
Abstract
Genetic instability is a hallmark of cancer cells. Homologous recombination (HR) plays a pivotal role in maintaining genome stability through its DNA repair and replication fork escort functions. Therefore, HR is classified as a tumour suppressor pathway. Consistently, many HR genes are mutated in cancer, especially in hereditary breast and ovarian cancer. However, although RAD51 controls the central steps of HR, no RAD51 mutations are associated with cancer predisposition, constituting the “RAD51 paradox”. One of the potential explanations for the “RAD51 paradox” is that mutations affecting mediator/accessory genes (such as BRCA1 or BRCA2) in cancer result in the absence of RAD51 on damaged DNA, leaving access to alternative exclusively mutagenic repair processes, such as single-strand annealing (SSA) or alternative end-joining (A-EJ), which can rescue some cell viability but also increase genetic instability. This raises the question of whether cancer predisposition actually results from HR deficiency itself or from alternative, nonconservative repair pathways. One study assessing this question in a mouse model revealed that decreasing RAD51 HR activity without stimulating SSA or A-EJ in vivo not only does not favour tumorigenesis but rather protects against it. These data suggest that RAD51-controlled HR is not a tumour suppressor but rather favours tumour progression. Cancer cells are highly proliferative, actively replicating their genomes, and are therefore subjected to high replication stress; pathways enabling them to cope with this massive replication stress, such as HR, should help them survive and proliferate, contrary to the belief dogma that HR acts as a tumour suppressor pathway. We propose that HR/RAD51, through its essential role in overcoming replication stress, should facilitate cancer progression as soon as early pretumorigenic hyperplasia states that trigger an active replication program, challenging commonly accepted views.
Similar content being viewed by others


Genome instability is a hallmark of aging and cancer cells [1,2,3]. Homologous recombination (HR), an evolutionarily conserved process, plays a crucial role in maintaining genome stability through the repair of DNA double-strand breaks (DSBs) and interstrand crosslinks (ICLs) and the protection and resumption of arrested replication forks [4, 5]. Therefore, HR is generally classified as a tumour suppressor pathway. Consistent with this, driver mutations in many HR genes, such as BRCA1 and BRCA2, are observed in cancer, especially in hereditary breast or ovary cancer [6, 7].
However, a comprehensive assessment of the roles of HR gene products and the consequences of their inactivation, not solely focused on HR itself, can lead to conclusions that challenge the commonly accepted idea that HR is a tumour-suppressing pathway. Indeed, most cancer cells are highly proliferative and therefore actively replicate their genome. However, the progression of replication forks is routinely hampered by both exogenous and endogenous factors. Consequently, cancer cells are subjected to considerable replicative stress. Therefore, mechanisms that allow cancer cells to cope with replication stress should favour tumour progression. Since HR plays a central role in overcoming replication stress, it should thus favour cancer cell survival and proliferation and, consequently, tumour progression. Here, we discuss this provocative point of view.
RAD51 plays a pivotal role in the key step of HR, i.e., the search for homologous sequences and strand exchange of homologous sequences (the step for which the mechanism was named). This role highlights an important concept and topic of this review—the “RAD51 paradox” [7]: the HR genes that are mutated in cancer encode accessory or mediator proteins, but despite the central role of RAD51 in HR, in contrast to that of other HR genes, the inactivation of RAD51 is not associated with cancer predisposition; rather, the overexpression of RAD51 is associated with a poor prognosis in different types of tumours [7,8,9,10,11,12,13,14].
The accessory/mediator proteins, whose genes are mutated in cancer, promote the loading of RAD51 and the stabilisation of the RAD51 filament, which then both induce HR and protect against alternative mutagenic repair processes. The inactivation of the mediator/accessory proteins results in the absence of RAD51 on damaged DNA, thus providing access to alternative nonconservative repair pathways such as single-strand annealing (SSA) or alternative end-joining (A-EJ) [15], which can preserve some cell viability but are exclusively mutagenic. This raises the following question: does cancer predisposition result from an inability to perform HR, from the activation of nonconservative repair processes, or from both mechanisms? Here, we will also discuss a mouse model that allows us to address the above question. Indeed, this mouse model expresses an inducible dominant negative form of RAD51 that specifically down-regulates HR [16] but still prevents the nonconservative repair pathway because it binds to the DNA [15, 16]. Remarkably, decreasing HR without stimulating alternative nonconservative pathways in vivo induced a pronounced premature aging phenotype but did not induce tumorigenesis and even, in contrast, protected against it [16]. This finding does not support the hypothesis that RAD51 and HR are a tumour suppressor pathway but rather that these factors might play a role in facilitating tumour progression in vivo.
The concepts and conclusions of the comment discussed here require that we first recall the main molecular mechanisms of the different repair pathways potentially involved, which have already been largely detailed in the literature, the “RAD51 paradox” and the RAD51 mouse model; then, we discuss them to explain why RAD51-mediated HR can be considered a tumour support pathway. By challenging the common view, the aim of this review is to redefine and determine the actual impact of RAD51 and HR on cancer predisposition/prevention, highlighting the importance of the balance between HR/RAD51 and alternative nonconservative repair pathways.
Main molecular steps of HR
HR-mediated DSB repair
The molecular role of RAD51 in HR can be described in the HR-mediated DSB repair model (Fig. 1), which illustrates the successive HR molecular steps [17]. Briefly, HR is initiated by single-strand resection of the DSB, generating 3’ single-strand DNA (ssDNA), and then RAD51 is loaded on the ssDNA by mediators such as BRCA1-BRCA2-PALB2, leading to the formation of the RAD51-ssDNA filament. Of note, BRCA1 plays an upstream role in the choice of the DSB repair pathway, favouring resection initiation; it is therefore considered a “resection licensing factor”. The other factors such as the RAD51 paralogs are “assisting factor on Rad51 stability and dynamics”. Since HR relies on sequence homology, its pivotal step is the search for DNA sequence homologies and the exchange of homologous sequences. This step is promoted by the RAD51-ssDNA filament, which thus represents the actual “active species” of HR.

First, the MRN complex (MRE11/RAD50/NBS1), in cooperation with the ataxia-telangiectasia mutated (ATM) kinase and the chromatin remodelling machinery, recognises DSBs and induces signalling pathways. The subsequent steps of HR can be summarised as follows: (1) The resection of the DSB generates a 3’ ssDNA stretch, which is coated with the replication protein RPA; (2) and (3) the mediators (BRCA1, BRCA2, PALB2) replace RPA by RAD51 on the ssDNA, forming the RAD51-ssDNA filament, which then promotes the search for homology and the invasion of a duplex DNA molecule harbouring homologous sequences; this corresponds to the central pivotal step of HR; (4) capture of the second DSB; (5) DNA synthesis is then primed using the invading 3’ ssDNA; and (6) the resolution of the HR intermediates leads to gene conversions associated or not with crossover or synthesis-dependent strand annealing (SDSA) or break-induced replication (BIR) [4, 96, 97]. Some of the main factors that are mutated in hereditary breast or ovary cancer are shown in the figure.
Recently it has been shown that in the absence of BRCA2, RAD51 may still be able to sustain some suboptimal recombination functions, massively antagonized by FIGNL1. Indeed, in the absence of BRCA2, RAD51 can be loaded onto ssDNA and promote some HR events when FIGNL1 is suppressed [18, 19]. FIGNL1 plays roles in modulating RAD51, by preventing its association to chromatin; loss of FIGNL1 allows thus RAD51 to load at DNA double-stranded breaks in BRCA2-deficient cells, rescuing some HR proficiency and viability upon exposure to Olaparib, a PARP inhibitor, or to cisplatin [18].
HR, a replication fork escort pathway
In addition to DSB repair, HR and RAD51 play essential roles in the protection and resumption of arrested replication forks. Replication fork progression is routinely obstructed by different endogenous factors, such as conflicts with transcription, regions that are difficult to replicate, nicks or gaps in the DNA matrix, molecules that covalently bind to DNA, and damages generated by endogenous reactive oxygen species (ROS) [20,21,22]. Reciprocally replication stress generates the production of ROS by the cell [23,24,25]. Exogenous stresses such as chemicals (notably chemotherapy) or ionizing radiation can also induce DNA damage that blocks the progression of replication forks. Notably, unrepaired DSBs lead to replication fork collapse, and reciprocally, the arrest of replication forks can generate DSBs [26].
In the absence of exogenous stress, defects in HR spontaneously affect replication dynamics [27, 28], notably because of the higher endogenous level of ROS [22]. Moreover, HR proteins are found on normal replicating forks [29].
HR actors are essential for both protecting arrested replication forks and resuming DNA synthesis at these forks (Fig. 2).

Different causes can lead to the arrest of replication fork progression, such as a blocking lesion (left panel) or a single-stranded nick or gap (middle panel). When a blocking lesion is reached (left panels), the reversion of the blocked replication fork can form a cruciform tetraplex structure frequently referred to as the “chicken foot” (left panels). RAD51 associated with or not associated with its mediators participates in fork reversion and, in addition, protects the reverted fork from degradation. Then, the resolution of the cruciform intermediate by a structure-specific endonuclease (for example, MUS81) generates a one-ended DSB. In the middle panels, when a progressing replication fork reaches a nick or a gap, this inevitably generates a broken fork with a one-ended DSB (middle panel). RAD51 loaded on the broken forks, arising from both the fork reversion (left panel) or from the nick/gap (middle panel), protects them from degradation and allows replication to resume through strand exchange with a homologous sequence, typically the sister chromatid (middle panel). As shown in the right panel, when the replication fork collapses, uncoupling with replication allows it to continue on the replicating sister chromatid. Then, fork regression generates single-stranded DNA (which corresponds to the molecule on which replication has been arrested). RAD51 loaded on this ssDNA molecule (1) protects it from degradation and (2) allows replication to resume after strand invasion of the downstream sequence, which is homologous.
RAD51 plays central roles in both these protection and restarting processes. Notably, RAD51 also plays roles that can be independent of its strand exchange activities and of other HR proteins (for review see) [30]. For example, extensive degradation initiated by MRE11 and EXO1/DNA2 can attack a reversed fork (Fig. 2, left panel). By loading RAD51 on the reversed arm, BRCA2 participates in fork protection against such degradation [31, 32]. Since replication fork reversal can also occur in BRCA2 KO cells [33,34,35], this finding suggests that RAD51 can act even in the absence of BRCA2 at such a step. One hypothesis is that when ssDNA tracts are short, mediators (such as BRCA2) are not required to load RAD51. Moreover, several studies have concluded that strand exchange and fork protection are independent activities (for a review, see) [30]. Indeed, the mutant form of RAD51-II3A is capable of promoting fork reversal, but it is unable to catalyse strand exchange [36]. Consistently, RAD51 cannot catalyse fork reversal alone in vitro [37], whereas it is capable of performing strand exchange, supporting the idea that the two processes are, at least in part, mechanistically different.
Recently, additional roles for RAD51 in the protection of abasic sites preventing replication fork breakage have been described [38].
Therefore, HR, particularly RAD51, plays essential roles in replication fork protection, reversion and the restart of arrested replication. Since replication forks are faced with a series of unavoidable endogenous obstructions, HR components, and RAD51 in particular, are essential for cells to replicate their genome and thus proliferate.
Since tumour cells are highly proliferative and thus actively replicate their genome, they are subjected to strong endogenous replication stress. In addition, they generally exhibit high levels of ROS, which generate replication stress [22], and HR enables cancer cells to cope with replication stress induced by endogenous ROS [22]. Thus, HR helps to manage replication stress, promoting cell survival, genome replication and, ultimately, proliferation. Therefore, according to this view, HR and RAD51 should favour tumour progression.
However, this hypothesis contradicts the fact that mutations in HR genes are found in tumours and even confer a predisposition to cancers, notably hereditary breast and ovarian cancers [7]. To address this contradiction, two points need to be considered first: (i) the mechanisms that compete with HR for DSB repair and (ii) the RAD51 paradox.
Competition between the different DSB repair pathways
Several processes can repair DSBs: on the one hand, canonical nonhomologous end-joining (C-NHEJ), which is dependent on KU80/70-Ligase4, and on the other hand, HR, single-strand annealing (SSA) and alternative end-joining (A-EJ), which are all initiated by ssDNA resection. Importantly, while HR and C-NHEJ are conservative repair processes [4, 39], both SSA and A-EJ are exclusively mutagenic since they systematically lead to deletion at the reseal junctions (Fig. 3); thus, they are nonconservative processes. We have proposed that the selection of the DSB repair process operates in two steps: first, the choice between C-NHEJ and ssDNA resection; second, on resected DNA, the choice between HR and nonconservative repair pathways (SSA, A-EJ) [30, 39,40,41] (Fig. 3):

The selection of the DSB repair process involves two successive steps: 1-C-NHEJ (KU80/70-ligase IV and partners), which seals DSBs and competes with single-strand resection, which generates 3’ ssDNA. 2- On resected single strands, there is competition between HR and nonconservative DSB repair (SSA and A-EJ). Both SSA and A-EJ occur via the annealing of complementary ssDNA exposed by the resection (long annealed sequence for SSA, short annealed sequences for A-EJ). Importantly, both SSA and A-EJ are unavoidably mutagenic events since the intervening sequence is always lost. When RAD51 is loaded on ssDNA (left panel), it promotes HR through its homology search and strand exchange activities and, in parallel, inhibits the annealing of complementary sequences, preventing SSA and A-EJ through DNA occupancy, independent of its HR activities [15]. In the absence of RAD51 (right panel), HR cannot occur, but damaged DNA becomes susceptible to mutagenic SSA and A-EJ.
RAD51 plays a pivotal role in the second step through two separable activities: (1) RAD51 fosters HR through its homology search and strand exchange activities, and in parallel, (2) RAD51 blocks the annealing step required for SSA and A-EJ through DNA occupancy, independent of its strand exchange activity [15]. Indeed, mutant forms of RAD51, such as yeast/mammalian chimeric SMRAD51, are unable to promote strand exchange but are capable of binding ssDNA, protecting against nonconservative SSA and A-EJ showing the separation of HR activity from protection against SSA and A-EJ [15, 16].
Consequently, the absence of RAD51 results in HR defects and, concomitantly, the stimulation of the mutagenic repair processes SSA and A-EJ. Therefore, mutations in genes encoding mediators and accessory proteins of HR (such as BRCA1 and BRCA2), which load RAD51 or stabilise the RAD51-ssDNA filament, result in the absence of the RAD51 protein on damaged DNA and consequently in the stimulation of the mutagenic repair pathways SSA and A-EJ [15, 42,43,44,45,46,47,48,49]. This should (i) compensate, at least in part, for the viability lost due to the HR defect and (ii) increase genetic instability.
This raises the question of whether cancer predisposition actually results from HR defects themselves, from the stimulation of nonconservative pathways, or both.
The “RAD51 paradox”
Many HR genes are mutated in cancer, particularly in hereditary breast or ovarian cancer (Table 1). The majority of the genes listed directly or indirectly control HR and DSB signalling (Table 1) [7]. These findings strongly suggest the importance of this pathway in cancer aetiology and support the concept that HR is a tumour suppressor pathway.
However, the fact that HR is mutated in cancer cells is contradictory with the fact that HR is essential for cell viability not only during embryonic and foetal development but also for in vitro cultured cell lines [50,51,52,53,54]. One possibility is that HR is not completely abolished and that residual HR activity would be sufficient to maintain cell viability. For example, several mutations of BRCA1 found in tumours reduce but do not completely abolish HR efficiency [55, 56]. Moreover, one can propose that a compensation mechanism(s) can rescue sufficient viability for cancer cells to survive and proliferate. For example, the re-priming of replication through PRIMPOL, downstream the arrested replication fork might resume replication [57].
HR-deficient cells are sensitive to PARP inhibitors [58, 59], which are now used in the clinic. However, resistance to PARP inhibitors occurs frequently, including in tumours that were not previously treated with anticancer agents [60,61,62,63,64]. Moreover, most of the cell lines generated from HR-deficient tumours have rescued efficient HR [65]. These findings underscore the importance and advantages of maintaining some HR activities or exploiting alternative compensatory processes in cancer cells.
Surprisingly, mutations in the gene encoding RAD51 are absent from the list in Table 1, even though it drives the active nucleoprotein molecule responsible for the HR process (i.e., the search for homology and strand exchange of homologous sequences). Indeed, despite extensive investigation, mutations in RAD51 have not been shown to be tumour driver mutations, in contrast to mediator/accessory HR genes. In contrast, the overexpression of RAD51 is associated with a poor prognosis [7, 9,10,11,12,13,14]. This phenomenon constitutes what we previously named “the RAD51 paradox” [7].
Moreover, defects in the HR activity of RAD51 can be dissociated from cancer predisposition. First, germ-line mutations in several HR genes lead to Fanconi anaemia (FA) syndrome. FA is a rare autosomal recessive syndrome associated with bone marrow failure, developmental malformations, and predispositions to acute myeloid leukaemia and cancers. The following HR genes have been found to be mutated in FA: BRCA1 (FA-S), BRCA2 (FA-D1), PALB2 (FA-N), RAD51C (FA-O), BRIP1 (FA-J) and XRCC2 (FA-U) [66]. Mutations in RAD51 have been described in FA-like patients, but while they exhibit many developmental defects, no cancer predisposition has been associated with these RAD51 mutations to date. This contrasts with the other HR genes mutated in FA but shows a similar situation as for breast and ovary cancer and again feeds the “RAD51 paradox” [66,67,68,69,70].
Second, mutations in RAD51 have been described in congenital mirror movement (CMM) syndrome, a hereditary neurodevelopmental disorder affecting pure unimanual and bimanual asymmetric coordinated movements [30, 71]. Remarkably, this syndrome is not associated with cancer predisposition, although the RAD51 mutation affects its capacity to perform strand exchange in vitro [72].
Collectively, these data show that defects in RAD51 do not actually lead to cancer predisposition, challenging the concept that RAD51 is actually a tumour suppressor gene. Note that RAD51 possesses roles that are independent of BRCA1 or BRCA2 and even independent of the strand exchange activity, the non-canonical roles of RAD51 [30], as discussed below. Moreover, these data suggest that alterations in accessory/mediator HR genes can be compensated for viability.
Non canonical roles of RAD51
First, the binding of RAD51 on ssDNA prevents nonconservative mutagenic DNA repair, independently of its strand exchange activity but relies on its ssDNA occupancy [15]. Second, at the arrested replication forks, RAD51 plays several noncanonical roles in the formation, protection, and management of fork reversal, allowing for the resumption of replication [30]. Indeed, replication fork reversal can occur in cells deficient for BRCA2 [33,34,35]. The recruitment of RAD51 on the forks was suggested to be mediated by the direct interaction with DNA polymerase α, RAD54 or RAD51C [32, 37, 73, 74]. Moreover, MCM8 and MCM9, which are involved in the pre-replication complex (pre-RC) formation, DNA replication elongation, have been shown to favour the recruitment of BRCA1 and RAD51 at stalled forks, protecting them from excessive degradation [75]. It is proposed that when the ssDNA tracts are short, no mediator (such as BRCA2) would be required to remove RPA from them. In line with this, fork reversal can occur in cells from a Fanconi patient that express a mutant form of RAD51 RAD51-T131P, which is unable to form RAD51 foci when DNA damage occurs (thus long filament) [35]. Moreover, a mutant form of RAD51, RAD51-II3A, which this unable to catalyze strand exchange, still remains capable of promoting fork reversal [36], exemplifying the dissociation of function between strand exchange (canonical role) and fork reversal activities. Third, overexpression of RAD51-K133R, mutated in the ATP binding/hydrolysis site and defective in HR, in BRCA2-deficient cells restores the resistance of the forks resistant to degradation [31].
Third, RAD51 has also been implicated in post-replicative repair by translesion synthesis in a mechanisms not related to its strand exchange activity [76].
Fourth, RAD51 also exhibits noncanonical roles in RNA-mediated processes. TERRA (telomeric repeat-containing RNA) is a long non-coding RNA that forms R-loops at telomeres [77,78,79]. RAD51 promotes the recruitment of TERRA onto telomeres, but BRCA2 does not play a key role in this process [80]. However, the mutant RAD51-II3A, cannot promote the recruitment of TERRA onto telomeres [80], putting this hypothesis into question. Reactive oxygen species (ROS) can induce the formation of R-loop at the transcription sites. In human cells, the complex RAD51/RAD52/CSB (Cockayne syndrome protein B) detects such R-loops [81], RAD51 playing a role in the R-loops dissolution, independently of BRCA1/2 and the classical HR pathway [81]. More generally, R-loops triggered by transcription conflicts appear to be resolved by RAD51 through mechanisms that are independent from the canonical role of RAD51 in repair [81,82,83].
Finally, the RAD51 pathogenic variants that have been described in the congenital mirror movement syndrome (see above), revealing an unexpected role in brain development, which might be independent of BRCA1 or BRCA2.
Therefore, we could propose that other unexplored noncanonical roles of RAD51 remain to be discovered and characterised. For example, we could speculate that RAD51 might play some unexpected roles at undamaged chromatin, controlling the homoeostasis equilibriums of the nucleus and more generally of the cell.
Hypotheses accounting for the “RAD51 paradox”
One hypothesis accounting, at least in part, for the “RAD51 paradox”, proposes that mutations of mediator/accessory genes result in the absence of RAD51 on damaged DNA, making it susceptible to further alternative nonconservative processes (see above). These alternative processes allow cancer cells to survive but at the cost of increased genetic instability. This hypothesis implies that inhibition of nonconservative pathways should be toxic in BRCA-deficient cells. The simultaneous association of several mechanisms could be responsible for the survival of BRCA2 deficiency. It is possible that all these mechanisms are necessary in parallel and that the inhibition of only one of them would be sufficient to lead to cell lethality in the context of BRCAness. Indeed, the inhibition of either PARP1 or Pol-θ, which play roles in A-EJ [84,85,86,87], or RAD52 that controls SSA [88], is toxic to HR-deficient cells in a synthetic-lethal manner [58, 59, 87, 89,90,91]. SSA requires homologous sequences, but repeat sequences are highly frequent in the human genome, therefore, SSA can occur in many places. In line with this, the EXO1 resection factor has been recently identified synthetic lethal with BRCA1, compared to wild-type or BRCA2-deficient tumours. Moreover, increased SSA-associated genomic scars have been described in BRCA-tumours [92, 93]. Collectively, these findings support the above hypothesis.
RAD51 mutations that permit its binding to DNA should not stimulate alternative mutagenic repair processes and, consequently, should not confer cancer predisposition. Moreover, RAD51 plays several roles, including roles independent of the mediator/accessory proteins (see above); therefore, the suppression of the RAD51 protein, which affects all RAD51 functions, might be too toxic for cancer cells, and alternative nonconservative repair processes would not be able to compensate for the loss of all these different functions.
Collectively, all the above data raise the question of whether cancer predisposition actually results from an inability to perform HR or from the stimulation of alternative nonconservative repair mechanisms.
In vivo specific inactivation of RAD51 HR activity suppressed tumour development
To address the above questions in vivo, we previously designed a mouse model in which RAD51 HR activity is decreased without stimulation of nonconservative pathways (see below).
In vivo mouse models are powerful models for addressing the above questions. Unfortunately, HR genes, including RAD51, are essential genes, and their knockout leads to early embryonic lethality. Many elaborate strategies have been designed to alter HR in vivo, for example, in specific tissues, and confirm the cancer predisposition of HR defects. However, all these models affect mediator/accessory genes, and none of them target RAD51 itself [50]. Therefore, these models confirm the conclusions from human cancer studies, but they have not addressed the questions related to the “RAD51 paradox”.
To overcome these development issues, we designed a mouse model with doxycycline-inducible expression of a RAD51 dominant-negative form (SMRAD51) [16], which poisons HR but does not stimulate alternative nonconservative pathways (SSA, A-EJ) because of its capacity to bind DNA and to inhibit the annealing of complementary strands [15, 16]. Indeed, SMRAD51 can bind ssDNA and assemble into foci. Importantly, the kinetics of assembly and disassembly of SMRAD51 are similar to those of wild-type RAD51. SMRAD51 inhibits strand exchange and HR but still protects against alternative mutagenic pathways SNA and A-EJ (in contrast with other RAD51 mutant forms); moreover, its ability to bind DNA allows it to protect arrested replication forks from degradation but blocks replication restart by strand exchange [15, 16]. Therefore, this mouse model constitutes a unique tool to separate the decrease in HR from the stimulation of alternative mutagenic pathways.
Decreasing HR through doxycycline supplementation led to rapid premature aging that resulted from exhaustion of the stem cell pool, precluding tissue renewal, and associated with systemic inflammation [16]. Remarkably, a decrease in RAD51 strand exchange activity induced by SMRAD51 expression, without SSA or A-EJ stimulation, did not stimulate oncogenesis, and no tumours were observed [16]. These findings do not support an actual tumour suppressor role for HR in vivo.
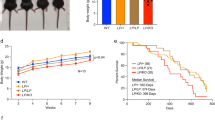
Moreover, we crossed our mouse model with a breast cancer predisposition model (PyMT, which expresses the mouse mammary tumour virus-polyomavirus middle T antigen). SMRAD51 expression in vivo does not favour tumorigenesis but rather decreases the number and size of breast tumours [16]. These findings indicate that not only HR and RAD51 are not tumour-suppressive factors but in fact favour tumour development in vivo. Notably, SMRAD51 expression led to increased levels of the DNA damage response markers γH2AX (which mainly detects DSBs) and pCHK1 (which are activated upon replication stress), which is consistent with HR defects, in hyperplastic as well as in carcinoma mammary cells [16]. Therefore, RAD51 likely functions at the early step of cancer progression, at pretumorigenic steps (hyperplasia), and should persist at tumorigenic steps (carcinoma). These conclusions are consistent with the replication fork escort role of HR since hyperplastic cells are highly proliferative and thus actively replicate their genome.
Noteworthy, the expression of SMRAD51 in MEFs leads to telomere shortening (data to be published). This can account for the premature ageing phenotype in mice, in addition to the attrition to the stem cell pools. Moreover, this could account for the anti-tumour phenotype in mice, since cancer cells need to maintain telomeres. Therefore, according to this function of telomere maintenance, this supports again a protumour role of functional RAD51.
However, the overexpression of wild-type RAD51 in a similar doxycycline-inducible mouse model did not promote tumorigenesis [16], suggesting that if RAD51 favours oncogenesis, it is not a true oncogene that promotes cancer initiation. Nevertheless, the effects of wild-type RAD51 overexpression on carcinogenesis remain to be explored in the PyMt model of breast cancer predisposition. Additionally, the impact of wild-type RAD51 overexpression following mutagenic genotoxic stresses or treatments that promote cancer initiation should also be addressed.
Collectively, these data suggest that RAD51 promotes progression in the early pretumorigenic state (hyperplasia) and persists at the carcinoma stages.
Discussion
The above data are summarised in Fig. 4A and discussed below.

A RAD51 plays a central role in HR, repairing damaged DNA and coping with replication stress. These findings support cell viability and proliferation, preventing premature aging. Indeed, the division of progenitor stem cells is required to maintain the stem cell pools, enabling tissue renewal. RAD51 and HR escort the replication of progenitor stem cells, helping maintain stem cell pools. In parallel, RAD51 on damaged DNA protects against mutagenic nonconservative repair processes such as SSA or A-EJ. The absence of RAD51 on damaged DNA impairs HR and concomitantly makes damaged DNA accessible for nonconservative repair. This allows partial rescue of cell viability but induces genetic instability. B During cancer progression, cells are highly proliferative, thus replicating their genome in the hyperplastic (nontumor) and carcinogenic steps (tumour). Therefore, they are subjected to high replication stress. HR, through its role as a replication fork escort, allows cells to cope with this high replication stress and thus should facilitate cancer progression as soon as the early nontumorigenic step (hyperplasia).
HR/RAD51, a pro-tumour pathway
The loading of Rad51 onto DNA is a prerequisite step for the protection and resumption of arrested replication forks. Therefore, HR/RAD51 is essential for the survival and proliferation of normal dividing cells as well as cancer dividing cells, which are confronted with a series of unavoidable endogenous obstacles that hinder the progression of replication forks during each cell division cycle.
Therefore, HR is essential during embryonic development, and the KO of HR genes leads to early embryonic lethality [50]. Moreover, HR/RAD51 plays a key role in somatic cells as a replication fork escort. In this context, HR plays a prime role in protection against premature aging by maintaining the pool of stem cells that need to divide [16].
The replication back-up role of HR should protect non cancer cells, exhibiting an apparent dual role for HR. Nonetheless, this protective role should also act on tumour cells. This should be even more essential for tumour cells than for primary cells since cancer cells are generally highly proliferative cells, actively replicating their genome and thus are subjected to intense replication stress (in contrast with non-transformed cells). Therefore, through this role on replication stress, HR should help tumour cells cope with this high replication stress. We propose that this is the main reason the pro-tumour role of HR. Thus, there are not two actual opposite faces of HR, but the same face that is an advantage both in non-cancer and in cancer cells. Accounting for its replication escort role, HR should facilitate cancer progression and thus act as a pro-tumour pathway.
The impact of Tp53 on HR is consistent with the above hypothesis. Indeed, the main role of Tp53, which is mutated at a very high frequency in tumours, is protection against cancer through the control of numerous processes, including cell cycle checkpoints, apoptosis and senescence. Therefore, Tp53 favours tumour suppressor pathways and prevents tumour drivers. The tumour suppressor Tp53 does not favour HR but rather restricts HR (for a review), see [94]. These findings suggest that HR is recognised by Tp53 as a tumour support pathway and thus should be restricted.
Dual roles of RAD51
On the one hand, RAD51 (and HR) protects genome stability through its roles in DNA repair and replication fork escort and against mutagenic pathways such as A-EJ and SSA. If we admit that genome stability maintenance prevents oncogenesis, RAD51 (and HR) plays a tumour-suppressive role. Nonetheless, this function of replication back-up is also beneficial for tumour cells. as discussed above. In addition, maintaining genome stability in cancer cells helps them to survive (high genetic instability is toxic) and to maintain the genetic formula that confers them their proliferative advantage. Finally, the protective role of HR may be counterbalanced by the fact that HR can also induce genome instability [95], which could favour cancer initiation.
On the other hand, RAD51 might favour cancer progression through its replication fork protection functions, making it a tumour driver (as discussed above). Therefore, RAD51 has opposite dual faces. As a double-edged sword, HR should thus be tightly controlled. However, HR inherently helps cancer cells proliferate, and consistent with its role in replication escort, HR should act as soon as the replication program is activated, even at the early step of the precancerous step (hyperplasia) of cancer progression.
Concluding remarks
Collectively, these data highlight the dark side of HR and, more specifically, RAD51 as a tumour supports at early pretumorigenic steps; the cancer predisposition associated with HR mutations might not result from HR defects per se but, at least in part, from the concomitant stimulation of alternative nonconservative pathways that compensate for defects in HR.
Figure 4B summarises the dual effects of HR: 1- Prior to cancer initiation, HR potentially functions as a tumour suppressor through its role in maintaining genome stability; however, this function could be counterbalanced by the capacity of HR to induce genetic instability [95]. 2- After initiation of the hyperproliferation program, HR promotes cell proliferation, though its replication fork functions, both at the precancerous stage (hyperplasia) and cancer stage (carcinoma).
Additional work might complete this comprehension, notably how cell-cycle and checkpoint control might impact the processes described above. Moreover, the impact, in vivo, of the genetic alteration of nonconservative repair pathways in different HR-deficient mouse models or to analyse the consequences of the stimulation of nonconservative repair pathways in the absence of a decrease in HR. In order to have a precise view, it is important to identify and characterise all noncanonical roles of RAD51, particularly those that are independent of BRCA1 and BRCA2. Elucidating the defect that causes the CMM syndrome is crucial and should be informative for both the pathology and for academic knowledge about brain development. Moreover, it remains to be explained how HR-deficient cancer cells can survive and proliferate. This might identify novel targets for synthetic lethality strategies in cancer therapy.
More specifically, these findings might have important implications for the design of anticancer strategies. Indeed, they suggest that targeting HR itself could be a promising strategy provided that alternative nonconservative pathways are not induced. Moreover, they identify nonconservative repair pathways that can compensate for HR defects as potential targets for synthetic lethality strategies alternative to PARP or Pol-θ inhibition.
By challenging common views, this review allows us to precisely redefine the actual impact of RAD51 and HR and the importance of balancing alternative nonconservative repair pathways on cancer predisposition/prevention.
References
Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31–46.
Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74.
López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. 2023;35:12–35.
Haber JE. Genome stability. DNA repair and recombination. Garland Science, Talor and Francis Group, New York and London, 2014.
Lim PX, Zaman M, Feng W, Jasin M. BRCA2 promotes genomic integrity and therapy resistance primarily through its role in homology-directed repair. Mol Cell. 2024;84:447–62.e10.
Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600.
Matos-Rodrigues G, Guirouilh-Barbat J, Martini E, Lopez BS. Homologous recombination, cancer and the ‘RAD51 paradox. NAR Cancer. 2021;3:zcab016.
Liu Y-C, Shen J. Meta-analysis of the association between overexpression of RAD51 family genes and prognosis and clinical features in breast cancer. Sci Rep. 2025;15:4229.
Tennstedt P, Fresow R, Simon R, Marx A, Terracciano L, Petersen C, et al. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int J Cancer. 2013;132:2118–26.
Maacke H, Opitz S, Jost K, Hamdorf W, Henning W, Krüger S, et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer. 2000;15:907–13.
Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–5.
Qiao GB, Wu YL, Yang XN, Zhong WZ, Xie D, Guan XY, et al. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br J Cancer. 2005;93:137–43.
Welsh JW, Ellsworth RK, Kumar R, Fjerstad K, Martinez J, Nagel RB, et al. Rad51 Protein Expression and Survival in Patients with Glioblastoma Multiforme. Int J Radiat Oncol Biol Phys. 2009;74:1251–5.
Li Y, Yu H, Luo RZ, Zhang Y, Zhang MF, Wang X, et al. Elevated expression of Rad51 is correlated with decreased survival in resectable esophageal squamous cell carcinoma. J Surg Oncol. 2011;104:617–22.
So A, Dardillac E, Muhammad A, Chailleux C, Sesma-Sanz L, Ragu S, et al. RAD51 protects against nonconservative DNA double-strand break repair through a nonenzymatic function. Nucleic Acids Res. 2022;50:2651–66.
Matos-Rodrigues G, Barroca V, Muhammad A, Dardillac E, Allouch A, Koundrioukoff S, et al. In vivo reduction of RAD51 -mediated homologous recombination triggers aging but impairs oncogenesis. EMBO J. 2023;42:1–21.
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35.
Kuthethur R, Acharya A, Sengodan SK. FIGNL1 inhibits homologous recombination in BRCA2 deficient cells by dissociating RAD51 filaments. biorxiv.org 2024. https://doi.org/10.1101/2024.11.03.621741v1.
Carver A, Yu Y, Yates LA, White T, Wang R, Lister K, et al. Molecular basis of FIGNL1 in dissociating RAD51 from DNA and chromatin. Science. 2024. https://doi.org/10.1126/science.adr7920.
Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radic Biol Med. 2002;33:1–14.
Magdalou I, Lopez BS, Pasero P, Lambert SAESAE. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol. 2014;30:154–64.
Wilhelm T, Ragu S, Magdalou I, Machon C, Dardillac E, Técher H, et al. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016;12:e1006007.
Ragu S., Dardillac, E. Caillat S, Ravanat JL, Lopez BS. Dysregulation of the low-level replication stress response intransformed cell lines. Sci Reports. 2025;15, https://doi.org/10.1038/s41598-025-05172.
Lopez BS. Specific Low/Endogenous Replication Stress Response Protects Genomic Stability via Controlled ROS Production in an Adaptive Way and Is Dysregulated in Transformed Cells. Cells. 2025;14:1183. https://doi.org/10.3390/cells14151183.
Ragu S, Droin N, Matos-Rodrigues G, Barascu A, Caillat S, Zarkovic G, et al. A noncanonical response to replication stress protects genome stability through ROS production, in an adaptive manner. Cell Death Differ. 2023;30:1349–65.
Saintigny Y, Delacôte F, Varès G, Petitot F, Lambert S, Averbeck D, et al. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–70.
Daboussi F, Courbet S, Benhamou S, Kannouche P, Zdzienicka M, Debatisse M, et al. A homologous recombination defect affects replication-fork progression in mammalian cells. J Cell Sci. 2008;121:162–6.
Wilhelm T, Magdalou I, Barascu A, Techer H, Debatisse M, Lopez BS. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc Natl Acad Sci USA. 2014;111:763–8.
Costes A, Lambert SA. Homologous Recombination as a Replication Fork Escort: Fork-Protection and Recovery. Biomolecules. 2012;1:39–71.
Thomas M, Dubacq C, Rabut E, Lopez BS, Guirouilh-Barbat J. Noncanonical Roles of RAD51. Cells. 2023;12:1169.
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42.
Berti M, Cortez D, Lopes M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol. 2020;21:633–51.
Berti M, Teloni F, Mijic S, Ursich S, Fuchs J, Palumbieri MD, et al. Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nat Commun. 2020;11:3531.
Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–7.
Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun. 2017;8:859.
Mason JM, Chan YL, Weichselbaum RW, Bishop DK. Non-enzymatic roles of human RAD51 at stalled replication forks. Nat Commun. 2019;10:4410.
Bugreev DV, Rossi MJ, Mazin AV. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011;39:2153–64.
Hanthi YW, Ramirez-Otero MA, Appleby R, De Antoni A, Joudeh L, Sannino V, et al. RAD51 protects abasic sites to prevent replication fork breakage. Mol Cell. 2024;84:3026–43.e11.
Bétermier M, Bertrand P, Lopez BS. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process?. PLoS Genet. 2014;10:e1004086.
Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–24.
So A, Le Guen T, Lopez BS, Guirouilh-Barbat J. Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells. FEBS J. 2017;284:2324–44.
Lambert S. Lopez BS. Characterization of mammalian RAD51 double strand break repair using non lethal dominant negative forms. EMBO J. 2000;19:3090–9.
Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–16.
Stark JM, Hu P, Pierce AJ, Moynahan ME, Ellis N, Jasin M. ATP hydrolysis by mammalian RAD51 has a key role during homology- directed DNA repair. J Biol Chem. 2002;277:20185–94.
Han J, Ruan C, Huen MSY, Wang J, Xie A, Fu C, et al. BRCA2 antagonizes classical and alternative nonhomologous end-joining to prevent gross genomic instability. Nat Commun. 2017;8:1470.
Ahrabi S, Sarkar S, Pfister SX, Pirovano G, Higgins GS, Porter ACG, et al. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 2016;44:5743–57.
Magin S, Papaioannou M, Saha J, Staudt C, Iliakis G. Inhibition of homologous recombination and promotion of mutagenic repair of DNA double-strand breaks underpins arabinoside-nucleoside analogue radiosensitization. Mol Cancer Ther. 2015;14:1424–33.
Stefanovie B, Hengel SR, Mlcouskova J, Prochazkova J, Spirek M, Nikulenkov F, et al. DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic Acids Res. 2020;48:694–708.
Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, Lukas C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 2016;23:714–21.
Matos-Rodrigues G, Martini E, Lopez BS. Mouse Models for Deciphering the Impact of Homologous Recombination on Tumorigenesis. Cancers. 2021;13:2083.
Feng W, Jasin M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat Commun 2017; 8. https://doi.org/10.1038/s41467-017-00634-0.
Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med. 2008;14:875–81.
Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol. 2010;17:1461–9.
Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: Past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–97.
Petitalot A, Dardillac E, Jacquet E, Nhiri N, Guirouilh-Barbat J, Julien P, et al. Combining Homologous Recombination and Phosphopeptide-binding Data to Predict the Impact of BRCA1 BRCT Variants on Cancer Risk. Mol Cancer Res 2018; 17: molcanres.0357.2017.
Diabate M, Islam MM, Nagy G, Banerjee T, Dhar S, Smith N, et al. DNA repair function scores for 2172 variants in the BRCA1 amino-terminus. PLoS Genet 2023; 19. https://doi.org/10.1371/journal.pgen.1010739.
Jahjah T, Singh JK, Gottifredi V, Quinet A. Tolerating DNA damage by repriming: Gap filling in the spotlight. DNA Repair 2024; 142. https://doi.org/10.1016/j.dnarep.2024.103758.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V, et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: A multi-institutional study. Clin Cancer Res. 2013;19:5485–93.
Bhin J, Paes Dias M, Gogola E, Rolfs F, Piersma SR, de Bruijn R et al. Multi-omics analysis reveals distinct non-reversion mechanisms of PARPi resistance in BRCA1- versus BRCA2-deficient mammary tumors. Cell Rep 2023; 42. https://doi.org/10.1016/j.celrep.2023.112538.
Domchek SM. Reversion mutations with clinical use of PARP inhibitors: Many genes, many versions. Cancer Discov. 2017;7:937–9.
Weigelt B, Comino-Méndez I, De Bruijn I, Tian L, Meisel JL, García-Murillas I, et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin Cancer Res. 2017;23:6708–20.
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–15.
Meijer TG, Martens JWM, Prager-van der Smissen WJC, Verkaik NS, Beaufort CM, van Herk S, et al. Functional Homologous Recombination (HR) Screening Shows the Majority of BRCA1/2-Mutant Breast and Ovarian Cancer Cell Lines Are HR-Proficient. Cancers. 2024;16:1–18.
Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35:909–23.
Geilmann S, Solstad R, Palmquist R, Daboub FJ, Botto LD, Grubb PH, et al. A novel RAD51 variant resulting in Fanconi anemia identified in an infant with multiple congenital anomalies. Clin Case Rep. 2023;11:e6810.
Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun 2015;8829–39.
Takenaka S, Kuroda Y, Ohta S, Mizuno Y, Hiwatari M, Miyatake S, et al. A Japanese patient with RAD51-associated Fanconi anemia. Am J Med Genet Part A. 2019;179:900–2.
Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell. 2015;59:478–90.
Trouillard O, Koht J, Gerstner T, Moland S, Depienne C, Dusart I, et al. Congenital Mirror Movements Due to RAD51: Cosegregation with a Nonsense Mutation in a Norwegian Pedigree and Review of the Literature. Tremor Other Hyperkinet Mov. 2016;6:424.
Trouillard O, Dupaigne P, Dunoyer M, Doulazmi M, Herlin MK, Frismand S, et al. Congenital mirror movements are associated with defective polymerisation of RAD51. J Med Genet. 2023;60:1116–26.
Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell. 2017;67:867–81.
Gildemeister OS, Sage JM, Knight KL. Cellular Redistribution of Rad51 in Response to DNA Damage: novel role for Rad51C. J Biol Chem. 2009;284:31945–52.
Griffin WC, McKinzey DR, Klinzing KN, Baratam R, Eliyapura A, Trakselis MA. A multi-functional role for the MCM8/9 helicase complex in maintaining fork integrity during replication stress. Nat Commun. 2022;13:5090.
Prado F. Non-recombinogenic functions of Rad51, BRCA2, and Rad52 in DNA damage tolerance. Genes. 2021;12:1550.
Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat-containing RNA and RNA surveillance factors at mammalian chromosome ends. Science (80-). 2007;318:798–801.
Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun. 2014;5:52220.
Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A, et al. Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell. 2017;170:72–85.e14.
Feretzaki M, Pospisilova M, Valador Fernandes R, Lunardi T, Krejci L, Lingner J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature. 2020;587:303–8.
Teng Y, Yadav T, Duan M, Tan J, Xiang Y, Gao B, et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat Commun. 2018;9:4115.
Chappidi N, Nascakova Z, Boleslavska B, Zellweger R, Isik E, Andrs M, et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol Cell. 2020;77:528–41.e8.
Marabitti V, Lillo G, Malacaria E, Palermo V, Pichierri P, Franchitto A. Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability. Cancers. 2020;12:389.
Audebert M, Salles B, Weinfeld M, Calsou P. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J Mol Biol. 2006;356:257–65.
Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82.
Wang Z, Song Y, Li S, Kurian S, Xiang R, Chiba T, et al. DNA polymerase (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J Biol Chem. 2019;294:3909.
Mateos-gomez PA, Gong F, Nair N, Miller KM, Lazzerini-denchi E, Sfeir A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–7.
Reddy G, Golub EI, Radding CM. Human Rad52 protein promotes single-strand DNA annealing followed by branch migration. Mutat Res Fundam Mol Mech Mutagen. 1997;377:53–59.
Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–54.
Feng W, Simpson DA, Carvajal-Garcia J, Price BA, Kumar RJ, Mose LE et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat Commun 2019; 10. https://doi.org/10.1038/s41467-019-12234-1.
Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108:686–91.
van de Kooij B, Schreuder A, Pavani R, Garzero V, Uruci S, Wendel TJ, et al. EXO1 protects BRCA1-deficient cells against toxic DNA lesions. Mol Cell. 2024;84:659–74.e7.
Setton J, Hadi K, Choo ZN, Kuchin KS, Tian H, Da Cruz Paula A, et al. Long-molecule scars of backup DNA repair in BRCA1- and BRCA2-deficient cancers. Nature. 2023;621:129–37.
Bertrand P, Saintigny Y, Lopez BS. p53’s double life: transactivation-independent repression of homologous recombination. Trends Genet. 2004;20:235–43.
Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS. Is homologous recombination really an error-free process?. Front Genet. 2014;5:175.
Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5:a012740.
Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. 2018;293:10524–35.
Acknowledgements
Thanks are due to all people having read and commented on the manuscript. This work was supported by grants from the Institut National du Cancer (PLBIO21-072, PLBIO24-196-2024-158), La Ligue Contre Le Cancer (ARN therapeutiques), ITMO Cancer (PCSI 2022), and Fondation ARC (ARCPJA2022060005157).
Author information
Authors and Affiliations
Contributions
Conceptualisation: BSL; funding acquisition: BSL; project administration: BSL; resources: BSL; writing the original draft: BSL; and writing – reviewing & editing: BSL.
Corresponding author
Ethics declarations
Competing interests
The author declares having no conflict of interest.
Ethics
Ethics approval was not needed because this manuscript reviews and discusses data that were already published elsewhere and for which ethics approvals were obtained. There are no novel experiments, methods and data.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lopez, B.S. RAD51-mediated homologous recombination is a pro-tumour driver pathway. Oncogene 44, 4006–4016 (2025). https://doi.org/10.1038/s41388-025-03583-x
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41388-025-03583-x
