
Abstract
Dopamine modulates corticostriatal plasticity in both the direct and indirect pathways of the cortico-striato-thalamo-cortical (CSTC) loops. These gradual changes in corticostriatal synaptic strengths produce long-lasting changes in behavioral responses. Under normal conditions, these mechanisms enable the selection of the most appropriate responses while inhibiting others. However, under dysregulated dopamine conditions, including a lack of dopamine release or dopamine signaling, these mechanisms could lead to the selection of maladaptive responses and/or the inhibition of appropriate responses in an experience-dependent and task-specific manner. In this review, we propose that preventing or reversing such maladaptive synaptic strengths and erasing such aberrant “memories” could be a disease-modifying therapeutic strategy for many neurological and psychiatric disorders. We review evidence from Parkinson’s disease, drug-induced parkinsonism, L-DOPA-induced dyskinesia, obsessive-compulsive disorder, substance use disorders, and depression as well as research findings on animal disease models. Altogether, these studies allude to an emerging theme in translational neuroscience and promising new directions for therapy development. Specifically, we propose that combining pharmacotherapy with behavioral therapy or with deep brain stimulation (DBS) could potentially cause desired changes in specific neural circuits. If successful, one important advantage of correcting aberrant synaptic plasticity is long-lasting therapeutic effects even after treatment has ended. We will also discuss the potential molecular targets for these therapeutic approaches, including the cAMP pathway, proteins involved in synaptic plasticity as well as pathways involved in new protein synthesis. We place special emphasis on RNA binding proteins and epitranscriptomic mechanisms, as they represent a new frontier with the distinct advantage of rapidly and simultaneously altering the synthesis of many proteins locally.
Similar content being viewed by others

The idea of altering memory or synaptic strength as therapy for neurological and psychiatric disorders has been explored previously. For example, reversing aberrant synaptic long-term depression (LTD) has been proposed as a therapy for Fragile X syndrome [1,2,3]. Altering fear memory during reconsolidation in post-traumatic stress disorder (PTSD) patients as a therapy represents another example [4, 5]. However, clinical trials based on these ideas have not been successful. Here we argue that circuit-specific correction of aberrant synaptic strength is essential for therapies based on such ideas. This review will focus on corticostriatal synaptic plasticity in the cortico-striato-thalamo-cortical (CSTC) loops. The reason for such a focus is that CSTC loops offer many good examples of how changes in synaptic strength can lead to both pathology and possible therapeutic solutions. These include clinical evidence of therapeutic benefits due to circuit-specific correction of aberrant synaptic strength, animal models of diseases with circuit-specific aberrant synaptic strength, and animal models of circuit specific correction of aberrant synaptic strength, although the existing literature lacks a systematic review devoted to such a focus. This review aims to fill such a gap. We will first briefly review CSTC loops, corticostriatal synaptic plasticity, and signaling mechanisms. Then we will review circuit-specific mechanisms and therapies relevant to various diseases.
Corticostriatal synaptic plasticity in the cortico-striato-thalamo-cortical loops and signaling mechanisms
According to the classic model of basal ganglia function [6,7,8,9], activity of D1 receptor-expressing striatal neurons in the direct ‘Go’ pathway (D1 neurons) increases excitation of cortical activity and facilitates movement. By contrast, activity of D2 receptor-expressing striatal neurons in the indirect ‘No-Go’ pathway (D2 neurons) inhibits cortical activity and movement [9,10,11]. However, recent studies suggest a more nuanced view of D2 neuron function; in vivo recording studies suggest that D2 neuron activity is also involved in initiating movement, discriminating between motor sequences, and altering motor sequences [12,13,14,15,16]. Nevertheless, direct manipulations of D2 neuron activity by optogenetics or chemogenetics clearly demonstrate their role in motor inhibition [9, 17]. Therefore, even though the circuit level function of the D2 neurons is to inhibit cortical neurons, D2 neuron activity is likely to play important roles in many motor acts and the expression of learned motor skills, as inhibition of specific cortical neurons through the D2 CSTC loops could always be important, even in movement initiation. On the other hand, specific alterations in striatal neuron activity or corticostriatal plasticity predominantly in the D1 or D2 pathway could lead to very distinct symptoms under certain pathological conditions, revealing the opposing yet cooperative roles of D1 and D2 loops, which will be discussed below.
At the cellular level, activation of dopamine receptors on striatal neurons modulates gating of ion channels and, therefore, acutely alters the intrinsic excitability of these neurons [18,19,20]. It is commonly understood in the field that activation of D1 receptors increases D1 neuron firing, whereas activation of D2 receptors decreases D2 neuron firing [19,20,21]. However, the literature on this topic, especially in vivo studies, is still limited. Moreover, there are also reports showing that striatal neurons form local synaptic connections through their local axon collaterals, thereby providing strong lateral inhibition on surrounding circuitry [22,23,24], suggesting a more complex picture of D1-D2 interactions at multiple levels.
Another important function of dopamine is to modulate corticostriatal plasticity in both the direct and indirect pathways [25,26,27,28,29]. Such a mechanism is able to produce cumulative and long-lasting changes in corticostriatal synapses which ensures persistent effects on behavior [30,31,32]. The role of dopamine in modulating corticostriatal plasticity fits well with the role of dopamine as the prediction error signal in reinforcement learning. Phasic increases and decreases of dopamine release relative to baseline are thought to encode positive and negative prediction error signals respectively [25, 33,34,35,36]. These “teaching signals” promote changes in corticostriatal synaptic strength, correcting errors in future responses. Therefore, the timing of regulated dopamine release accompanying activity at the relevant corticostriatal synapses is essential for causing changes in specific synapses to reinforce only the most relevant motor acts in a specific task while inhibiting the others.
It is often hypothesized that the D1 receptor is more sensitive to phasic increase in dopamine release (positive reward prediction error) but is not sensitive to phasic decrease in dopamine release (negative reward prediction error) [33, 37, 38]. This is presumably because the D1 receptor has low affinity for dopamine and is not activated at the baseline condition [33]. In contrast, the D2 receptor is more sensitive to phasic decrease in dopamine release (negative reward prediction error) but is not sensitive to phasic increase in dopamine release (positive reward prediction error) because the D2 receptor has high affinity for dopamine and is already saturated at the baseline dopamine level [33]. However, this hypothesis has been challenged for the lack of evidence on D1 versus D2 receptor affinity for dopamine under in vivo conditions [39,40,41,42]. We present below an alternative hypothesis: the effects of phasic dopamine signaling (prediction errors) on learning need to be consolidated, which requires new protein synthesis stimulated by high cAMP levels in D1 and D2 neurons.
Intracellularly, both the dopamine D1 and D2 receptors are strongly coupled to the cAMP pathway [43, 44]. Dopamine mainly stimulates cAMP production in D1 neurons and inhibits cAMP production in D2 neurons. Therefore, cAMP is elevated in D1 neurons during phasic increase in dopamine release (positive reward prediction error). In contrast, cAMP is elevated in D2 neurons during phasic decrease in dopamine release (negative reward prediction error). Thus, we hypothesize that reward prediction error signals elevate cAMP level in D1 neurons, promote LTP and new protein synthesis there and consolidate specific motor memories after learning associated with positive reward prediction error signals. Meanwhile, negative reward prediction error signals elevate cAMP level in D2 neurons, promote LTP and new protein synthesis there and consolidate specific motor memories after learning associated with negative reward prediction error signals.
The striatum is unique in the expression of the calcium/calmodulin (CaCaM)-independent adenylyl cyclase type 5 (AC5) [45,46,47]. This is distinct from other brain regions such as the hippocampus and cortex that predominantly express the CaCaM-activated cyclase, AC1 [48,49,50]. There is little or no AC1 expression in the adult striatum [45,46,47]. Therefore, cAMP production in adult striatum can be highly modulated by G-protein coupled receptors, relying less on CaCaM. This may explain why dopamine signaling plays such a dominant role in the induction and directionality of corticostriatal plasticity [27,28,29, 51]. Studies, including ours, suggest that the direction and magnitude of plasticity in D1 and D2 neurons are regulated by both the afferent activity and intracellular cAMP [28, 51,52,53]. For example, high concentrations of dopamine reduce cAMP via D2 receptor activation and facilitate LTD in the indirect pathway [51,52,53]. Conversely, low dopamine levels increase intracellular cAMP, favoring LTP in the indirect pathway [28, 51]. Based on the above hypothesis, LTP is likely more important in memory consolidation in the striatum, whereas LTD is more likely to play a role in short term memory or indirectly affects memory consolidation through its interactions with LTP.
Taken together, with the above mechanisms, dopamine activation of the D1 receptor during a specific motor response will favor corticostriatal LTP in the direct pathway and therefore future selection of such a response under the same context. In contrast, lack of dopamine activation of the D2 receptor during a specific motor response will favor corticostriatal LTP in the indirect pathway and therefore future inhibition of such a response under the same context. While these mechanisms are important for normal response selection and inhibition, under certain pathological conditions, the same mechanisms can become maladaptive; for instance, the almost complete lack of dopamine in Parkinson’s disease (PD), or unregulated dopamine release in L-DOPA-induced dyskinesia. In this review, we hypothesize that aberrant corticostriatal LTP in the D1 pathway is a key contributor to L-DOPA-induced dyskinesia, obsessive-compulsive disorder, and substance use disorders. In contrast, aberrant corticostriatal LTP in the D2 pathway is at least partially responsible for PD motor symptoms, drug-induced parkinsonism, and depression. Preventing or reversing aberrant corticostriatal LTP in the respective pathways could be therapeutic. We propose that combining pharmacotherapy with behavioral therapy or with deep brain stimulation (DBS) could potentially cause desired changes in selected circuits and synapses, and reverse aberrant corticostriatal plasticity. If successful, one significant advantage of correcting aberrant synaptic plasticity is that the therapeutic effects could be long lasting even after cessation of treatment. Potential molecular targets for the pharmacotherapy component of such therapeutic approaches include the cAMP pathway, synaptic proteins involved in synaptic plasticity, and pathways involved in new protein synthesis.
It is important to point out that the above discussions are limited to the role of phasic increase (reward prediction error) or decrease (negative prediction error) in dopamine release during learning. The role of phasic changes in dopamine is only meaningful if we also understand the role of tonic dopamine. Moreover, phasic decrease in dopamine signaling is certainly dependent on tonic dopamine signaling that precedes it. It is likely that phasic and tonic dopamine release are regulated differently and play distinct roles. At baseline condition, dopamine neurons fire spontaneously and asynchronously at low frequency [54, 55]. Because of the potent GABAergic inhibition, not all dopamine neurons fire spontaneously in the basal condition [56]. Additionally, not all action potentials lead to dopamine release. Only a small percentage of synaptic vesicles belong to the readily releasable pool which is only slowly replenished [57, 58]. Therefore, the tonic extracellular dopamine level is relatively stable and low. In contrast, dopamine neuron burst firing can generate phasic, short and fast dopamine transients. Moreover, burst firing is often synchronized across dopamine neurons which can overwhelm dopamine transporter’s reuptake activity, cause a strong phasic dopamine release, and potentially recruit additional distant receptors [42, 54, 55, 59, 60]. While tonic and phasic signals are generally considered to be distinct dopamine signaling mechanisms, the exact nature of tonic dopamine and its function are still not well defined in the literature. In addition to the above view of tonic dopamine caused by baseline spontaneous asynchronous low frequency dopamine neuron firing, some researchers view tonic dopamine as accumulation of phasic dopamine, therefore reflecting net rate of rewards [56]. In this view, tonic dopamine serves the important function of determining the optimal rate of responding of the animal in a particular environment, implying a tight coupling between motivational states and tonic dopamine [61]. Others also emphasize the potential negative impact of tonic dopamine on phasic dopamine. Tonic dopamine may blunt phasic dopamine signals due to either reduced contrast from the elevated baseline dopamine level or a reduction in receptor sensitivity [62, 63].
Related to the above topic, dopamine release may not always reflect dopamine neuron firing. One important mechanism is that striatal cholinergic interneurons can directly cause dopamine release independent of dopamine neuron firing [64]. These cholinergic interneurons, often referred to as tonically active neurons in primate striatum, fire spontaneously, and this spontaneous firing has been linked to acetylcholine release [65,66,67,68]. Furthermore, cortical and thalamic glutamatergic inputs help synchronize the firing of these cholinergic interneurons while dopamine inhibits acetylcholine transients, indicating that glutamate and dopamine serve as distinct, yet complementary, regulatory forces shaping cholinergic interneuron function and, in turn, physiological responses in the striatum [69,70,71]. However, under in vivo conditions, studies also suggest that dopamine dynamics and reward encoding may not depend on acetylcholine release [70, 71]. Extracellular dopamine and acetylcholine levels fluctuate and do not arise from direct local interactions between them within the striatum [70, 71]. These findings underscore the complexity of striatal signaling and highlight the need for additional research to reconcile these diverse observations.
Striatal cholinergic interneurons have an important impact on both corticostriatal LTD and LTP. The D2 receptor dependence of LTD induction in both D1 and D2 neurons seems due to D2 receptors on cholinergic interneurons [72] while deletion of D2 receptors on D2 striatal neurons has more limited impact on LTD induction [73]. In in vivo studies, it was reported that corticostriatal LTP is dependent on the coincidence of phasic dopamine activation and pauses in cholinergic interneurons [74, 75].
It is also conceivable that dopamine signaling could be subcellularly localized. However, our understanding is still very limited in this regard. There are only a few examples and suggestive evidence. Phosphodiesterase 10 A (PDE10A), the major cAMP PDE in mouse striatum, is localized at the plasma membrane and in dendritic spines close to postsynaptic densities and is associated with the A kinase anchoring protein (AKAP150), PKA, NR2A, NR2B, and PSD95. Affinity of PDE10A to the signaling complexes formed around AKAP150 could be reduced by PDE10A phosphorylation [76]. The regional distribution of DARPP-32 in the rat brain follows the general pattern of dopaminergic innervation; it appears to be concentrated in D1 neurons where it is localized in cell bodies, dendrites, axons, and nerve terminals [77]. Live imaging and computational models suggest maximal effects on cAMP production in secondary dendrites, due to segmental decrease of dendrite diameter. Thus, signaling from dendrites to nucleus is not inversely proportional to the distance [78]. With the development of many new tools, we expect to see much better understanding of subcellularly localized dopamine signaling in the near future.
All the above discussions also suggest that the traditional emphasis on dopamine signaling through volume transmission needs to be revised. Due to technical limitations, it was not possible to accurately measure extracellular dopamine levels close to the synapse or capture its dynamics in behaving animals. However, recently developed genetically-encoded dopamine sensors have dramatically improved the spatiotemporal resolution in measuring extracellular dopamine [79,80,81,82,83].
Aberrant corticostriatal synaptic plasticity in neurological and psychiatric disorders: loss of control
L-DOPA-induced dyskinesia (LID)
We will start with LID, the primary detrimental side-effect of PD therapy [84]. Although it has been extensively studied, the underlying mechanism is not well understood [85]. In our view, it is one of the best examples that can be explained by aberrant synaptic plasticity in the CSTC loops. LID refers to the abnormal involuntary movements produced by chronic dopamine replacement therapies in advanced stage PD patients [84]. In early-stage PD, L-DOPA is very effective in controlling PD motor symptoms. This is likely because the remaining dopamine terminals are still able to support regulated dopamine release with precise timing, and L-DOPA, as a dopamine precursor, helps to compensate for the loss of dopamine neurons and terminals [86,87,88,89]. In advanced stage PD, however, partially restored dopamine release due to L-DOPA therapy is very different from regulated dopamine release. Due to the significant loss of striatal dopamine terminals and the conversion of L-DOPA to dopamine by non-dopamine neurons, dopamine release is no longer physiological, lacking input dependent and properly regulated firing patterns [87, 90]. Moreover, it has been well documented in animal models of PD that the brain’s capacity for storage and clearance of dopamine is greatly impaired [89]. Therefore, in advanced stage PD, administration of L-DOPA will result in dopamine production and release, but it is no longer regulated with precise timing.
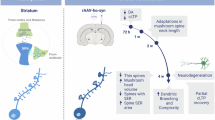
How will the effects of regulated and unregulated dopamine release on motor control differ? Striatum dependent learning is correlated with changes in corticostriatal synaptic strength [25, 91,92,93]. Phasic dopamine release is regarded as the prediction error signal, and therefore the teaching signal that causes changes in corticostriatal synaptic strength. Consequently, the CSTC loops can make predictions better in the future and facilitate the selection of the most appropriate responses [33] (Fig. 1). Therefore, the timing of regulated dopamine release is essential for causing changes in specific synapses to reinforce the relevant motor acts in a specific task while inhibiting the others [33,34,35,36]. In contrast, due to conversion of L-DOPA to dopamine by non-dopamine neurons in advanced stage PD upon L-DOPA treatment, unregulated dopamine release will likely cause changes in synapses that reinforce motor acts irrelevant to the specific task in our opinion (Fig. 1). Chronically, such aberrant synaptic plasticity could lead to many unwanted motor acts (dyskinesia). Indeed, studies on animal models that examine cellular mechanisms of LID have found aberrant corticostriatal plasticity [94]. In one example, LTP could be de-potentiated in non-dyskinetic rats but not in dyskinetic rats, and the D1 pathway was implicated [95]. In another example, a sub-population of D1 neurons showed abnormally high firing rates evoked by L-DOPA in dyskinetic mice [96]. In a related study from the same group, it was found that a subset of striatal D1 receptor expressing neurons (potentially memory engram cells specific for this type of dyskinesia) were mostly responsible for the dyskinesia as activation of these neurons induced dyskinesia even in the absence of L-DOPA while silencing of these neurons were therapeutic [97].

Dopamine modulates corticostriatal plasticity in both the direct “Go” (green) and indirect “NoGo” (red) pathways. Such modulation is usually limited to active corticostriatal synapses (task-specific). Gradual changes in corticostriatal synaptic strengths produce cumulative and long-lasting changes in response selection. Normal phasic dopamine release with precise timing causes changes in corticostriatal synaptic strengths in both “Go” and “NoGo” pathways so that the CSTC loops can facilitate the selection of the most appropriate responses (LTP in the “Go” pathway in task-relevant synapses) and inhibit the rest. Under no dopamine condition (e.g., in PD), excessive corticostriatal LTP in the “NoGo” pathway will lead to experience-dependent and task-specific inhibition. In contrast, unregulated dopamine release (e.g., in advanced stage PD upon L-DOPA treatment) will cause changes in synapses that reinforce motor acts irrelevant to the specific task. Chronically, such aberrant synaptic plasticity could lead to many unwanted motor acts (e.g., dyskinesia).
Obsessive-compulsive disorder (OCD)
OCD is characterized by intrusive thoughts and repetitive behaviors [98,99,100,101]. Functional neuroimaging studies have consistently revealed aberrant activity within the CSTC loops in individuals with OCD [102, 103]. The frontal cortex, striatum, globus pallidus, substantia nigra and thalamus all belong to the CSTC loops that connect discrete parts of the striatum and cortex [6]. These loops are central to both goal-directed actions and habits, which are shaped by striatum dependent learning with changes in corticostriatal synaptic strength as the underlying mechanism [104,105,106,107,108]. The sensorimotor loop, connecting the dorsolateral striatum (DLS) and the sensorimotor cortical areas, plays a more important role in habits and habit learning. In contrast, the more ventral loop, connecting the dorsomedial striatum (DMS) and the association cortices, serves functions related to more flexible goal-directed actions and reward learning. Human behavior and imaging studies have shown an impaired balance between goal-directed behavior and habit learning in OCD patients [109, 110].
Studies on animal models suggest that mutations in synaptic proteins can cause aberrant corticostriatal synaptic plasticity and lead to repetitive, stereotyped, habitual motor behaviors [111, 112]. The otherwise normal role of synaptic proteins in corticostriatal plasticity could become maladaptive with certain mutations and lead to aberrant corticostriatal synaptic plasticity, pathological habitual motor acts and impaired behavioral flexibility. For example, SAP90/PSD95-associated protein 3 (SAPAP3) is a postsynaptic scaffolding protein at excitatory synapses that is highly expressed in the striatum. Mice with genetic deletion of Sapap3 exhibit increased compulsive grooming behavior which are alleviated by a selective serotonin reuptake inhibitor [111], a first-line OCD treatment. Sapap3-mutant mice display defects in cortico-striatal synapses [112]. In the central striatum, postsynaptic responses to inputs from the secondary motor area (M2) were significantly higher in strength and reliability in mutants compared to wild-types, suggesting that increased M2-striatal inputs may contribute to both striatal hyperactivity and compulsive behaviors [112]. Furthermore, lentiviral-mediated expression of Sapap3 in the striatum rescues the synaptic and behavioral defects [111]. DBS that corrects such aberrant synaptic strength in the CSTC loops has been found to be therapeutic in these animal models [113].
Conversely, experimentally induced aberrant corticostriatal synaptic plasticity has been shown to induce OCD-like behaviors in animal models. For example, repeated optogenetic stimulation of the orbitofrontal cortex (OFC)-ventromedial striatum (VMS) projection was reported to progressively increase grooming that persisted even after stimulation cessation [114]. The progressive increase in grooming was correlated with a progressive increase in evoked firing of postsynaptic VMS cells. Furthermore, both increased grooming and evoked firing were reversed by chronic fluoxetine [114]. These data further support the causation of CSTC circuit dysregulation in OCD. However, these studies do not tell us whether abnormal D1 or D2 pathway activity in any of the specific CSTC loops would predict OCD-like behaviors.
Similar to OCD, studies of Tourette syndrome have also suggested that dysfunction in the basal ganglia circuit and corticostriatal synaptic plasticity may contribute to the disorder [115,116,117,118], though there is little direct evidence on synaptic plasticity.
Substance use disorders
Substance use disorders are increasingly recognized as a synaptic disease with maladaptive appetitive associative learning as one fundamental aspect [25, 119,120,121,122,123,124]. Instrumental action-outcome learning is important in establishing drug-seeking behavior [125,126,127,128,129,130]. Pavlovian associative learning is important in making otherwise neutral environmental cues acquire strong incentive values; and drug craving can be induced by conditioned environmental cues [34, 129, 131,132,133].
Almost all addictive drugs are known to either directly or indirectly increase dopamine signaling. The mesolimbic dopamine system, which originates from the ventral tegmental area (VTA) and projects mainly to the nucleus accumbens, is especially implicated in substance use disorders [119,120,121,122,123,124, 134]. Repeated drug exposure often leads to long-lasting changes in synaptic strengths in the VTA and the nucleus accumbens, such as LTP [25, 26, 122, 124, 135,136,137,138,139]. These changes are thought to contribute to both the development and the persistence of substance use disorders. Like the dorsal striatum, the nucleus accumbens are largely composed of neurons that express dopamine D1 receptors or D2 receptors [140, 141]. Drug induced increases in synaptic strength in the D1 receptor expressing direct pathway have been found to be correlated with drug seeking behaviors [142,143,144,145]. In contrast, the D2 receptor expressing-indirect pathway is often implicated in extinction or aversive learning [14, 146, 147]. Our and other groups’ studies have demonstrated that dopamine signaling through AC5 and the cAMP pathway plays a central role in synaptic plasticity and appetitive learning [43, 45, 51, 124, 148, 149]. While we emphasize D1 and D2 neuron’s distinct roles here, we do not rule out the possibility that both D1 and D2 neuron’s firing patterns can still be well correlated with behaviors in both acquisition and extinction of appetitive and aversive learning since activities of both neurons may be important in the expression of specific behaviors.
Aberrant corticostriatal synaptic plasticity in neurological and psychiatric disorders: lack of movement or motivation
Parkinson’s disease (PD)
The contribution of aberrant corticostriatal synaptic plasticity to PD motor symptoms has been highlighted by multiple studies [28, 31, 150]. In 6-OHDA lesioned mice, a model of PD, the induction of corticostriatal LTD in the indirect pathway by a high-frequency stimulation protocol (HFS) was impaired. Correcting this impaired LTD was therapeutic [150]. In addition, in mice lacking D2 receptor activation, HFS that would normally induce LTD instead induced LTP [151]. Therefore, when dopamine levels decrease as PD progresses, it favors corticostriatal LTP induction in the indirect “NoGo” pathway and facilitates motor inhibition. However, it’s not clear in those studies if corticostriatal synaptic plasticity is important to motor learning or motor performance.
Studies in our laboratory have led us to develop a new framework [32, 152,153,154,155] that challenges the traditional view, which suggests that dopamine neuron denervation causes an acute imbalance between the direct and indirect pathways, leading to impaired motor performance [7, 8]. We have shown in animal models that the combination of dopamine deficiency and motor experience lead to aberrant corticostriatal LTP in the indirect pathway. Consequently, these animals gradually develop task-specific, experience-dependent motor inhibition and deteriorating motor performance—a ‘use it and lose it’ phenomenon that we call “aberrant inhibitory motor learning” [32, 152,153,154,155] (Fig. 1). This framework fits with the cellular level function of dopamine as the reward prediction error signal in modulating corticostriatal plasticity in learning [25,26,27,28,29]: responses that are reinforced by dopamine will be selected more in the future whereas responses that are not reinforced by dopamine will be inhibited in the future [32, 33]. In the pathological condition of Parkinson’s disease, dopamine deficiency or dopamine receptor blockade can cause any motor response to undergo experience-dependent, task-specific deterioration. The task specificity corresponds to the fact that corticostriatal plasticity in specific synapses is dependent on specific cortical glutamatergic inputs at that time.
Drug-induced parkinsonism (DIP)
DIP is the second most common cause of parkinsonism after idiopathic PD [156,157,158,159,160,161]. Antipsychotic drugs that block the dopamine D2 receptors are the most important and best characterized cause of DIP [156,157,158,159,160,161,162]. It was estimated that DIP prevalence might even approach that of idiopathic PD if the use of antipsychotic drugs in the aging population continues to rise [160]. DIP is characterized mainly by rigidity and bradykinesia, similar to the main motor symptoms of idiopathic PD, while other common idiopathic PD symptoms such as tremor and gait instability as well as many nonmotor symptoms are less prominent in DIP [156,157,158, 163, 164]. The differences between DIP and idiopathic PD suggest that dopamine D2 receptor blockade in DIP mainly causes rigidity and bradykinesia, while degeneration of dopamine neurons in the midbrain and other places (e.g., the enteric nervous system) in PD lead to motor, as well as nonmotor symptoms.
In DIP, parkinsonism lasts for weeks to months after cessation of antipsychotic treatment [165]. The persistence of these effects even in the absence of dopamine receptor blockade suggests that synaptic plasticity is a key contributing factor [30, 166]. The commonly used animal model for DIP is haloperidol-induced catalepsy. Rats or mice treated with haloperidol initially show mild akinesia and rigidity (i.e., catalepsy). However, repeated administration of haloperidol leads to more severe catalepsy (sensitization) [30, 167,168,169,170]. Importantly, haloperidol-induced catalepsy and its sensitization are dopamine D2 receptor dependent; and the sensitization effect is also context dependent [30, 167, 168]. Moreover, adenosine A2A antagonists can significantly protect against the development of catalepsy [169, 170]. A2A antagonists can reduce cAMP levels in D2 neurons and prevent the development of corticostriatal LTP [28], suggesting the role of corticostriatal LTP in the indirect pathway in DIP [30]. Taken together, these observations suggest that D2 receptor blockade will increase cAMP levels in D2 neurons; under conditions of specific cortical glutamatergic inputs (i.e., motor experience), LTP will develop at these excitatory synapses onto D2 neurons in the indirect pathway [32, 152] (Fig. 1). Such aberrant LTP will lead to task-specific inhibition and gradual worsening of motor function. In this model, motor performance deterioration in DIP requires both dopamine deficiency and motor experience.
Depression
Anhedonia, reduced motivation, and lack of energy are common in depression, and are often linked to a dysfunctional dopamine reward pathway [171, 172]. Human brain imaging studies suggest that depression is associated with impaired brain signals for reward learning and reward sensitivity [173]. One meta-analysis compared healthy controls, current or past major depression or bipolar disorder patients, and used reinforcement learning models to isolate reward sensitivity and learning rate [174]. Results suggest that depression and anhedonia reduced reward sensitivity more than learning rate.
Additionally, studies of patients with known dopamine system dysfunction have revealed high rates of depression diagnoses; nearly 40% of PD patients suffer from depression. This comorbidity is strongly associated with decreased activity in the limbic component of the CSTC loops [175]. In contrast, L-DOPA administration is known to lead to impaired impulse controls, which is associated with increased limbic CSTC circuit activity [176]. Together, this evidence suggests a role for dopamine in bidirectionally mediating these limbic and associative circuits.
Beyond clinical observation, animal models of depression allow for direct examination of synaptic changes within the CSTC loops. Chronic restraint stress in mice decreases the strength of excitatory synapses onto nucleus accumbens D1 neurons [177]. Social defeat stress and anhedonia increase excitatory input frequency onto nucleus accumbens D2 neurons, while simultaneously reducing excitatory transmission onto D1 neurons and diminishing their dendritic complexity [178, 179].
While the above studies highlight the importance of dopamine in depression, it is also important to consider how other neurotransmitters, particularly serotonin, may act on these same CSTC loops. Serotonin is the primary neurotransmitter implicated in depression, as evidenced by the serotonergic nature of most antidepressants. Nevertheless, serotonin can alter dopamine signaling through its effect on the activity of dopamine neurons and striatal neurons. More recent studies suggest the role of serotonin in depression is related to encoding appetitive or aversive outcomes [180]. Using fast-scan cyclic voltammetry recording from the striatum of human patients with PD [181], it was found that transiently increased serotonin was mostly correlated with negative reward prediction errors. Furthermore, trial-to-trial serotonin fluctuations were mostly correlated with protective choices (loss aversion) made by subjects following negative reward prediction errors. If this is true, lower serotonin could result in impaired protective mechanisms following negative outcomes [182]. In mice, it has been reported that serotonin neurons develop a slow ramp-up response to the reward-predicting cue, and ultimately remain responsive to the reward, i.e., they respond to both expected and unexpected rewards [183, 184]. In comparison, dopamine neurons increase their response to the cue but reduce their response to the reward when the animal gradually learns to predict the reward based on the cue, consistent with the reward prediction error hypothesis discussed above. The activities of both types of neurons are modulated by reward values whereas stressors substantially reduce the response strength of both neuron types in the nucleus accumbens [183, 184]. However, studies by another group on mice reported that different subsets of serotonin neurons modulated their responses differently in the dorsal raphe, either to reward or punishment, either phasically or tonically [185]. These studies exemplified the challenges in understanding the exact information encoded by serotonin under different environmental conditions, and whether serotonin dysfunction is the main mechanism for depression [186]. Nevertheless, the recent findings on the role of serotonin in appetitive and aversive learning suggest its either direct role in modulating synaptic plasticity in CSTC loops or its indirect role through affecting dopamine signaling.
Targeting synaptic plasticity as a therapeutic approach
The above discussion highlights the potential role of maladaptive corticostriatal synaptic plasticity in various psychiatric and neurological disorders, suggesting that preventing or reversing aberrant corticostriatal synaptic strength is indeed a promising therapeutic approach. However, such an approach has not been systematically explored. In the following section, we will discuss some limited examples that include pharmacotherapy, DBS or their combination that target synaptic plasticity in the striatum to treat substance use disorders, PD, OCD, and depression. Table 1 summarizes some of the published studies, although it’s not an exhaustive list.
Substance use disorders
Correcting aberrant synaptic strength (e.g., by DBS) has been proposed as novel treatments for substance use disorders. In animal models, optogenetic approaches have shown therapeutic potential by restoring circuit function through the reversal of specific forms of synaptic plasticity [187,188,189]. In one example, cocaine potentiates excitatory transmission in D1-receptor-expressing striatal neurons in mice via ERK signaling, with a time course paralleling locomotor sensitization. Depotentiation of cortical-nucleus accumbens inputs by optogenetic stimulation in vivo restored normal transmission and abolished cocaine-induced locomotor sensitization [142]. Moreover, low-frequency DBS of the nucleus accumbens combined with dopamine D1 receptor blockade abolished cocaine induced behavioral sensitization [143].
Similarly, repeated alcohol consumption in mice resulted in LTP in the D1-receptor expressing striatal neurons [190], and chemogenetic excitation of these neurons in vivo promoted alcohol consumption behavior. In another example, LTP was induced by pairing presynaptic glutamatergic stimulation with optogenetic postsynaptic depolarization in the dorsomedial striatum. Such experimentally induced LTP in D1 striatal neurons in vivo caused a long-lasting increase in alcohol-seeking behavior, while experimentally induced LTD decreased alcohol-seeking behavior [145]. Repeated morphine administration potentiated excitatory transmission in D1 striatal neurons. In vivo optogenetic stimulation of infralimbic cortex-accumbens shell inputs reversed such pathophysiology and blocked reinstatement of morphine-evoked conditioned place preference [144].
Currently, DBS applications for substance use disorder treatment in humans are still limited, but cases have been reported in which DBS alleviated cocaine, methamphetamine, opioids, alcohol, and tobacco abuse [191,192,193,194]. For example, DBS in the nucleus accumbens on patients with alcohol addiction significantly decreased the alcohol craving and consumption [195,196,197]. In another 30-month longitudinal study, nucleus accumbens DBS in a patient with cocaine addiction was able to reduce the drug craving and achieve long-term abstinence [198]. While these cases highlighted the potential of DBS in treating substance use disorders, double-blinded studies are needed to assess treatment efficacy without bias.
Parkinson’s disease (PD)
Dopamine replacement therapy usually causes an immediate improvement in motor function, known as the short-duration response (SDR), followed by a long-duration response (LDR) that continues to improve motor function and develops slowly over days to weeks [199]. The phenomenon of LDR supports our hypothesis that PD motor symptoms are at least partially due to aberrant corticostriatal synaptic strength, and that preventing or correcting such aberrant corticostriatal synaptic strength could be therapeutic. Our animal data and model suggest that dopamine-dependent corticostriatal plasticity in the indirect pathway, and retention of such corrected synaptic strength for some time in the absence of dopamine, are the mechanisms underlying LDR [32, 152,153,154,155]. Conversely, experience-dependent aberrant plasticity explains the gradual loss of the LDR after continued cessation of therapy [32, 152,153,154,155].
In one clinical study, PD patients learned a speed-accuracy task in the “on” state (during L-DOPA treatment) and then were tested in the “off” state. Their performance progressively worsened when subsequently tested during the “off” state [200], suggesting gradual loss of the LDR after cessation of therapy. In another study using existing data from more than 350 PD patients, the hypothesis that a dopamine-dependent motor learning mechanism underlies the LDR was tested [201]. To measure LDR, the performance in finger-tapping before daily L-DOPA doses (in the absence of SDR) was compared among three time points: before the initiation of L-DOPA therapy, 9 weeks and 40 weeks after initiation of therapy. Even though there was no difference between dominant and nondominant hands in SDR, more LDR associated improvement was observed in the dominant compared to nondominant hand, and this effect was dose dependent. These data again support our aberrant corticostriatal plasticity hypothesis for PD motor symptoms.
Non-dopamine replacement therapy that targets signaling molecules for corticostriatal plasticity (e.g., the cAMP pathway and downstream signaling molecules) also supports our aberrant corticostriatal plasticity hypothesis. A2A antagonists have been approved by the FDA to be used in combination with L-DOPA therapy. They can prolong LDR in PD patients but are not effective if used alone [202,203,204]. This aligns with the known effects of A2A antagonists in preventing aberrant corticostriatal LTP in the indirect pathway induced by dopamine deficiency [28, 51].
LDR in PD therapy suggests the under-appreciated potential of preventing and reversing aberrant corticostriatal synaptic strength as therapy. It remains to be demonstrated if direct manipulation of corticostriatal plasticity through DBS or through the combination of DBS and pharmacotherapy could be used as a novel therapy for PD. Although DBS is already successfully used in treating PD, it is limited to acute therapeutic effects; PD symptoms usually quickly return if DBS stops. What we propose here is to alter corticostriatal synaptic strength instead. If successful, such therapeutic effects should be long lasting.
Obsessive compulsive disorder (OCD) and depression
Selective serotonin reuptake inhibitors (SSRIs) have demonstrated efficacy in reducing OCD symptoms which is often attributed to modulating serotonin levels and normalizing neurotransmission within the CSTC circuitry [205, 206]. Cognitive-behavioral therapy is another cornerstone of OCD treatment in which exposure and response prevention techniques are used to gradually expose individuals to feared situations or thoughts while preventing the accompanying compulsive behaviors [205, 206].
However, approximately 10% of patients do not respond to either of those treatments. With treatment-resistant patients, DBS has shown promise. The most common targets include the ventral capsule/ventral striatum (VC/VS), the anterior limb of the internal capsule (ALIC), and the subthalamic nucleus (STN) [207]. Stimulation of these regions within the CSTC circuitry aims to normalize neural activity, thus alleviating OCD symptoms. Meta-analysis of therapeutic effects of DBS among OCD patients found that such treatments significantly reduced OCD symptoms [208,209,210].
DBS has also been tested in animal models. As discussed above, deletion of Sapap3 in mice results in excessive grooming and a selective deficit in behavioral response inhibition. Optogenetic stimulation of the lateral orbitofrontal cortex and its terminals in the striatum normalized striatal neuron activity and restored the behavioral response inhibition [113].
Similar to OCD, major depression is most commonly treated with SSRIs. There is limited literature on DBS treatment of major depression, but it has shown its promise [211]. DBS significantly alleviates depressive symptoms in treatment-resistant depression (TRD) patients by targeting the subcallosal cingulate gyrus (SCG), VC/VS, medial forebrain bundle (MFB), and nucleus accumbens core [212]. One study reviewed both preclinical and clinical findings [213]. In preclinical studies, stimulation parameters and neuroanatomical locations could influence DBS-related therapeutic effects, suggesting that the modulatory effects of monoamine neurotransmitters could be the reason for such therapeutic effects. In clinical studies, DBS in the SCG, ALIC and MFB yielded relatively consistent antidepressant response rates [213]. Interestingly, acute responses to DBS often do not predict the long-term therapeutic effects, suggesting that synaptic plasticity may play an important role in the long-term but not the acute therapeutic effects [211]. In animal model studies, chronic stress decreases the strength of hippocampus–accumbens synapses and impairs LTP whereas antidepressant treatment can reverse such aberrant changes [214].
Erasing “bad memories” during reconsolidation as a therapeutic approach
The above examples are limited to diseases that are at least partially attributable to abnormal basal ganglia functions; and we discussed some examples in which preventing or reversing aberrant corticostriatal synaptic strength represents promising therapeutic approaches.
A related approach is erasing “bad memories” during reconsolidation even though many of the studies are outside of the basal ganglia field. Under the reconsolidation hypothesis, when a memory is recalled, it becomes labile and needs to be reconsolidated which provides a window of opportunity for memory alteration [5, 215,216,217]. One often discussed example of such an idea is PTSD although clinical trials have not been successful [218, 219]. PTSD is characterized by the re-experiencing of a traumatic event through intrusive memories [98]. The reconsolidation of traumatic memories involves the strengthening of specific synaptic connections in the brain, and disrupting these connections during the process of memory reconsolidation can potentially weaken the traumatic memory and reduce symptoms [5, 215, 216]. Pharmacological interventions and behavioral techniques have been used, including the beta-adrenergic antagonist propranolol. Propranolol, administered immediately after a traumatic event or before memory retrieval, can reduce the incidence of PTSD symptoms [4, 220, 221]. In addition, many preclinical studies have conducted and have provided proof of principle for such approaches [222,223,224,225], and for mechanisms of synaptic plasticity involved in extinction learning and exposure therapy which gradually weaken the memory and PTSD symptoms [226].
Targeting reconsolidation could be a promising approach for correcting maladaptive memories in basal ganglia-related disorders as well [227]. In human studies, interfering with the reconsolidation of nicotine-associated memories using propranolol decreased craving for smoking [228]. In preclinical studies, NMDA receptor antagonist MK-801 was able to impair drug-seeking related memory reconsolidation and therefore reduce relapse to drug-seeking behaviors in animal models [229,230,231].
Molecular targets to consider
Targeting signaling pathways involved in synaptic plasticity
If targeting specific aberrant synaptic plasticity is a potential therapeutic approach for neurological and psychiatric disorders, then it is possible to take advantage of the known molecular mechanisms involved in synaptic plasticity. Not surprisingly, in the above discussion, whether to induce corticostriatal LTP or LTD as a therapy, or to prevent memory reconsolidation, drugs associated with specific molecular targets are usually used, alone or in combination with behavioral therapy or with DBS.
There is a vast literature on neurotransmitter receptors, intracellular signaling molecules, synaptic proteins, cytoskeletal proteins, and cell adhesion molecules involved in synaptic plasticity and dendritic remodeling, e.g., NMDA, AMPA, mGluR1/5, CaMKII, AC1/8, AC5, PKA, deltaFosB, ARC, Homer, BDNF, CREB, PSD-95, Shank, Neuroligins, DISC1 and Dynamin etc. There are already many comprehensive review papers on them [217, 232,233,234,235,236,237,238,239,240,241,242]. Both NMDA and mGluR1/5 have been tested as therapeutic targets as discussed in the above examples.
One potential disadvantage of targeting these molecules alone is that they are less likely to be selective to the underlying pathological mechanisms as they are also crucial for normal synaptic mechanisms involved in learning, memory, and other functions. This may partially explain why clinical trials based on these ideas have not been successful [243,244,245,246,247]. DBS alone has similar limitations. Even though DBS has been successful in treating PD, the therapeutic effect stops when the treatment stops. We propose that combining behavioral therapy or DBS with molecular targets offers unique advantages by combining task-relevant circuit selectivity with specific molecular targets to achieve more specificity in correcting aberrant synaptic plasticity. Another potential advantage of such therapies is that the therapeutic effects could be long-lasting even after cessation of treatment, as corrected synaptic strengths would take time to become aberrant again.
Targeting new protein synthesis, epigenetic mechanisms, RNA binding proteins (RBPs) and RNA modifications
Based on the memory consolidation idea discussed above, one unique class of molecular targets are those regulating protein synthesis. It is known that new protein synthesis is required for long-term changes in synaptic strength and for converting short-term memory to long-term memory [248, 249], including plasticity involved in the basal ganglia [250,251,252]. Therefore, blocking protein synthesis can block reconsolidation of old memories or consolidation of new memories, either could be potentially therapeutic.
Synaptic activity can rapidly change the synthesis rates of both specific proteins and the overall proteome [253, 254]. Protein synthesis inhibitors or genetic manipulations that affect protein synthesis have been shown to cause decay of LTP or LTD, blocking the animal’s ability to remember after learning in both explicit and implicit memory tasks [255,256,257,258,259]. Indeed, therapeutic approaches have been proposed by taking advantage of protein synthesis inhibition and interfering with memory reconsolidation [5, 260, 261].
All protein synthesis starts from gene expression. Gene expression studies have revealed some of the important pathways involved in learning and memory [262, 263]. Epigenetic mechanisms of gene expression regulation can potentially induce long-lasting changes in the brain that may underlie persistent behavioral abnormalities [263]. Many studies have shown that manipulations of histone acetylation are effective treatment in animal models of substance use disorders and depression [264,265,266,267].
Although important, the above mechanisms affect gene expression at transcriptional level, which lacks synaptic specificity. One neuron could have thousands of synapses, but plasticity is often synapse specific. Moreover, the distance between synapses and the soma creates a fundamental challenge for the neuron. A substantial amount of work has shown that local protein synthesis in dendrites is often required, among other mechanisms, for long-term synaptic plasticity [254, 255, 268, 269]. Some mRNA transcripts are selectively localized to dendrites, suggesting mechanisms that control their distribution and translation [270,271,272,273,274].
The key factors that regulate translation temporally and spatially are those that control RNA transport, localization, translation, and degradation. RNA binding proteins (RBPs) are one of the major mechanisms for such regulations [254, 255, 268, 269, 275]. One important example is the Fragile X mental retardation protein (FMRP) [1, 276]. FMRP was first identified as a key protein related to Fragile X syndrome. Loss of FMRP leads to the disorder that causes intellectual retardation and Autism [277]. Functionally, FMRP is an RBP that modulates synaptic plasticity by regulating local translation [276]. Fmr1 mutant mice show an increased mGluR-dependent LTD in the hippocampus [278]. Furthermore, reducing mGluR5 expression or inhibiting mGluR5 activity in animal models of Fragile X alleviates the disease phenotype [2, 3]. However, clinical trials have not been successful [279, 280]. Another interesting example is disrupting up-frameshift protein (UPF2), a nonsense-mediated mRNA decay (NMD) component downstream of protein translation. Neuron-specific disruption of UPF2 in adult mice was shown to impair synaptic plasticity, learning, and memory, as well as cause perseverative behavior [281].
How do RBPs recognize and bind to specific targets? The affinities of RNAs to RBPs are often regulated by RNA modifications [282]. Although there are many, we want to emphasize N6-Methyladenosine (m6A) RNA methylation as an example because it is especially abundant and important in this regard [283,284,285,286,287,288,289,290]. Studies of m6A so far embody the concept of “epitranscriptome” and epitranscriptomic regulation. Studies in the past a few years indicate that the m6A modification of RNAs affects almost every phase of mRNA metabolism and function, including RNA transport, localization, splicing, nuclear export, stability, and translational efficiency [283,284,285,286,287,288,289,290]. m6A is highly enriched in mRNAs in the brain, especially in the adult brain, and it is present in over 4500 mRNAs in the mouse brain [291]. Notably, behavioral training has been shown to increase m6A levels in the prefrontal cortex of mice [292].
The functional significance of m6A RNA methylation is exerted by three groups of proteins: “writers” (methyltransferases) that install, “erasers” (demethylases) that remove, and “readers” that are RBPs that recognize m6A and determine the cellular fate of the modified RNA [283, 290, 293,294,295,296,297,298]. The identification of these key players in the m6A pathway has opened the door to functional studies of m6A.
METTL14 is one essential subunit of the m6A methyltransferase complex [283]. In our published studies, Mettl14 gene deletion in dopamine receptor expressing neurons severely impaired motor learning [299]. In another study, the other essential subunit of the m6A methyltransferase complex, METTL3, has been found to regulate the efficacy of hippocampus-dependent memory consolidation by promoting the translation of immediate early genes during memory formation [300]. The m6A demethylase FTO is highly expressed in the brain and may be dynamically regulated [301]. In human genetic studies, mutations in the FTO gene are strongly linked to obesity and diabetes; FTO polymorphisms are also implicated in attention-deficit/hyperactivity disorder, Alzheimer’s disease, and abnormal brain volumes [301]. Animal studies indicate that the FTO knockout mice have an abnormal behavioral and electrophysiological response to cocaine [302].
m6A reader proteins are special RBPs with diverse functions. YTH-domain containing reader proteins (YTHDF1, 2 and 3) are found at the dendrites of mouse hippocampal neurons, and loss of these proteins results in dysfunction of synaptic transmission [303]. In Drosophila, YTHDF reader proteins are also found to be critical to memory formation [304]. These data suggest that the m6A methylated mRNA near the synapse can potentially be recognized and regulated by these reader proteins. Among them, YTHDF1 is especially important to neurons. YTHDF1 plays important roles in promoting synaptic protein synthesis (e.g., CaMKIIα) in response to stimuli, as well as in synaptic plasticity and learning [305]. Interestingly, studies also reveal that FMRP preferentially binds to m6A-containing RNA and represses protein synthesis through interactions with YTHDF1 [306,307,308,309]. Therefore, if FMRP indirectly inhibits the translation of YTHDF1 target transcripts through this interaction, it may have significantly more target transcripts than previously recognized. This suggests the broader importance of FMRP as a translation repressor in the brain, and that therapies targeting RBPs must be tailored to specific cell types or administered within certain time windows associated with aberrant synaptic plasticity, or both, in order to minimize side effects.
By deleting YTHDF1 selectively in D1 and D2 striatal neurons [295, 296], our own studies have found that YTHDF1 deficiency in D1 neurons selectively impaired the acquisition of motor skill learning whereas YTHDF1 deficiency in D2 neurons virtually eliminated inhibitory motor learning in PD models and haloperidol-induced catalepsy in drug-induced parkinsonism models. Moreover, YTHDF1 deficiency mimics METTL14 deficiency in a cell type specific manner [296], suggesting that YTHDF1 is the main m6A reader protein that mediates m6A’s function in learning.
Therefore, targeting protein synthesis pathways (e.g., inhibiting YTHDF1) to prevent memory consolidation or reconsolidation could be therapeutic. There are many potential molecular targets to consider. Among them are the well-studied pathways involved in gene expression regulation including epigenetic mechanisms. Here, we argue that RBPs and epitranscriptomic mechanisms represent a new frontier and have the advantage of rapidly altering local protein synthesis in synapses. We expect that this new research direction will yield significant opportunities for the development of novel therapies.
Conclusion
There are many neurological and psychiatric disorders in which aberrant synaptic plasticity has been implicated. These studies represent an exciting new theme in translational neuroscience and promising new directions for therapy development. However, merely labeling a disease condition as aberrant plasticity is not helpful. We need to understand the specific neural pathways and synapses affected by the pathology; we need to know how specific alterations (strengthening, weakening or other modulations) in synaptic strengths of specific circuits lead to predictable behavioral outcomes based on known circuit function. Only then can we harness the power of synaptic plasticity as therapy rather than fall victim to the devastating consequences of aberrant synaptic plasticity.
One potential advantage of correcting aberrant synaptic plasticity is that the therapeutic effects could be long lasting even after cessation of treatment, as corrected synaptic strengths would take time to become aberrant again. It is conceivable that such approaches, if they work as predicted, could have fewer side effects. Even if the acute treatment itself has side effects, they would be short-lived whereas the corrected synaptic strength and the associated therapeutic effects would be relatively long-lasting.
One of the most important advances in modern neuroscience research is the development of optogenetic and chemogenetic tools, which enable precise manipulation of neural circuits and synapses to produce specific behavioral changes [310,311,312]. While these tools are not yet practical for human therapy, they nevertheless have shown us the promise of direct and selective circuit manipulations compared to DBS, which often lacks cell-type specificity. An intriguing possibility in humans is the combination of pharmacotherapies with direct brain circuit manipulations or specific behaviors, which could potentially be far more selective in correcting aberrant synaptic plasticity than either approach alone. Such approaches fit with the known procedures used in inducing LTP or LTD. They also fit with the idea of combining specific behaviors with drugs that interfere with memory reconsolidation such that the specific memories will become labile and prone to alteration. In this regard, we see a very promising future in combining decades of research on molecular and pharmacological targets with the more recent research on direct circuit manipulations.
References
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–7.
Krueger DD, Bear MF. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu Rev Med. 2011;62:411–29.
Dolen G, Carpenter RL, Ocain TD, Bear MF. Mechanism-based approaches to treating fragile X. Pharmacol Ther. 2010;127:78–93.
Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–8.
Nader K. Reconsolidation and the dynamic nature of memory. Cold Spring Harb Perspect Biol. 2015;7:a021782.
Alexander GE, Delong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–81.
Albin RL, Young AB, Penney JB. The functional anatomy of disorders of the basal ganglia. Trends Neurosci. 1995;18:63–4.
DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64:20–4.
Kravitz AV, Freeze BS, Parker PRL, Kay K, Thwin MT, Deisseroth K, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–U7.
Oldenburg IA, Sabatini BL. Antagonistic but not symmetric regulation of primary motor cortex by basal ganglia direct and indirect pathways. Neuron. 2015;86:1174–81.
Lee HJ, Weitz AJ, Bernal-Casas D, Duffy BA, Choy M, Kravitz AV, et al. Activation of direct and indirect pathway medium spiny neurons drives distinct brain-wide responses. Neuron. 2016;91:412–24.
Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–42.
Tecuapetla F, Jin X, Lima SQ, Costa RM. Complementary contributions of striatal projection pathways to action initiation and execution. Cell. 2016;166:703–15.
Zalocusky KA, Ramakrishnan C, Lerner TN, Davidson TJ, Knutson B, Deisseroth K. Nucleus accumbens D2R cells signal prior outcomes and control risky decision-making. Nature. 2016;531:642–6.
Li H, Jin X. Multiple dynamic interactions from basal ganglia direct and indirect pathways mediate action selection. eLife. 2023;12:RP87644.
Geddes CE, Li H, Jin X. Optogenetic editing reveals the hierarchical organization of learned action sequences. Cell. 2018;174:32–43.e15.
Freeze BS, Kravitz AV, Hammack N, Berke JD, Kreitzer AC. Control of basal ganglia output by direct and indirect pathway projection neurons. J Neurosci. 2013;33:18531–9.
Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, et al. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–95.
Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215.
Surmeier DJ, Ding J, Day M, Wang ZF, Shen WX. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–35.
Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–66.
Burke DA, Rotstein HG, Alvarez VA. Striatal local circuitry: a new framework for lateral inhibition. Neuron. 2017;96:267–84.
Lemos JC, Friend DM, Kaplan AR, Shin JH, Rubinstein M, Kravitz AV, et al. Enhanced GABA transmission drives bradykinesia following loss of dopamine D2 receptor signaling. Neuron. 2016;90:824–38.
Taverna S, Ilijic E, Surmeier DJ. Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. J Neurosci. 2008;28:5504–12.
Reynolds JNJ, Wickens JR. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw. 2002;15:507–21.
Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–9.
Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–61.
Shen WX, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–51.
Surmeier DJ, Plotkin J, Shen WX. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol. 2009;19:621–8.
Wiecki TV, Riedinger K, von Ameln-Mayerhofer A, Schmidt WJ, Frank MJ. A neurocomputational account of catalepsy sensitization induced by D2 receptor blockade in rats: context dependency, extinction, and renewal. Psychopharmacology (Berl). 2009;204:265–77.
Wiecki TV, Frank MJ. Neurocomputational models of motor and cognitive deficits in Parkinson’s disease. Prog Brain Res. 2010;183:275–97.
Zhuang X, Mazzoni P, Kang UJ. The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nat Rev Neurol. 2013;9:248–56.
Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in Motivational Control: Rewarding, Aversive, and Alerting. Neuron. 2010;68:815–34.
Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–63.
Brzosko Z, Schultz W, Paulsen O. Retroactive modulation of spike timing-dependent plasticity by dopamine. eLife. 2015;4:e09685.
Yagishita S, Hayashi-Takagi A, Ellis-Davies GC, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science. 2014;345:1616–20.
Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci. 2010;30:14273–83.
Nair AG, Gutierrez-Arenas O, Eriksson O, Jauhiainen A, Blackwell KT, Kotaleski JH. Modeling intracellular signaling underlying striatal function in health and disease. Prog Mol Biol Transl Sci. 2014;123:277–304.
Richfield EK, Penney JB, Young AB. Anatomical and affinity state comparisons between dopamine D1 and D2 receptors in the rat central nervous system. Neuroscience. 1989;30:767–77.
Hunger L, Kumar A, Schmidt R. Abundance compensates kinetics: similar effect of dopamine signals on D1 and D2 receptor populations. J Neurosci. 2020;40:2868–81.
Skinbjerg M, Sibley DR, Javitch JA, Abi-Dargham A. Imaging the high-affinity state of the dopamine D2 receptor in vivo: fact or fiction? Biochem Pharmacol. 2012;83:193–8.
Liu C, Goel P, Kaeser PS. Spatial and temporal scales of dopamine transmission. Nat Rev Neurosci. 2021;22:345–58.
Iwamoto T, Okumura S, Iwatsubo K, Kawabe J, Ohtsu K, Sakai I, et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem. 2003;278:16936–40.
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: From structure to function. Physiol Rev. 1998;78:189–225.
Kheirbek MA, Britt JP, Beeler JA, Ishikawa Y, McGehee DS, Zhuang X. Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J Neurosci. 2009;29:12115–24.
Matsuoka I, Suzuki Y, Defer N, Nakanishi H, Hanoune J. Differential expression of type I, II, and V adenylyl cyclase gene in the postnatal developing rat brain. J Neurochem. 1997;68:498–506.
Onda T, Hashimoto Y, Nagai M, Kuramochi H, Saito S, Yamazaki H, et al. Type-specific regulation of adenylyl cyclase. Selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J Biol Chem. 2001;276:47785–93.
Villacres EC, Wu Z, Hua W, Nielsen MD, Watters JJ, Yan C, et al. Developmentally expressed Ca2+-sensitive adenylyl cyclase activity is disrupted in the brains of type I adenylyl cyclase mutant mice. J Biol Chem. 1995;270:14352–7.
Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, et al. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci. 1995;92:220–4.
Impey S, Wayman G, Wu Z, Storm DR. Type I adenylyl cyclase functions as a coincidence detector for control of cyclic AMP response element-mediated transcription: synergistic regulation of transcription by Ca 2+ and isoproterenol. Mol Cell Biol. 1994;14:8272–81.
Augustin SM, Beeler JA, McGehee DS, Zhuang X. Cyclic AMP and afferent activity govern bidirectional synaptic plasticity in striatopallidal neurons. J Neurosci. 2014;34:6692–9.
Calabresi P, Fedele E, Pisani A, Fontana G, Mercuri NB, Bernardi G, et al. Transmitter release associated with long-term synaptic depression in rat corticostriatal slices. Eur J Neurosci. 1995;7:1889–94.
Ochi M, Inoue H, Koizumi S, Shibata S, Watanabe S. Long-term enhancement of dopamine release by high frequency tetanic stimulation via a N-methyl-D-aspartate-receptor-mediated pathway in rat striatum. Neuroscience. 1995;66:29–36.
Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–32.
Sulzer D, Cragg SJ, Rice ME. Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia. 2016;6:123–48.
Goto Y, Otani S, Grace AA. The Yin and Yang of dopamine release: a new perspective. Neuropharmacology. 2007;53:583–7.
Pan PY, Ryan TA. Calbindin controls release probability in ventral tegmental area dopamine neurons. Nat Neurosci. 2012;15:813–5.
Turner TJ. Nicotine enhancement of dopamine release by a calcium-dependent increase in the size of the readily releasable pool of synaptic vesicles. J Neurosci. 2004;24:11328–36.
Venton BJ, Zhang H, Garris PA, Phillips PE, Sulzer D, Wightman RM. Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. J Neurochem. 2003;87:1284–95.
Heien ML, Wightman RM. Phasic dopamine signaling during behavior, reward, and disease states. CNS Neurol Disord Drug Targets. 2006;5:99–108.
Niv Y. Cost, benefit, tonic, phasic: what do response rates tell us about dopamine and motivation? Ann N Y Acad Sci. 2007;1104:357–76.
Grace AA. The tonic/phasic model of dopamine system regulation: its relevance for understanding how stimulant abuse can alter basal ganglia function. Drug Alcohol Depend. 1995;37:111–29.
Budygin EA, Bass CE, Grinevich VP, Deal AL, Bonin KD, Weiner JL. Opposite consequences of tonic and phasic increases in accumbal dopamine on alcohol-seeking behavior. iScience. 2020;23:100877.
Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64.
Aosaki T, Kimura M, Graybiel AM. Temporal and spatial characteristics of tonically active neurons of the primate’s striatum. J Neurophysiol. 1995;73:1234–52.
Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–9.
Yorgason JT, Zeppenfeld DM, Williams JT. Cholinergic interneurons underlie spontaneous dopamine release in nucleus accumbens. J Neurosci. 2017;37:2086–96.
Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41.
Straub C, Tritsch NX, Hagan NA, Gu C, Sabatini BL. Multiphasic modulation of cholinergic interneurons by nigrostriatal afferents. J Neurosci. 2014;34:8557–69.
Chantranupong L, Beron CC, Zimmer JA, Wen MJ, Wang W, Sabatini BL. Dopamine and glutamate regulate striatal acetylcholine in decision-making. Nature. 2023;621:577–85.
Krok AC, Maltese M, Mistry P, Miao X, Li Y, Tritsch NX. Intrinsic dopamine and acetylcholine dynamics in the striatum of mice. Nature. 2023;621:543–9.
Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–52.
Augustin SM, Chancey JH, Lovinger DM. Dual dopaminergic regulation of corticostriatal plasticity by cholinergic interneurons and indirect pathway medium spiny neurons. Cell Rep. 2018;24:2883–93.
Reynolds JNJ, Avvisati R, Dodson PD, Fisher SD, Oswald MJ, Wickens JR, et al. Coincidence of cholinergic pauses, dopaminergic activation and depolarisation of spiny projection neurons drives synaptic plasticity in the striatum. Nat Commun. 2022;13:1296.
Zucca S, Zucca A, Nakano T, Aoki S, Wickens J. Pauses in cholinergic interneuron firing exert an inhibitory control on striatal output in vivo. eLife. 2018;7:e32510.
Russwurm C, Koesling D, Russwurm M. Phosphodiesterase 10A is tethered to a synaptic signaling complex in striatum. J Biol Chem. 2015;290:11936–47.
Ouimet CC, Miller PE, Hemmings HC Jr., Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3’:5’-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci. 1984;4:111–24.
Li L, Gervasi N, Girault JA. Dendritic geometry shapes neuronal cAMP signalling to the nucleus. Nat Commun. 2015;6:6319.
Lin L, Gupta S, Zheng WS, Si K, Zhu JJ. Genetically encoded sensors enable micro- and nano-scopic decoding of transmission in healthy and diseased brains. Mol Psychiatry. 2021;26:443–55.
Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong WH, et al. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science. 2018;360:eaat4422.
Sun F, Zeng J, Jing M, Zhou J, Feng J, Owen SF, et al. A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell. 2018;174:481–96.e19.
Patriarchi T, Mohebi A, Sun J, Marley A, Liang R, Dong C, et al. An expanded palette of dopamine sensors for multiplex imaging in vivo. Nat Methods. 2020;17:1147–55.
Zhuo Y, Luo B, Yi X, Dong H, Miao X, Wan J, et al. Improved green and red GRAB sensors for monitoring dopaminergic activity in vivo. Nat Methods. 2024;21:680–91.
Bastide MF, Meissner WG, Picconi B, Fasano S, Fernagut P-O, Feyder M, et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog Neurobiol. 2015;132:96–168.
Borgkvist A, Lieberman OJ, Sulzer D. Synaptic plasticity may underlie l -DOPA induced dyskinesia. Curr Opin Neurobiol. 2018;48:71–8.
Dauer W, Przedborski S. Parkinson’s disease. Neuron. 2003;39:889–909.
Mosharov EV, Borgkvist A, Sulzer D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov Disord. 2015;30:45–53.
Cumming P, Borghammer P. Molecular Imaging and the Neuropathologies of Parkinson’s Disease. Springer Berlin Heidelberg; 2011. p. 117–48.
Lee J, Zhu WM, Stanic D, Finkelstein DI, Horne MH, Henderson J, et al. Sprouting of dopamine terminals and altered dopamine release and uptake in Parkinsonian dyskinaesia. Brain. 2008;131:1574–87.
Munoz A, Lopez-Lopez A, Labandeira CM, Labandeira-Garcia JL. Interactions between the serotonergic and other neurotransmitter systems in the basal ganglia: role in Parkinson’s disease and adverse effects of L-DOPA. Front Neuroanat. 2020;14:26.
Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–45.
Tang K-C, Low MJ, Grandy DK, Lovinger DM. Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci. 2001;98:1255–60.
Costa RM. Plastic corticostriatal circuits for action learning: what’s dopamine got to do with it? Ann N Y Acad Sci. 2007;1104:172–91.
Calabresi P, Ghiglieri V, Mazzocchetti P, Corbelli I, Picconi B. Levodopa-induced plasticity: a double-edged sword in Parkinson’s disease? Philos Trans R Soc B: Biol Sci. 2015;370:20140184.
Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA–induced dyskinesia. Nat Neurosci. 2003;6:501–6.
Ryan MB, Bair-Marshall C, Nelson AB. Aberrant striatal activity in parkinsonism and levodopa-induced dyskinesia. Cell Rep. 2018;23:3438–46.e5.
Girasole AE, Lum MY, Nathaniel D, Bair-Marshall CJ, Guenthner CJ, Luo L, et al. A subpopulation of striatal neurons mediates levodopa-induced dyskinesia. Neuron. 2018;97:787–95.e6.
Association AP. Diagnostic and Statistical Manual of Mental Disorders. DSM-V; 2013. 5th editon.
Robbins TW, Vaghi MM, Banca P. Obsessive-compulsive disorder: puzzles and prospects. Neuron. 2019;102:27–47.
Simmler LD, Ozawa T. Neural circuits in goal-directed and habitual behavior: implications for circuit dysfunction in obsessive-compulsive disorder. Neurochem Int. 2019;129:104464.
Ting JT, Feng G. Glutamatergic synaptic dysfunction and obsessive-compulsive disorder. Curr Chem Genomics. 2008;2:62–75.
Rotge J-Y, Langbour N, Guehl D, Bioulac B, Jaafari N, Allard M, et al. Gray matter alterations in obsessive–compulsive disorder: an anatomic likelihood estimation meta-analysis. Neuropsychopharmacology. 2010;35:686–91.
Pauls DL, Abramovitch A, Rauch SL, Geller DA. Obsessive–compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci. 2014;15:410–24.
Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–76.
Dolan RJ, Dayan P. Goals and habits in the brain. Neuron. 2013;80:312–25.
Balleine BW. The meaning of behavior: discriminating reflex and volition in the brain. Neuron. 2019;104:47–62.
Balleine BW, O’Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35:48–69.
Wickens JR, Horvitz JC, Costa RM, Killcross S. Dopaminergic mechanisms in actions and habits: Fig. 1. J Neurosci. 2007;27:8181–3.
Voon V, Reiter A, Sebold M, Groman S. Model-based control in dimensional psychiatry. Biol Psychiatry. 2017;82:391–400.
Gillan CM, Papmeyer M, Morein-Zamir S, Sahakian BJ, Fineberg NA, Robbins TW, et al. Disruption in the balance between goal-directed behavior and habit learning in obsessive-compulsive disorder. Am J Psychiatry. 2011;168:718–26.
Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding J-D, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900.
Wan Y, Ade KK, Caffall Z, Ilcim Ozlu M, Eroglu C, Feng G, et al. Circuit-selective striatal synaptic dysfunction in the Sapap3 knockout mouse model of obsessive-compulsive disorder. Biol Psychiatry. 2014;75:623–30.
Burguière E, Monteiro P, Feng G, Graybiel AM. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science. 2013;340:1243–6.
Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, et al. Repeated cortico-striatal stimulation generates persistent OCD-Like behavior. Science. 2013;340:1234–9.
Stillman AA, Krsnik ZE, Sun J, RašIn M-R, State MW, ŠEstan N, et al. Developmentally regulated and evolutionarily conserved expression of SLITRK1 in brain circuits implicated in Tourette syndrome. J Comp Neurol. 2009;513:21–37.
Worbe Y, Marrakchi-Kacem L, Lecomte S, Valabregue R, Poupon F, Guevara P, et al. Altered structural connectivity of cortico-striato-pallido-thalamic networks in Gilles de la Tourette syndrome. Brain. 2015;138:472–82.
Pogorelov V, Xu M, Smith HR, Buchanan GF, Pittenger C. Corticostriatal interactions in the generation of tic-like behaviors after local striatal disinhibition. Exp Neurol. 2015;265:122–8.
Delorme C, Salvador A, Valabregue R, Roze E, Palminteri S, Vidailhet M, et al. Enhanced habit formation in Gilles de la Tourette syndrome. Brain. 2016;139:605–15.
Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–79.
Di Chiara G. A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol. 1998;12:54–67.
Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–9.
Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47:61–79.
Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci. 2001;2:695–703.
Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–98.
Carelli RM. Nucleus accumbens cell firing and rapid dopamine signaling during goal-directed behaviors in rats. Neuropharmacology. 2004;47:180–9.
Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM. Dopamine operates as a subsecond modulator of food seeking. J Neurosci. 2004;24:1265–71.
Day JJ, Roitman MF, Wightman RM, Carelli RM. Associative learning mediates dynamic shifts in dopamine signaling in the nucleus accumbens. Nat Neurosci. 2007;10:1020–8.
Pascoli V, Terrier J, Hiver A, Luscher C. Sufficiency of mesolimbic dopamine neuron stimulation for the progression to addiction. Neuron. 2015;88:1054–66.
Schultz W, Apicella P, Ljungberg T. Responses of monkey dopamine neurons to reward and conditioned-stimuli during successive steps of learning a delayed-response task. J Neurosci. 1993;13:900–13.
Stuber GD, Roitman MF, Phillips PEM, Carelli RM, Wightman RM. Rapid dopamine signaling in the nucleus accumbens during contingent and noncontingent cocaine administration. Neuropsychopharmacology. 2005;30:853–63.
Dickinson A, Smith J, Mirenowicz J. Dissociation of pavlovian and instrumental incentive learning under dopamine antagonists. Behav Neurosci. 2000;114:468–83.
Saunders BT, Robinson TE. The role of dopamine in the accumbens core in the expression of Pavlovian-conditioned responses. Eur J Neurosci. 2012;36:2521–32.
Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, et al. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469:53–7.
Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–94.
Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–31.
Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–90.
Lovinger DM, Tyler E. Synaptic transmission and modulation in the neostriatum. Int Rev Neurobiol. 1996;39:77–111.
Smith-Roe SL, Kelley AE. Coincident activation of NMDA and dopamine D1 receptors within the nucleus accumbens core is required for appetitive instrumental learning. J Neurosci. 2000;20:7737–42.
Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, et al. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–7.
Gong SC, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, et al. Targeting cre recombinase to specific neuron Populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27:9817–23.
Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320.
Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–5.
Creed M, Pascoli VJ, Luscher C. Addiction therapy. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science. 2015;347:659–64.
Hearing MC, Jedynak J, Ebner SR, Ingebretson A, Asp AJ, Fischer RA, et al. Reversal of morphine-induced cell-type–specific synaptic plasticity in the nucleus accumbens shell blocks reinstatement. Proc Natl Acad Sci. 2016;113:757–62.
Ma T, Cheng Y, Roltsch Hellard E, Wang X, Lu J, Gao X, et al. Bidirectional and long-lasting control of alcohol-seeking behavior by corticostriatal LTP and LTD. Nat Neurosci. 2018;21:373–83.
Wise RA. Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotox Res. 2008;14:169–83.
Soares-Cunha C, De Vasconcelos NAP, Coimbra B, Domingues AV, Silva JM, Loureiro-Campos E, et al. Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol Psychiatry. 2020;25:3241–55.
Kheirbek MA, Beeler JA, Ishikawa Y, Zhuang X. A cAMP pathway underlying reward prediction in associative learning. J Neurosci. 2008;28:11401–8.
Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–59.
Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–7.
Calabresi P, Saiardi A, Pisani A, Baik J-H, Centonze D, Mercuri NB, et al. Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci. 1997;17:4536–44.
Beeler JA, Frank MJ, McDaid J, Alexander E, Turkson S, Bernandez MS, et al. A role for dopamine-mediated learning in the pathophysiology and treatment of Parkinson’s disease. Cell Rep. 2012;2:1747–61.
Beeler JA, Cao ZFH, Kheirbek MA, Ding YM, Koranda J, Murakami M, et al. Dopamine-dependent motor learning insight into levodopa’s long-duration response. Ann Neurol. 2010;67:639–47.
Koranda JL, Krok AC, Xu J, Contractor A, McGehee DS, Beeler JA, et al. Chronic nicotine mitigates aberrant inhibitory motor learning induced by motor experience under dopamine deficiency. J Neurosci. 2016;36:5228–40.
Cheung THC, Ding Y, Zhuang X, Kang UJ. Learning critically drives parkinsonian motor deficits through imbalanced striatal pathway recruitment. Proc Natl Acad Sci. 2023;120:e2213093120.
Friedman JH. Viewpoint: challenges in our understanding of neuroleptic induced parkinsonism. Parkinsonism Relat D. 2014;20:1325–8.
Shin HW, Chung SJ. Drug-induced parkinsonism. J Clin Neurol. 2012;8:15–21.
Susatia F, Fernandez HH. Drug-induced parkinsonism. Curr Treat Option Ne. 2009;11:162–9.
Jimenez-Jimenez FJ, Orti-Pareja M, Ayuso-Peralta L, Gasalla T, Cabrera-Valdivia F, Vaquero A, et al. Drug-induced parkinsonism in a movement disorders unit: A four-year survey. Parkinsonism Relat Disord. 1996;2:145–9.
Mena MA, de Yebenes JG. Drug-induced parkinsonism. Expert Opin Drug Saf. 2006;5:759–71.
Kuzuhara S. Drug-induced parkinsonism. Nihon Rinsho. 1997;55:112–7.
Seeman P, Tallerico T. Antipsychotic drugs which elicit little or no Parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol Psychiatry. 1998;3:123–34.
Kim JS, Youn J, Shin H, Cho JW. Nonmotor symptoms in drug-induced parkinsonism and drug-naive Parkinson disease. Can J Neurol Sci. 2013;40:36–41.
Morley JF, Pawlowski SM, Kesari A, Maina I, Pantelyat A, Duda JE. Motor and non-motor features of Parkinson’s disease that predict persistent drug-induced Parkinsonism. Parkinsonism Relat D. 2014;20:738–42.
d’Errico A, Strippoli E, Vasta R, Ferrante G, Spila Alegiani S, Ricceri F. Use of antipsychotics and long-term risk of parkinsonism. Neurol Sci. 2022;43:2545–53.
Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, et al. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–22.
Amtage J, Schmidt WJ. Context-dependent catalepsy intensification is due to classical conditioning and sensitization. Behav Pharmacol. 2003;14:563–7.
Boulay D, Depoortere R, Oblin A, Sanger DJ, Schoemaker H, Perrault G. Haloperidol-induced catalepsy is absent in dopamine D-2 but maintained in dopamine D-3 receptor knock-out mice. Eur J Pharmacol. 2000;391:63–73.
Gongora-Alfaro JL, Moo-Puc RE, Villanueva-Toledo JR, Alvarez-Cervera FJ, Bata-Garcia JL, Heredia-Lopez FJ, et al. Long-lasting resistance to haloperidol-induced catalepsy in male rats chronically treated with caffeine. Neurosci Lett. 2009;463:210–4.
Trevitt J, Vallance C, Harris A, Goode T. Adenosine antagonists reverse the cataleptic effects of haloperidol: Implications for the treatment of Parkinson’s disease. Pharmacol Biochem Be. 2009;92:521–7.
Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9.
Pizzagalli DA. Depression, stress, and anhedonia: toward a synthesis and integrated model. Annu Rev Clin Psychol. 2014;10:393–423.
Chen C, Takahashi T, Nakagawa S, Inoue T, Kusumi I. Reinforcement learning in depression: a review of computational research. Neurosci Biobehav Rev. 2015;55:247–67.
Huys QJ, Pizzagalli DA, Bogdan R, Dayan P. Mapping anhedonia onto reinforcement learning: a behavioural meta-analysis. Biol Mood Anxiety Disord. 2013;3:12.
Cummings JL. Depression and Parkinson’s disease: a review. Am J Psychiatry. 1992;149:443–54.
Vriend C, Pattij T, van der Werf YD, Voorn P, Booij J, Rutten S, et al. Depression and impulse control disorders in Parkinson’s disease: two sides of the same coin? Neurosci Biobehav Rev. 2014;38:60–71.
Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–9.
Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. 2015;77:212–22.
Fox ME, Chandra R, Menken MS, Larkin EJ, Nam H, Engeln M, et al. Dendritic remodeling of D1 neurons by RhoA/Rho-kinase mediates depression-like behavior. Mol Psychiatry. 2020;25:1022–34.
Nord CL, Lawson RP, Huys QJM, Pilling S, Roiser JP. Depression is associated with enhanced aversive Pavlovian control over instrumental behaviour. Sci Rep. 2018;8:12582.
Moran RJ, Kishida KT, Lohrenz T, Saez I, Laxton AW, Witcher MR, et al. The protective action encoding of serotonin transients in the human brain. Neuropsychopharmacology. 2018;43:1425–35.
Dayan P, Huys QJM. Serotonin, inhibition, and negative mood. PLoS Computational Biol. 2008;4:e4.
Zhong W, Li Y, Feng Q, Luo M. Learning and stress shape the reward response patterns of serotonin neurons. J Neurosci. 2017;37:8863–75.
Li Y, Zhong W, Wang D, Feng Q, Liu Z, Zhou J, et al. Serotonin neurons in the dorsal raphe nucleus encode reward signals. Nat Commun. 2016;7:10503.
Cohen JY, Amoroso MW, Uchida N. Serotonergic neurons signal reward and punishment on multiple timescales. eLife. 2015;4:e06346.
Moncrieff J, Cooper RE, Stockmann T, Amendola S, Hengartner MP, Horowitz MA. The serotonin theory of depression: a systematic umbrella review of the evidence. Mol Psychiatry. 2023;28:3243–56.
Luscher C, Pascoli V, Creed M. Optogenetic dissection of neural circuitry: from synaptic causalities to blue prints for novel treatments of behavioral diseases. Curr Opin Neurobiol. 2015;35:95–100.
Creed M. Current and emerging neuromodulation therapies for addiction: insight from pre-clinical studies. Curr Opin Neurobiol. 2018;49:168–74.
Lüscher C. The emergence of a circuit model for addiction. Annu Rev Neurosci. 2016;39:257–76.
Cheng Y, Huang CCY, Ma T, Wei X, Wang X, Lu J, et al. Distinct synaptic strengthening of the striatal direct and indirect pathways drives alcohol consumption. Biol Psychiatry. 2017;81:918–29.
Salling MC, Martinez D. Brain stimulation in addiction. Neuropsychopharmacology. 2016;41:2798–809.
Wang TR, Moosa S, Dallapiazza RF, Elias WJ, Lynch WJ. Deep brain stimulation for the treatment of drug addiction. Neurosurg Focus. 2018;45:E11.
Chang R, Peng J, Chen Y, Liao H, Zhao S, Zou J, et al. Deep brain stimulation in drug addiction treatment: research progress and perspective. Front Psychiatry. 2022;13:858638.
Yuen J, Kouzani AZ, Berk M, Tye SJ, Rusheen AE, Blaha CD, et al. Deep brain stimulation for addictive disorders—where are we now? Neurotherapeutics. 2022;19:1193–215.
Kuhn J, Lenartz D, Huff W, Lee S, Koulousakis A, Klosterkoetter J, et al. Remission of alcohol dependency following deep brain stimulation of the nucleus accumbens: valuable therapeutic implications? J Neurology, Neurosurg Psychiatry. 2007;78:1152–3.
Müller U, Sturm V, Voges J, Heinze HJ, Galazky I, Büntjen L, et al. Nucleus accumbens deep brain stimulation for alcohol addiction – safety and clinical long-term results of a pilot trial. Pharmacopsychiatry. 2016;49:170–3.
Heinze HJ, Heldmann M, Voges J, Hinrichs H, Marco-Pallares J, Hopf JM, et al. Counteracting incentive sensitization in severe alcohol dependence using deep brain stimulation of the nucleus accumbens: clinical and basic science aspects. Front Hum Neurosci. 2009;3:22.
Goncalves-Ferreira A, do Couto FS, Campos AR, Neto LPL, Goncalves-Ferreira D, Teixeira J. Deep brain stimulation for refractory cocaine dependence. Biol Psychiatry. 2016;79:E87–E9.
Nutt JG, Carter JH, Woodward WR. Long-duration response to levodopa. Neurology. 1995;45:1613–6.
Anderson ED, Horak FB, Lasarev MR, Nutt JG. Performance of a motor task learned on levodopa deteriorates when subsequently practiced off. Mov Disord. 2014;29:54–60.
Kang UJ, Auinger P, ELLDOPA PSG. Activity enhances dopaminergic long-duration response in Parkinson disease. Neurology. 2012;78:1146–9.
LeWitt PA, Guttman M, Tetrud JW, Tuite PJ, Mori A, Chaikin P, et al. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol. 2008;63:295–302.
Tao YQ, Liang GB. Efficacy of adenosine A2A receptor antagonist istradefylline as augmentation for Parkinson’s disease: a meta-analysis of randomized controlled trials. Cell Biochem Biophys. 2015;71:57–62.
Mizuno Y, Kondo T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov Disord. 2013;28:1138–41.
Koran LM, Hanna GL, Hollander E, Nestadt G, Simpson HB, American Psychiatric A. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry. 2007;164:5–53.
Simpson HB, Foa EB, Liebowitz MR, Huppert JD, Cahill S, Maher MJ, et al. Cognitive-behavioral therapy vs risperidone for augmenting serotonin reuptake inhibitors in obsessive-compulsive disorder. JAMA Psychiatry. 2013;70:1190.
Kohl S, Schönherr DM, Luigjes J, Denys D, Mueller UJ, Lenartz D, et al. Deep brain stimulation for treatment-refractory obsessive compulsive disorder: a systematic review. BMC Psychiatry. 2014;14:214.
Cruz S, Gutierrez-Rojas L, Gonzalez-Domenech P, Diaz-Atienza F, Martinez-Ortega JM, Jimenez-Fernandez S. Deep brain stimulation in obsessive-compulsive disorder: results from meta-analysis. Psychiatry Res. 2022;317:114869.
Alonso P, Cuadras D, Gabriëls L, Denys D, Goodman W, Greenberg BD, et al. Deep Brain Stimulation for obsessive-compulsive disorder: a meta-analysis of treatment outcome and predictors of response. PLoS ONE. 2015;10:e0133591.
Huys D, Kohl S, Baldermann JC, Timmermann L, Sturm V, Visser-Vandewalle V, et al. Open-label trial of anterior limb of internal capsule-nucleus accumbens deep brain stimulation for obsessive-compulsive disorder: insights gained. J Neurol Neurosurg Psychiatry. 2019;90:805–12.
Widge AS. Closing the loop in psychiatric deep brain stimulation: physiology, psychometrics, and plasticity. Neuropsychopharmacology. 2024;49:138–49.
Zhou C, Zhang H, Qin Y, Tian T, Xu B, Chen J, et al. A systematic review and meta-analysis of deep brain stimulation in treatment-resistant depression. Prog Neuropsychopharmacol Biol Psychiatry. 2018;82:224–32.
Dandekar MP, Fenoy AJ, Carvalho AF, Soares JC, Quevedo J. Deep brain stimulation for treatment-resistant depression: an integrative review of preclinical and clinical findings and translational implications. Mol Psychiatry. 2018;23:1094–112.
LeGates TA, Kvarta MD, Tooley JR, Francis TC, Lobo MK, Creed MC, et al. Reward behaviour is regulated by the strength of hippocampus-nucleus accumbens synapses. Nature. 2018;564:258–62.
LeDoux JE. The day I told Karim Nader, “Don’t do the study”. Brain Res Bull. 2022;189:1–3.
Ferrara NC, Kwapis JL, Trask S. Memory retrieval, reconsolidation, and extinction: exploring the boundary conditions of post-conditioning cue exposure. Front Synaptic Neurosci. 2023;15:1146665.
Centonze D, Siracusano A, Calabresi P, Bernardi G. Removing pathogenic memories: a neurobiology of psychotherapy. Mol Neurobiol. 2005;32:123–32.
Steenen SA, van Wijk AJ, van der Heijden GJ, van Westrhenen R, de Lange J, de Jongh A. Propranolol for the treatment of anxiety disorders: systematic review and meta-analysis. J Psychopharmacol. 2016;30:128–39.
Roullet P, Vaiva G, Very E, Bourcier A, Yrondi A, Dupuch L, et al. Traumatic memory reactivation with or without propranolol for PTSD and comorbid MD symptoms: a randomised clinical trial. Neuropsychopharmacology. 2021;46:1643–9.
Pigeon S, Lonergan M, Rotondo O, Pitman RK, Brunet A. Impairing memory reconsolidation with propranolol in healthy and clinical samples: a meta-analysis. J Psychiatry Neurosci. 2022;47:E109–E22.
Brunet A, Orr SP, Tremblay J, Robertson K, Nader K, Pitman RK. Effect of post-retrieval propranolol on psychophysiologic responding during subsequent script-driven traumatic imagery in post-traumatic stress disorder. J Psychiatr Res. 2008;42:503–6.
Haubrich J, Nader K. Memory Reconsolidation. Springer International Publishing; 2016. p. 151–76.
Gamache K, Pitman RK, Nader K. Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology. 2012;37:2789–96.
Haubrich J, Bernabo M, Nader K. Noradrenergic projections from the locus coeruleus to the amygdala constrain fear memory reconsolidation. eLife. 2020;9:e57010.
Monfils M-H, Cowansage KK, Klann E, Ledoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–5.
Davis M, Myers KM. The role of glutamate and gamma-aminobutyric acid in fear extinction: clinical implications for exposure therapy. Biol Psychiatry. 2002;52:998–1007.
Noel X. A critical perspective on updating drug memories through the integration of memory editing and brain stimulation. Front Psychiatry. 2023;14:1161879.
Lin X, Deng J, Yuan K, Wang Q, Liu L, Bao Y, et al. Neural substrates of propranolol-induced impairments in the reconsolidation of nicotine-associated memories in smokers. Transl Psychiatry. 2021;11:441.
Exton-Mcguinness MTJ, Patton RC, Sacco LB, Lee JLC. Reconsolidation of a well-learned instrumental memory. Learn Mem. 2014;21:468–77.
Tedesco V, Mutti A, Auber A, Chiamulera C. Nicotine-seeking reinstatement is reduced by inhibition of instrumental memory reconsolidation. Behav Pharmacol. 2014;25:725–31.
Exton-McGuinness MTJ, Drame ML, Flavell CR, Lee JLC. On the resistance to relapse to cocaine-seeking following impairment of instrumental cocaine memory reconsolidation. Front Behav Neurosci. 2019;13:242.
Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26.
Lüscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–50.
Sheng M, Ertürk A. Long-term depression: a cell biological view. Philos Trans R Soc B: Biol Sci. 2014;369:20130138.
Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–47.
Cang J, Feldheim DA. Developmental mechanisms of topographic map formation and alignment. Annu Rev Neurosci. 2013;36:51–77.
Oswald Steward PW. Local synthesis of proteins at synaptic sites on dendrites: role in synaptic plasticity and memory consolidation? Neurobiol Learn Mem. 2002;78:508–27.
Parrish JZ, Emoto K, Kim MD, Jan YN. Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Annu Rev Neurosci. 2007;30:399–423.
Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97.
Horton AC, Ehlers MD. Secretory trafficking in neuronal dendrites. Nat Cell Biol. 2004;6:585–91.
Arriagada-Diaz J, Prado-Vega L, Diaz AMC, Ardiles AO, Gonzalez-Jamett AM. Dynamin superfamily at pre- and postsynapses: master regulators of synaptic transmission and plasticity in health and disease. Neuroscientist. 2022;28:41–58.
Yang YR, Liu JJ. Structural LTP: Signal transduction, actin cytoskeleton reorganization, and membrane remodeling of dendritic spines. Curr Opin Neurobiol. 2022;74:102534.
Iwata Y, Nakajima S, Suzuki T, Keefe RS, Plitman E, Chung JK, et al. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta-analysis of double-blind randomized controlled trials. Mol Psychiatry. 2015;20:1151–60.
Hackos DH, Lupardus PJ, Grand T, Chen Y, Wang TM, Reynen P, et al. Positive allosteric modulators of GluN2A-Containing NMDARs with distinct modes of action and impacts on circuit function. Neuron. 2016;89:983–99.
Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–54.
Litman RE, Smith MA, Doherty JJ, Cross A, Raines S, Gertsik L, et al. AZD8529, a positive allosteric modulator at the mGluR2 receptor, does not improve symptoms in schizophrenia: a proof of principle study. Schizophr Res. 2016;172:152–7.
Salih H, Anghelescu I, Kezic I, Sinha V, Hoeben E, Van Nueten L, et al. Pharmacokinetic and pharmacodynamic characterisation of JNJ-40411813, a positive allosteric modulator of mGluR2, in two randomised, double-blind phase-I studies. J Psychopharmacol. 2015;29:414–25.
Kandel ER. Neuroscience - The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–8.
Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14.
Hernandez PJ, Sadeghian K, Kelley AE. Early consolidation of instrumental learning requires protein synthesis in the nucleus accumbens. Nat Neurosci. 2002;5:1327–31.
Jedynak J, Hearing M, Ingebretson A, Ebner SR, Kelly M, Fischer RA, et al. Cocaine and amphetamine induce overlapping but distinct patterns of AMPAR plasticity in nucleus accumbens medium spiny neurons. Neuropsychopharmacology. 2016;41:464–76.
Scheyer AF, Wolf ME, Tseng KY. A protein synthesis-dependent mechanism sustains calcium-permeable AMPA receptor transmission in nucleus accumbens synapses during withdrawal from cocaine self-administration. J Neurosci. 2014;34:3095–100.
Di Prisco GV, Huang W, Buffington SA, Hsu CC, Bonnen PE, Placzek AN, et al. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat Neurosci. 2014;17:1073–82.
Jiang CG, Schuman EM. Regulation and function of local protein synthesis in neuronal dendrites. Trends Biochem Sci. 2002;27:506–13.
Holt CE, Schuman EM. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron. 2013;80:648–57.
Haubrich J, Bernabo M, Baker AG, Nader K. Impairments to consolidation, reconsolidation, and long-term memory maintenance lead to memory erasure. Annu Rev Neurosci. 2020;43:297–314.
Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–6.
Peng J-Y, Li B-M. Protein synthesis is essential not only for consolidation but also for maintenance and post-retrieval reconsolidation of acrobatic motor skill in rats. Mol Brain. 2009;2:12.
Longo F, Mancini M, Ibraheem PL, Aryal S, Mesini C, Patel JC, et al. Cell-type-specific disruption of PERK-eIF2α signaling in dopaminergic neurons alters motor and cognitive function. Mol Psychiatry. 2021;26:6427–50.
Huang W, Placzek AN, Viana Di Prisco G, Khatiwada S, Sidrauski C, Krnjevic K, et al. Translational control by eIF2alpha phosphorylation regulates vulnerability to the synaptic and behavioral effects of cocaine. eLife. 2016;5:e12052.
Placzek AN, Molfese DL, Khatiwada S, Viana Di Prisco G, Huang W, Sidrauski C, et al. Translational control of nicotine-evoked synaptic potentiation in mice and neuronal responses in human smokers by eIF2alpha. eLife. 2016;5:e12056.
Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–37.
Walker DM, Cates HM, Heller EA, Nestler EJ. Regulation of chromatin states by drugs of abuse. Curr Opin Neurobiol. 2015;30:112–21.
Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, et al. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat Neurosci. 2013;16:434–40.
Covington HE, Vialou VF, Laplant Q, Ohnishi YN, Nestler EJ. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci Lett. 2011;493:122–6.
Covington HE, Maze I, Laplant QC, Vialou VF, Ohnishi YN, Berton O, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–60.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–25.
Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58.
Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, et al. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–93.
Perez J, Schuman E. Subcellular RNA-seq for the analysis of the dendritic and somatic transcriptomes of single neurons. BIO-PROTOCOL. 2022;12:e4278.
Fusco CM, Desch K, Dörrbaum AR, Wang M, Staab A, Chan ICW, et al. Neuronal ribosomes exhibit dynamic and context-dependent exchange of ribosomal proteins. Nat Commun. 2021;12:6127.
Glock C, Biever A, Tushev G, Nassim-Assir B, Kao A, Bartnik I, et al. The translatome of neuronal cell bodies, dendrites, and axons. Proc Natl Acad Sci. 2021;118:e2113929118.
Das S, Singer RH, Yoon YJ. The travels of mRNAs in neurons: do they know where they are going? Curr Opin Neurobiol. 2019;57:110–6.
Kosik KS. Life at low copy number: how dendrites manage with so few mRNAs. Neuron. 2016;92:1168–80.
Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Gene Dev. 2009;23:1–11.
Bhakar AL, Dölen G, Bear MF. The Pathophysiology of Fragile X (and What It Teaches Us about Synapses). Annu Rev Neurosci. 2012;35:417–43.
Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Primers. 2017;3:17065.
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci. 2002;99:7746–50.
Hagerman RJ, Des-Portes V, Gasparini F, Jacquemont S, Gomez-Mancilla B. Translating molecular advances in fragile X syndrome into therapy: a review. J Clin Psychiatry. 2014;75:e294–307.
Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3:64ra1.
Notaras M, Allen M, Longo F, Volk N, Toth M, Li Jeon N, et al. UPF2 leads to degradation of dendritically targeted mRNAs to regulate synaptic plasticity and cognitive function. Mol Psychiatry. 2020;25:3360–79.
Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat Rev Genet. 2021;22:119–31.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15:293–306.
Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798.
Jia GF, Fu Y, Zhao X, Dai Q, Zheng GQ, Yang Y, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Liu N, Dai Q, Zheng GQ, He C, Parisien M, Pan T. N-6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Wang X, He C. Reading RNA methylation codes through methyl-specific binding proteins. RNA Biol. 2014;11:669–72.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Zhu TT, Roundtree IA, Wang P, Wang X, Wang L, Sun C, et al. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014;24:1493–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46.
Widagdo J, Zhao Q-Y, Kempen M-J, Tan MC, Ratnu VS, Wei W, et al. Experience-dependent accumulation of N6-Methyladenosine in the prefrontal cortex is associated with memory processes in mice. J Neurosci. 2016;36:6771–7.
Sikorski V, Selberg S, Lalowski M, Karelson M, Kankuri E. The structure and function of YTHDF epitranscriptomic m(6)A readers. Trends Pharmacol Sci. 2023;44:335–53.
Flamand MN, Tegowski M, Meyer KD. The proteins of mRNA modification: writers, readers, and erasers. Annu Rev Biochem. 2023;92:145–73.
Sendinc E, Shi Y. RNA m6A methylation across the transcriptome. Mol Cell. 2023;83:428–41.
Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. 2023;24:143–60.
Murakami S, Jaffrey SR. Hidden codes in mRNA: control of gene expression by m(6)A. Mol Cell. 2022;82:2236–51.
Moshitch-Moshkovitz S, Dominissini D, Rechavi G. The epitranscriptome toolbox. Cell. 2022;185:764–76.
Koranda JL, Dore L, Shi H, Patel MJ, Vaasjo LO, Rao MN, et al. Mettl14 is essential for epitranscriptomic regulation of striatal function and learning. Neuron. 2018;99:283–92.e5.
Zhang Z, Wang M, Xie D, Huang Z, Zhang L, Yang Y, et al. METTL3-mediated N6-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 2018;28:1050–61.
Satterlee JS, Basanta-Sanchez M, Blanco S, Li JB, Meyer K, Pollock J, et al. Novel RNA Modifications in the Nervous System: Form and Function. J Neurosci. 2014;34:15170–7.
Hess ME, Hess S, Meyer KD, Verhagen LAW, Koch L, Bronneke HS, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–U96.
Merkurjev D, Hong W-T, Iida K, Oomoto I, Goldie BJ, Yamaguti H, et al. Synaptic N6-methyladenosine (m6A) epitranscriptome reveals functional partitioning of localized transcripts. Nat Neurosci. 2018;21:1004–14.
Kan L, Ott S, Joseph B, Park ES, Dai W, Kleiner RE, et al. A neural m6A/Ythdf pathway is required for learning and memory in Drosophila. Nat Commun. 2021;12:1458.
Shi H, Zhang X, Weng Y-L, Lu Z, Liu Y, Lu Z, et al. m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. 2018;563:249–53.
Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 2018;27:3936–50.
Zou Z, Wei J, Chen Y, Kang Y, Shi H, Yang F, et al. FMRP phosphorylation modulates neuronal translation through YTHDF1. Mol Cell. 2023;83:4304–17.e8.
Wen K, Shi Z, Yu P, Mo L, Sullere S, Yang V, et al. Opposing motor memories in the direct and indirect pathways of the basal ganglia. bioRxiv. [Preprint]. 2024 https://www.biorxiv.org/content/10.1101/2024.02.26.582159v1.full.
Shi ZWK, Zou Z, Fu W, Guo K, Sammudin NH, Ruan X, Sullere S, Wang S, Zhang X, Thinakaran G, He C, Zhuang X YTHDF1 mediates translational control by m6A mRNA methylation in adaptation to environmental challenges. BioRxiv. [Preprint]. 2024 https://doi.org/10.1101/2024.08.07.607063.
Boyden ES. Optogenetics and the future of neuroscience. Nat Neurosci. 2015;18:1200–1.
Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412.
Dong S, Rogan SC, Roth BL. Directed molecular evolution of DREADDs: a generic approach to creating next-generation RASSLs. Nat Protoc. 2010;5:561–73.
Funding
This work was supported in part by research grants from NIDA (R01DA044997) and NINDS (R01NS095374).
Author information
Authors and Affiliations
Contributions
XZ conceived the review. ZS, KW and XZ gathered references and wrote the first draft. NHS and NL edited the following two revisions. All authors wrote, reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, Z., Wen, K., Sammudin, N.H. et al. Erasing “bad memories”: reversing aberrant synaptic plasticity as therapy for neurological and psychiatric disorders. Mol Psychiatry 30, 3209–3225 (2025). https://doi.org/10.1038/s41380-025-03013-0
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41380-025-03013-0
