cases with limited material available for IHC. We had the opportunity to test CK20 in multiple tumor blocks of our wholly embedded cohort and found at least focal CK20 expression in all of them; moreover, CK7 resulted always negative in our cases. We also observed at least focal cathepsin K expression in 3 cases, already described by Palsgrove et al. [9]. As these authors suggested, such finding can be explained by the lack of function of TSC, leading to constitutive activation of mTORC1 and dysregulation of multiple cellular pathways. Specifically, increased mTORC signaling secondary to TSC mutations may increase the expression of cathepsin K, a lysosomal cysteine protease typically overexpressed in perivascular epithelioid cell tumors (PEComas) [15,16,17] and pulmonary lymphangioleiomyomatosis [18], also harboring TSC genes inactivation and mTOR overexpression, as well as MiT family translocation RCCs [19,20,21]. Interestingly, we found a diffuse positivity for S100A1, which may point to oncocytoma in the differential diagnosis [22]. Parvalbumin can be of help in the differential diagnosis, since it is expressed by the majority chromophobe carcinomas [23], while resulted negative in all of our ESC RCC cases. We underline the importance of immunohistochemical testing for FH and SDHB protein expression, especially in the younger population, as it has been demonstrated that more than three-fourths of previously unclassified oncocytic pediatric RCCs are either fumarate hydratase-deficient, succinate dehydrogenase-deficient, or ESC RCC harboring TSC1 or TSC2 mutations [5].
From the molecular point of view, the first data on TSC mutations in ESC RCC were provided by two works from the University of Chicago and the Johns Hopkins University. In the first paper, Parilla et al. demonstrated biallelic TSC2 mutations in two cases of ESC RCC occurring in adult women (33 and 65 years) and confirmed the somatic nature of these alterations in one case [6]. In the second paper, Palsgrove et al. reported mutations in TSC1 or TSC2 in 6 of 6 ESC RCC in adult patients and, notably, in 8 of 9 ESC RCC found in younger patients (<35 years), expanding the clinical spectrum of ESC RCC to the young population [9]. Although they could not perform TSC analysis on normal tissue in most cases, such mutations were considered to be bona fide somatic on clinical grounds (absence of renal AML, skin lesions and seizures). Importantly, these authors confirmed the presence of somatic TSC mutations also in a case with metastases, demonstrating the malignant potential of ESC RCC. Notably, the latter case responded to treatment with an mTOR inhibitor, underlying the importance of TSC mutations also as predictive markers. In another important study, Mehra et al. found somatic bi-allelic loss of the TSC1 or TSC2 gene in 6 of 7 ESC RCC cases: of these, 3 cases showed biallelic mutation, while the other 3 cases showed a mutation plus loss of heterozygosity. Similarly to our data in the sporadic cases, such alterations were not identified in germline DNA obtained from adjacent non-neoplastic renal parenchyma [8]. As pointed out by these authors, although accumulated evidence, even though indirect, supports the clonal nature of the TSC1/2 mutations, multi-regional analysis has never been performed so far [8]. At variance with their study, in our work we were able to extensively analyze our sporadic ESC RCC in multiple tumor blocks. In all cases, we found the same pathogenetic TSC1 mutation throughout all the tissue samples, providing strong evidence of the clonal nature of TSC alteration in ESC RCC. Differently from Mehra et al., we did not find mutations in TSC2 gene.
TSC mutations have also been reported in other RCC types, including RCC with leiomyomatous stroma [24]; moreover, mTOR activation has been shown in both primary and metastatic clear cell renal cell carcinoma [25]. Thus, the detection of TSC mutations in RCC does not establish the diagnosis of ESC RCC per se [9]. However, TSC bi-allelic loss within RCC is a rare event, occurring in only 2 out of 39 RCC cases harboring TSC mutations that had been found within The Cancer Genome Atlas cohort (TCGA; n = 1316 patients) [8, 10]. In this regard, CNVs analysis demonstrated a bi-allelic event in the mutated TSC1 loci in two out of three sporadic ESC RCC cases. Such finding was not completely unexpected, given the high allele fraction reported for mutated TSC1 in both cases. The same was true for the TSC-associated ESC RCC. Regarding ESC RCC3, no copy number alterations were observed, suggesting that there could be a second event that could not be detected with our NGS panel, including promoter methylation. Besides TSC mutations, it should be kept in mind that it is possible that mutations in other members of the MTOR pathway may result in the same phenotype. In this regard, we report that a second mutation other than TSC1 was found in our ESC RCC1 case. Although its significance remains unknown, such frameshift mutation could result in MTOR loss of function. However, given the diffuse Cathepsin K expression observed in ESC RCC1, we postulate that such MTOR mutation only affected one allele, leaving the other allele intact. This hypothesis is supported by the fact that we were not able to demonstrate loss of heterozygosity for the mutated MTOR locus in our experiments.
Chen et al. reported mutations in TSC2 or MTOR genes in a group of 7 renal tumors characterized by nested architecture with tumor cells showing eosinophilic and vacuolated cytoplasm and frequent calcification [26], suggesting that activating mutations of MTOR gene can lead to similar tumor phenotype as biallelic TSC2 inactivating mutations. At variance with ESC RCC, these tumors showed a CK20‒/CK7‒ phenotype, were predominantly nested and lacked admixed aggregates of foamy histiocytes and lymphocytes. More recently, Tjota et al. reported a series of 18 cases of renal eosinophilic tumors with unusual morphology and alterations of TSC1, TSC2 or MTOR genes and divided them into 3 groups. Neoplasms belonging to the third group showed solid, cystic and papillary architecture, CK20 and vimentin expression, were negative for CK7 in all but one case and harbored TSC1 or TSC2 alterations [27]. Such cases appear to us to be ESC RCC on a morphological, immunophenotypical and molecular ground. The remaining tumors showed a solid architecture, vimentin−/CK20−/CK7+ phenotype and harbored TSC2 (group 1) and TSC1, TSC2 or MTOR (group 2) alterations. The expression of vimentin and, although focal, of CK20 that we demonstrated in all tumor blocks examined together with negative CK7 can help distinguishing ESC RCC from other eosinophilic neoplasms including those described by Chen et al. [26] as well as those reported by Tjota et al. in group 1 and 2, which in turn share similar features with the recently described “high grade oncocytic renal tumors” (HOT) [28] and low grade oncocytic tumors of kidney (LOT) [29], respectively.
Recently, The Genitourinary Pathology Society (GUPS) proposed the name “eosinophilic vacuolated tumor” (EVT) for the entity initially described by He et al. [28] and Chen et al. [26] as “high grade oncocytic renal tumors” and “sporadic RCC with eosinophilic and vacuolated cytoplasm”, respectively. Of note, all documented cases of EVT so far have shown indolent behavior, at variance with ESC RCC which demonstrated metastatic potential in rare cases [13]. The macroscopic, morphological and immunophenotypical differences between ESC RCC and EVT underscore the fact that mutation of TSC and/or MTOR genes do not appear to be specific for any entity but may help in the differential diagnosis if proven to be either biallelic/germinal, or within a constellation of morphology and IHC profile.
A limitation of this study is the small number of the cases included. It must be kept in mind, however, that ESC RCC is a rare neoplasms with an estimated incidence of 0.2% only 1 to 2 cases documented through focused review in the majority of institutions, some representing large uropathology practices [4].
In summary, we performed a multiregional tumor sampling NGS analysis and an extensive immunohistochemical study of both sporadic and TSC-associated ESC RCCs, proving that TSC loss is a clonal event in these neoplasms which should be considered as a distinct entity given their morphologic, immunophenotypical and molecular characteristics. Further studies are needed to determine the relationship between ESC RCC and the spectrum of sporadic RCC characterized by TSC1/TSC2 or MTOR alterations.
Data availability
The data in this manuscript are available from the corresponding author upon reasonable request.
References
Yang P, Cornejo KM, Sadow PM, Cheng L, Wang M, Xiao Y, et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014;38:895–909.
Guo J, Tretiakova MS, Troxell ML, Osunkoya AO, Fadare O, Sangoi AR, et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014;38:1457–67.
Trpkov K, Hes O, Bonert M, Lopez JI, Bonsib SM, Nesi G, et al. Eosinophilic, solid, and cystic renal cell carcinoma: clinicopathologic study of 16 unique, sporadic neoplasms occurring in women. Am J Surg Pathol. 2016;40:60–71.
Trpkov K, Abou-Ouf H, Hes O, Lopez JI, Nesi G, Comperat E, et al. Eosinophilic solid and cystic renal cell carcinoma (ESC RCC): further morphologic and molecular characterization of ESC RCC as a distinct entity. Am J Surg Pathol. 2017;41:1299–308.
Li Y, Reuter VE, Matoso A, Netto GJ, Epstein JI, Argani P. Re-evaluation of 33 “unclassified” eosinophilic renal cell carcinomas in young patients. Histopathology. 2018;72:588–600.
Parilla M, Kadri S, Patil SA, Ritterhouse L, Segal J, Henriksen KJ, et al. Are sporadic eosinophilic solid and cystic renal cell carcinomas characterized by somatic tuberous sclerosis gene mutations? Am J Surg Pathol. 2018;42:911–7.
McKenney JK, Przybycin CG, Trpkov K, Magi-Galluzzi C. Eosinophilic solid and cystic renal cell carcinomas have metastatic potential. Histopathology. 2018;72:1066–7.
Mehra R, Vats P, Cao X, Su F, Lee ND, Lonigro R, et al. Somatic Bi-allelic loss of TSC genes in eosinophilic solid and cystic renal cell carcinoma. Eur Urol. 2018;74:483–6.
Palsgrove DN, Li Y, Pratilas CA, Lin MT, Pallavajjalla A, Gocke C, et al. Eosinophilic solid and cystic (ESC) renal cell carcinomas harbor TSC mutations: molecular analysis supports an expanding clinicopathologic spectrum. Am J Surg Pathol. 2018;42:1166–81.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49.
Calio A, Brunelli M, Segala D, Pedron S, Doglioni C, Argani P, et al. VEGFA amplification/increased gene copy number and VEGFA mRNA expression in renal cell carcinoma with TFEB gene alterations. Mod Pathol. 2019;32:258–68.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70.
Trpkov K, Williamson SR, Gill AJ, Adeniran AJ, Agaimy A, Alaghehbandan R, et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol. 2021. https://doi.org/10.1038/s41379-021-00737-6.
Tretiakova MS. Eosinophilic solid and cystic renal cell carcinoma mimicking epithelioid angiomyolipoma: series of 4 primary tumors and 2 metastases. Hum Pathol. 2018;80:65–75.
Martignoni G, Bonetti F, Chilosi M, Brunelli M, Segala D, Amin MB, et al. Cathepsin K expression in the spectrum of perivascular epithelioid cell (PEC) lesions of the kidney. Mod Pathol. 2012;25:100–11.
Caliò A, Mengoli MC, Cavazza A, Rossi G, Ghimenton C, Brunelli M, et al. Cathepsin K expression in clear cell “sugar” tumor (PEComa) of the lung. Virchows Arch. 2018;473:55–59.
Martignoni G, Pea M, Zampini C, Brunelli M, Segala D, Zamboni G, et al. PEComas of the kidney and of the genitourinary tract. Semin Diagn Pathol. 2015;32:140–59.
Chilosi M, Pea M, Martignoni G, Brunelli M, Gobbo S, Poletti V, et al. Cathepsin-k expression in pulmonary lymphangioleiomyomatosis. Mod Pathol. 2009;22:161–6.
Martignoni G, Pea M, Gobbo S, Brunelli M, Bonetti F, Segala D, et al. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol. 2009;22:1016–22.
Martignoni G, Gobbo S, Camparo P, Brunelli M, Munari E, Segala D, et al. Differential expression of cathepsin K in neoplasms harboring TFE3 gene fusions. Mod Pathol. 2011;24:1313–9.
Caliò A, Brunelli M, Segala D, Pedron S, Tardanico R, Remo A, et al. t(6;11) renal cell carcinoma: a study of seven cases including two with aggressive behavior, and utility of CD68 (PG-M1) in the differential diagnosis with pure epithelioid PEComa/epithelioid angiomyolipoma. Mod Pathol. 2018;31:474–87.
Rocca PC, Brunelli M, Gobbo S, Eccher A, Bragantini E, Mina MM, et al. Diagnostic utility of S100A1 expression in renal cell neoplasms: an immunohistochemical and quantitative RT-PCR study. Mod Pathol. 2007;20:722–8.
Martignoni G, Pea M, Chilosi M, Brunelli M, Scarpa A, Colato C, et al. Parvalbumin is constantly expressed in chromophobe renal carcinoma. Mod Pathol. 2001;14:760–7.
Shah RB, Stohr BA, Tu ZJ, Gao Y, Przybycin CG, Nguyen J, et al. “Renal cell carcinoma with leiomyomatous stroma” harbor somatic mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): clinicopathologic and molecular characterization of 18 sporadic tumors supports a distinct entity. Am J Surg Pathol. 2020;44:571–81.
Schultz L, Chaux A, Albadine R, Hicks J, Kim JJ, De Marzo AM, et al. Immunoexpression status and prognostic value of mTOR and hypoxia-induced pathway members in primary and metastatic clear cell renal cell carcinomas. Am J Surg Pathol. 2011;35:1549–56.
Chen Y-B, Mirsadraei L, Jayakumaran G, Al-Ahmadie HA, Fine SW, Gopalan A, et al. Somatic mutations of TSC2 or MTOR characterize a morphologically distinct subset of sporadic renal cell carcinoma with eosinophilic and vacuolated cytoplasm. Am J Surg Pathol. 2019;43:121–31.
Tjota M, Chen H, Parilla M, Wanjari P, Segal J, Antic T. Eosinophilic renal cell tumors with a TSC and MTOR gene mutations are morphologically and immunohistochemically heterogenous. Am J Surg Pathol. 2020;44:943–54.
He H, Trpkov K, Martinek P, Isikci OT, Maggi-Galuzzi C, Alaghehbandan R, et al. “High-grade oncocytic renal tumor”: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018;473:725–38.
Trpkov K, Williamson SR, Gao Y, Martinek P, Cheng L, Sangoi AR, et al. Low‐grade oncocytic tumour of kidney (CD117‐negative, cytokeratin 7‐positive): a distinct entity? Histopathology. 2019;75:174–84.
Funding
This study was partially supported by the Italian Association for Cancer Research (AIRC) grants 5X1000 2018 Id. 21147 (LM), IG 2017 Id. 19920 (LM).
Author information
Authors and Affiliations
Contributions
EM organized the data and wrote the manuscript; GS, SL, SS performed molecular analysis; MM performed immunohistochemistry; AC, MB performed FISH; SG helped in the organization of results; PV, NT provided reagents; DS provided part of the illustrations; GJN provided edits on the manuscript; LM provided funding; GZ provided facility for molecular analysis; GM provided samples and oversaw all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
The study was conducted in accordance with the ethical standards of the institutional research committee (authorization 1745CESC on behalf of MeTTOs group) and with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Munari, E., Settanni, G., Caliò, A. et al. TSC loss is a clonal event in eosinophilic solid and cystic renal cell carcinoma: a multiregional tumor sampling study. Mod Pathol 35, 376–385 (2022). https://doi.org/10.1038/s41379-021-00816-8
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41379-021-00816-8
This article is cited by
-
mTOR eosinophilic renal cell carcinoma: a distinctive tumor characterized by mTOR mutation, loss of chromosome 1, cathepsin-K expression, and response to target therapy
Virchows Archiv (2023)
-
TFE3 and TFEB-rearranged renal cell carcinomas: an immunohistochemical panel to differentiate from common renal cell neoplasms
Virchows Archiv (2022)
