
Abstract
Schizophrenia spectrum disorders (SSD) involve disturbances in the integration of perception, emotion and cognition. The corticolimbic system is an interacting set of cortical and subcortical brain regions critically involved in this process. Understanding how neural circuitry and molecular mechanisms within this corticolimbic system may contribute to the development of not only positive symptoms but also negative and cognitive deficits in SSD has been a recent focus of intense research, as the latter are not adequately treated by current antipsychotic medications and are more strongly associated with poorer functioning and long-term outcomes. This review synthesises recent developments examining corticolimbic dysfunction in the pathophysiology of SSD, with a focus on neuroimaging advances and related novel methodologies that enable the integration of data across different scales. We then integrate how these findings may inform the identification of novel therapeutic and preventive targets for SSD symptomatology. A range of pharmacological interventions have shown initial promise in correcting corticolimbic dysfunction and improving negative, cognitive and treatment-resistant symptoms. We discuss current challenges and opportunities for improving the still limited translation of these research findings into clinical practice. We argue how our knowledge of the role of corticolimbic dysfunction can be improved by combining multiple research modalities to examine hypotheses across different spatial and temporal scales, combining neuroimaging with experimental interventions and utilising large-scale consortia to advance biomarker identification. Translation of these findings into clinical practice will be aided by consideration of optimal intervention timings, biomarker-led patient stratification, and the development of more selective medications.
Similar content being viewed by others
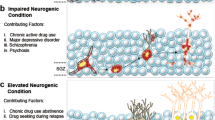
Introduction
Schizophrenia spectrum disorders (SSD) encompass a broad range of symptoms beyond the commonly recognised positive symptoms (i.e. hallucinations and delusions), affecting cognitive and negative domains. Understanding the neural circuitry involved in the development of these symptoms is crucial for improving treatments and long-term outcomes. The corticolimbic system is an interacting set of cortical and subcortical brain regions, including the medial prefrontal cortex (mPFC), anterior cingulate cortex (ACC), nucleus accumbens, hippocampus and amygdala [1] (Fig. 1). This circuitry is critical for facilitating interactions between emotion, cognition and decision-making functions, all of which are affected in SSD [2].

A Abnormalities have been identified in corticolimbic structure, functional activation and connectivity in SSD. B Diagram displaying a model for interactions between corticolimbic brain circuits and mechanisms leading to symptom domains. Hippocampal hyperactivity and alterations of rhythmicity cause downstream effects within the nucleus accumbens, ventral pallidum and ventral tegmental area, resulting in increased striatal dopamine signalling and positive symptoms. It may also cause negative symptoms by affecting emotional processing and reward outcomes via the amygdala, and impacting decision-making through the medial prefrontal, anterior cingulate and orbitofrontal cortices. Increased excitatory output from the hippocampus to the dorsolateral prefrontal cortex may lead to reduced synaptic density and disruption to the oscillatory activity necessary for cognitive functions.
Recent advancements in neuroimaging have significantly enhanced our understanding of the corticolimbic system’s role in SSD. For instance, ultra-high field magnetic resonance imaging (MRI) offers precise examination of corticolimbic structure, function and connectivity at subfield levels [3]. Moreover, higher-resolution imaging enhances resolution for magnetic resonance spectroscopy (MRS), facilitating more reliable and specific measurements of relevant metabolites [4]. Multimodal imaging can combine structural, functional and molecular data, yielding a more comprehensive view of corticolimbic dysfunction across different spatial and temporal scales [5]. Additionally, large-scale consortia that share multimodal data address the limitations encountered in previous studies, such as small sample sizes, heterogeneous protocols and reproducibility issues [6, 7]. These large-scale studies provide the required power for multivariate data-driven methods, such as machine learning, allowing inferences to be made at the subgroup [8] or individual level [9], helping to circumvent the limitations of traditional analytic approaches that compare patients to healthy controls at the group level to untangle clinical and biological heterogeneity in SSD. Another innovative approach is normative modelling, which involves comparing individuals to a reference model in order to make inferences at the individual level [10]. Integrating these methodologies enriches our understanding of corticolimbic dysfunction and holds promise for a more personalised approach to SSD management by identifying biomarkers at the individual level. Furthermore, combining advanced neuroimaging methods with drug administration studies has the potential to translate corticolimbic system findings into clinically relevant therapeutic targets. This approach uses pharmacological agents to target specific mechanisms and analyses the impact on broader activation and signalling pathways [11]. This enhances our comprehension of the neurobiological mechanisms underlying SSD symptoms and has promise in predicting individual medication response [11].
To achieve a better understanding of the mechanisms leading to positive, negative and cognitive symptom manifestations, this review synthesises recent developments in the study of corticolimbic dysfunction in the pathophysiology of SSD with a focus on neuroimaging advances and approaches to bridge evidence across different scales of description (cellular/molecular, circuit, whole-brain imaging and behaviour). Finally, the discussion addresses how these methodologies and findings can be leveraged to inform the identification of novel therapeutic and preventative targets in SSD.
Mechanisms of corticolimbic dysfunction in SSD
The corticolimbic system relies on coordinated interactions between the PFC and limbic regions. The hippocampus and amygdala are highly interconnected limbic structures located in the medial temporal lobe which act at the interface between cognition and emotion [12]. The hippocampus plays a key role in memory and associative learning [1], while the amygdala is involved in processing emotion and biological salience [1]. These structures bidirectionally influence each other, with the amygdala modulating hippocampal-dependent memory encoding and consolidation for emotional stimuli, and the anterior hippocampus regulating the amygdala’s response to emotional stimuli [12]. The PFC integrates sensory and limbic inputs to regulate cognitive-emotional processes and guide goal-directed behaviours [2]. Disruptions to multiple regions in these complex circuits contribute to the emotional and cognitive dysregulation observed in SSD.
Among these regions, the hippocampus has been consistently identified as a central hub of abnormality within the corticolimbic system in SSD [13, 14]. Neuroimaging studies have demonstrated reductions in volume [15], and increases in resting cerebral blood volume [16] and flow [17, 18]. Resting hippocampal hyperactivity has also been found in individuals at clinically high risk for psychosis (CHR-P) [19,20,21] and healthy people with psychosis-like experiences [22]. This resting hyperactivity impacts function as there is reduced task-related recruitment of the anterior hippocampus during memory and visual stimulation in early psychosis [17]. Hippocampal hyperactivity has also been associated with negative and cognitive symptom severity [23], and predicts transition to psychosis in CHR-P [24, 25]. In terms of connectivity, reduced resting functional connectivity has been identified between the hippocampus and limbic and frontal regions across the SSD spectrum [26], indicating broader changes to corticolimbic circuitry.
Regarding mechanisms underlying corticolimbic dysfunction, several lines of evidence suggest it results from an imbalance in excitatory and inhibitory signalling [27]. A balanced interplay between glutamatergic neurons and gamma-aminobutyric acid (GABA) interneurons, termed excitation/inhibition balance, is required for the development of microcircuits and larger neural networks [27]. There are several families of glutamatergic receptors, including N-methyl-D-aspartic acid (NMDAR), α-amino-3-hydroxy-5-methylisoxazole-4-proprionic acid (AMPAR), kainite and metabotropic receptors (mGluR). There are also diverse GABAergic interneuron cell-types, identified by the presence of calcium-binding proteins (e.g. calbindin, parvalbumin), neuropeptides (e.g. somatostatin, cholecystokinin) or morphological appearance (e.g. chandelier, basket) [28]. This complexity leads to multiple possible mechanisms that may disrupt excitation/inhibition balance and, consequently, hippocampal and broader corticolimbic system disruption.
Among GABAergic interneuron cell-types, parvalbumin-expressing interneurons (PVI) are key to maintaining excitation/inhibition balance and shaping cortical circuitry during development [29]. Alterations in PVI are well-established in SSD by post-mortem examination, including decreased density of PVI in the hippocampus [30, 31] and decreased parvalbumin protein/mRNA expression in the dorsolateral PFC (dlPFC) [32]. NMDAR hypofunction has been implicated in these alterations, with pre-clinical evidence that NMDAR disruption can lead to oxidative stress [33] and reduced PVI density in the hippocampus and PFC [34]. Parvalbumin is a calcium-binding protein which enables high frequency firing by shortening the post-spike after hyperpolarisation [35]. However, this high metabolic demand renders these neurons more susceptible to oxidative stress [36] (an imbalance between free radicals and antioxidant defence). Severe long-term imbalances can result in cell damage and death [37], while smaller imbalances reversibly alter the function of redox-sensitive proteins, impacting neurotransmission, cellular proliferation and differentiation [38]. Several interacting factors lead to feedforward loops exacerbating oxidative stress; this includes: NMDAR hypofunction [39], antioxidant deficits [40], mitochondrial dysfunction [41] and neuroinflammation [42]. These cascading effects, if not compensated, can lead to progressive hippocampal dysfunction and broader corticolimbic abnormalities [37].
Consequences of corticolimbic dysfunction
Corticolimbic system abnormalities are proposed to drive diverse symptom domains in SSD [43, 44] (Fig. 1). More specifically, hippocampal hyperactivity causes downstream effects within the nucleus accumbens, ventral pallidum and ventral tegmental area, resulting in increased striatal dopamine signalling and positive symptoms. Additionally, hippocampal hyperactivity and dysrhythmicity may lead to negative symptoms through disruptions of amygdala activity, impacting emotional salience and reward processing, and with the mPFC, ACC and orbitofrontal cortex, impacting reward anticipation and value-based decisions. Finally, increased excitatory output from the hippocampus indirectly to the dlPFC may lead to disruption of the oscillatory activity critical for cognitive functions. This section explores the neuroimaging evidence for this model and explores how this may lead to different SSD symptom domains.
Positive symptoms
Positive symptoms have been linked extensively to the dopamine system: elevated pre-synaptic striatal dopamine synthesis capacity and release is a robust finding in SSD, and all current antipsychotics target dopamine D2 receptors to some degree [45]. Numerous pre-clinical studies ([44] for overview) have examined the involvement of corticolimbic circuitry in dysregulating dopaminergic system function. Here, we provide a brief overview of these findings focusing on neuroimaging evidence.
Pre-clinical studies suggest that the spontaneous activity of midbrain dopamine neurons is regulated by corticolimbic regions [44], which is important for controlling the response to context and environmental conditions. Dopaminergic signalling can be tonic or phasic in nature, and these two forms of signalling are highly interlinked [44]. Tonic signalling refers to baseline spontaneous firing, which is regulated by glutamatergic afferents from the hippocampus and amygdala [46]. Phasic signalling refers to rapid bursts of action potentials which is important for determining salience and reinforcement of learning [47]. Phasic signalling relies on prior depolarisation from tonic activity [48]. For instance, in threatening contexts, the hippocampus causes a downstream increase in tonic dopaminergic activity, which causes greater phasic dopamine release and rapid focus on stimuli [47]. In SSD, disrupted modulation of the dopaminergic system may result in excessive tonic activity leading to higher phasic signalling, resulting in hyper-responsivity to irrelevant stimuli and impaired salience detection [49].
Complex interactions between multiple corticolimbic regions are involved in regulating tonic dopaminergic signalling. For instance, ventral tegmental area neurons are sustained in a hyperpolarised state by ventral pallidum GABAergic inputs, which keeps them below the firing threshold [50]. This is regulated by excitatory projections from the hippocampus to the nucleus accumbens [51], which increases inhibition of the ventral pallidum and, therefore, increases dopaminergic signalling from the ventral tegmental area to the associative striatum [52]. The amygdala activates the ventral pallidum, reducing dopamine activity between the ventral tegmental area and ventral striatum [43]. Imaging studies have investigated the functional alterations that contribute to the dysregulation of this intricate system, disrupting dopaminergic signalling and leading to aberrant salience.
Inappropriately attributing salience to neutral or irrelevant stimuli has been demonstrated across SSD stages and is a contributing mechanism in the formation of positive symptoms [53]. Imaging studies using salience and reward prediction error paradigms have identified functional alterations in the corticolimbic system in SSD. For instance, in oddball paradigms, antipsychotic-naïve first-episode psychosis patients showed reduced activation in the amygdala, VTA, ACC and striatum in response to negative emotional salience [54]. Similar findings are seen in CHR-P individuals using the same paradigm [55]. A positron emission tomography (PET) study using a [15O]H2O tracer with emotionally salient visual images reported impaired ventral striatum response to salient stimuli and elevated baseline activity in the ventral striatum and the amygdala, which inversely correlated with overall symptom severity [56]. Finally, prediction error, the mismatch between predicted and actual outcomes, is closely linked to salience. Studies with reward prediction error paradigms reveal abnormalities in the striatum, midbrain, limbic regions and mPFC in first-episode psychosis [57, 58] and CHR-P [59]. Given its association with reward processing and negative symptoms, we discuss this further in the following section.
PET studies using radiolabelled L-dihydroxyphenylalanine (L-DOPA), the dopamine precursor, have reported increased dopamine synthesis capacity in the associative striatum in CHR-P individuals [60, 61] and those with SSD [62]. A study with SSD individuals during a relapse of psychotic symptoms found increased intrinsic activity in the dorsal striatum, which correlated with positive symptom severity [63]. Multimodal imaging combining PET and MRS in CHR-P individuals found a negative correlation between hippocampal glutamate levels and striatal dopamine synthesis capacity, this was particularly marked in the CHR-P individuals who later transitioned to psychosis [64]. This suggests that the relationship between hippocampal glutamate and striatal dopamine may be a risk marker for psychosis transition in CHR-P.
Disrupted control of dopamine signalling due to excessive phasic release may result in diminished functional connectivity between the cortex and the associative striatum, thereby impairing integration from emotional, cognitive and motor areas [65]. fMRI studies have found decreased functional connectivity between the associative striatum and the ACC and dlPFC in psychosis-like experiences [66], CHR-P [67], first-episode psychosis and SSD [68]. Interestingly, improvements in positive symptoms during antipsychotic treatment have been associated with increased functional connectivity between the striatum and the ACC, dlPFC and limbic regions [69]. This improved functional connectivity may provide a biomarker for symptom improvements with antipsychotic treatment.
There is evidence that the dopamine system also contributes to cognitive and negative symptoms. Increased striatal dopamine and disrupted connectivity between the frontal cortex and associative striatum and/or disruption of cortical dopamine may contribute to cognitive impairments [70]. The striatum regulates reward-related behaviours, and impaired reward processing is associated with negative symptoms [71, 72], as discussed in the following section.
Negative symptoms
Negative symptoms have been classified into five key constructs within two overarching factors: diminished expression (blunted affect and alogia) and amotivation (asociality, avolition and anhedonia) [73]. Approximately 40–60% of patients with SSD experience prominent negative symptoms, which are also frequently present in early psychosis and in CHR-P [74]. Multiple corticolimbic regions have been associated with negative symptoms, including the hippocampus, amygdala, mPFC, orbitofrontal cortex, ACC, striatum and insula [75, 76]. Depending on the specific location of dysfunction within these circuits, a range of symptomatology may be explained, including diminished expression and amotivation.
The hippocampus regulates affective states and shares strong reciprocal connectivity with the amygdala, a key structure in emotion perception [77], emotional significance processing [78], emotional regulation [79] and salience evaluation [80]. Neuroimaging studies consistently implicate the amygdala in SSD, showing reduced volumes in multiple meta-analyses [81, 82] and in large consortium studies [83, 84]. Lower amygdala volume is observed in CHR-P individuals and in unaffected relatives [85, 86], suggesting a potential neurodevelopmental and/or genetic origin. There are alterations in structural and resting functional connectivity between the amygdala and corticolimbic regions. This includes reduced fractional anisotropy in the uncinate fasciculus [87], linking the amygdala to the mPFC/ orbitofrontal cortex, although some studies have only found this in patients with prominent negative symptoms [88]. Age-related alterations in amygdala resting functional connectivity reveal a nuanced trajectory: during late childhood and early adolescence, there is reduced functional connectivity with the putamen/occipital cortex [89], subsequently there is an accelerated decline in functional connectivity with the ventrolateral PFC, striatum and thalamus [89]. Reductions in resting functional connectivity between the amygdala and orbitofrontal cortex have been observed across SSD stages, and this correlates with overall symptom severity [87, 90]. Meta-analyses have found that orbitofrontal cortex cortical thinning is associated with negative symptom severity [91].
Functional neuroimaging studies examining cognitive-emotional processing in SSD have primarily used facial emotion recognition tasks. There is evidence for facial emotion processing dysfunction in first-episode psychosis [92], in SSD [93] and in first-degree relatives [94]. However, findings in CHR-P have been mixed with some studies finding a significant difference for emotion identification but not discrimination [95], and others finding poorer recognition only for negative emotions [96]. Meta-analyses have demonstrated reduced amygdala activation in response to emotional stimuli in first-episode psychosis [97] and in chronic SSD [98, 99]. The findings in CHR-P have again been inconsistent [97], possibly due to sample heterogeneity and methodological inconsistencies. Reduced amygdala activation represents reduced recruitment when processing emotionally salient stimuli and/or increased response to neutral stimuli. Dysfunctional PFC engagement, notably the ACC and dlPFC, is evident in SSD and psychosis risk during tasks involving emotional stimuli appraisal [100], subjective affect regulation [101] or inhibition of emotional distractions [102].
The “emotional paradox” in SSD describes a disconnect between reported emotional experiences and observed behaviours [103]. While individuals with SSD often report intact emotional experiences [104], studies indicate reduced engagement in pleasurable activities [105]. Functional neuroimaging, particularly using paradigms such as the Monetary Incentive Delay task [106], has examined neural responses to reward anticipation and outcomes. While most studies report reduced ventral striatum activation during anticipatory reward [107], findings are inconsistent and are influenced by antipsychotic medication and negative symptom severity [108, 109]. Neural responses to reward outcomes vary, with some studies showing reduced activation in the ventral striatum, mPFC, and orbitofrontal cortex [109, 110], while others find no significant differences [111]. A recent meta-analysis found reduced activation in the ACC, amygdala, insula and striatum during anticipation [112]. Striatal activation decreases correlated with increased negative symptoms [112]. During reward outcomes, there was increased activation in the striatum, insula, amygdala, hippocampus and right parahippocampal gyrus, alongside decreased activation in the mPFC and dlPFC [112]. Decreased mPFC activation correlated with increased positive symptoms [112].
Deficits in value-based decision-making may be due to difficulty integrating information and updating value estimations [113]. Studies using probabilistic selection paradigms have revealed reinforcement learning deficits in SSD [72]. Interestingly, while preference for previously rewarded stimuli was reduced, patients could learn to avoid stimuli associated with negative outcomes, and this was particularly pronounced in those with severe negative symptoms [72]. Compared to healthy controls, SSD participants have difficulty integrating value information when making behavioural choices [71]. In controls, valued actions were associated with increased caudate and mPFC activation, whereas patients exhibited significantly less caudate activity, which correlated with measures of avolition and alogia [71].
Overall, the evidence suggests that corticolimbic brain changes including the amygdala, hippocampus, PFC and striatum lead contribute to negative symptoms in SSD by impairing emotional processing and motivation.
Cognitive deficits
Cognitive dysfunction in SSD impacts multiple cognitive domains, often precedes psychosis onset [114] and is closely associated with poorer social and occupational functioning [115]. The PFC has a well-established role in supporting higher-order cognitive processes. Studies have examined the cellular and local network changes which may result in altered regional PFC activity and impaired recruitment of wider task-relevant networks, resulting in cognitive symptoms.
The PFC receives dense glutamatergic projections from the hippocampus [116]. Hippocampal hyperactivity, therefore, increases excitatory input to the PFC and this increased tonic activity can lead to reductions in synaptic density [117]. For instance, post-mortem analyses in SSD have shown reduced density of basilar dendritic spines on layer III pyramidal neurons in the dlPFC [118] and decreased synaptophysin protein, a marker of axon terminals, in the PFC and hippocampus [119].
Gamma oscillations are a potential translational bridge between these cellular/molecular alterations and cognitive symptoms. The coordinated firing of neuronal populations creates rhythmic brain activity, which is reflected as oscillations in extracellular brain potentials [120]. In SSD, electroencephalogram (EEG) and magnetoencephalography (MEG) has shown differences in oscillatory activity during cognitive tasks, including in theta, alpha and gamma ranges [121, 122]. In particular, impaired task-evoked gamma oscillatory activity has been found during cognitive control [123, 124] and working memory tasks [125, 126] in first-episode psychosis and chronic SSD, regardless of medication status.
Synaptic interactions between interneurons and pyramidal cells are involved in establishing and maintaining brain oscillations [127]. PVI strongly impact gamma modulation in pre-clinical studies [128]. Excitation of PVI by pyramidal neurons provides coordinated feedback inhibition to multiple pyramidal neurons [120]. Following inhibition, pyramidal neurons are more likely to fire in coordination, thereby generating oscillations [120]. PVIs also synapse onto themselves, which may help synchronise oscillations [129].
Understanding the mechanisms underlying gamma oscillation alterations and their cognitive consequences is a focus of pre-clinical and human research. Optogenetics, involving the introduction of light-sensitive proteins into specific cells to regulate their activity (for review of methods, see [130]), has demonstrated that inhibiting PVI in the PFC supresses gamma oscillations [131]. Rhythmic light stimulation at the gamma frequency enhanced interneuron-driven gamma oscillations and normalised rule-shifting task performance [132]. Furthermore, misaligned light stimulation disrupts gamma synchronisation and impairs task performance [133]. In humans, transcranial direct current stimulation, which modulates cortical excitability via low-current scalp electrodes, increases gamma oscillatory power and enhances cognition when applied to the dlPFC [134]. Collectively, these findings suggest that modifying PFC interneuron function alters oscillatory activity and impacts cognitive function.
PFC GABA and glutamate function in SSD has been assessed using neuroimaging techniques. MRS meta-analyses found lower glutamate in the mPFC [135, 136] but no significant difference in GABA levels in the mPFC or dlPFC [137]. A large meta-analysis reported higher glutamate to creatine ratio in the mPFC; this was positively associated with increased total symptom severity and antipsychotic dose was negatively associated with mPFC glutamate levels [138]. This indicates that higher levels of glutamate in the mPFC are associated with higher symptom severity but that these levels may be reduced with antipsychotic medication. While higher GABA levels predicted better working memory performance in healthy controls [138], the opposite was found in SSD [139]. This suggests that GABAergic increases may serve a compensatory role for disrupted connectivity between inhibitory interneurons and pyramidal cells. A small EEG and MRS study found that peak gamma frequency during the encoding stage of a working memory task correlated with dlPFC GABA levels and task performance [125]. While MRS can provide regional quantification of biochemistry in vivo, this approach measures extracellular and intracellular metabolite concentrations across all tissue types. Hence, these findings cannot provide higher-resolution insights into neurophysiological changes.
PET can be used to measure GABA regulation with benzodiazepine site-binding radiotracers (e.g. [11C]flumazenil). In healthy participants, [11C]flumazenil binding increased after blockade of the GABA membrane transporter with tiagabine, which significantly correlated with gamma oscillatory power during a working memory task [140]. Another study reported positive correlations between tiagabine-induced change in binding and delay-related gamma oscillatory power in healthy participants, whereas SSD participants exhibited reduced capacity to increase extracellular GABA [141]. This reduced capacity was associated with decreased gamma oscillatory power during the cognitive task, particularly in antipsychotic-naïve individuals. Baseline [11C]flumazenil binding was raised in antipsychotic-naïve individuals, possibly reflecting early compensatory GABAA receptor upregulation.
Overall, findings point to the key role of hippocampal hyperactivity in increasing excitatory input to the PFC, causing cellular, molecular and network alterations. Gamma oscillations have emerged as a translational link between these alterations and cognitive symptoms, with disruption observed across SSD stages. Neuroimaging studies investigating PFC GABA and glutamate reveal associations with gamma oscillatory power, treatment response and cognitive deficits. These findings underscore the intricate relationship between these neurophysiological alterations and cognitive dysfunction in SSD.
Targeting corticolimbic circuit dysfunction
There is a persisting unmet need to develop new therapeutic options to treat the negative, cognitive and treatment-resistant symptoms of SSD, and the corticolimbic system shows promise as a target for upstream interventions. This section outlines investigated targets and the effects on molecular and wider circuit functioning via neuroimaging modalities.
Targeting excitation/inhibition balance
As discussed above, deficits in hippocampal PVI and increased pyramidal neuron activity can lead to excitation/inhibition imbalances across the network. Pharmacological approaches to rebalance excitation/inhibition include modulation of glutamatergic or GABAergic mechanisms (Fig. 2) [142].

PVI primarily exert inhibitory effects via synapses onto the perisomatic region of pyramidal cells. Gamma-aminobutyric acid (GABAA) receptors on the postsynaptic membrane receive GABA signalling from the PVI. A possible mechanism to compensate for the loss of PVI function is to increase postsynaptic GABA transmission via GABAA receptor modulation. PVI receive input via N-methyl-D-aspartic acid receptors (NMDAR) and α-amino-3-hydroxy-5-methylisoxazole-4-proprionic acid receptors (AMPAR). Another possible mechanism leading to excitation/inhibition imbalances is the hypofunction of NMDAR on PVI, leading to reduced inhibition of pyramidal cells and increased glutaminergic transmission. A potential method to correct this is to increase the activity of the NMDAR. Allostatic modulation of AMPAR facilitates the removal of magnesium from NMDAR and, therefore, may also improve NMDAR signalling. Another possible approach is to reduce pre-synaptic glutamate release from pyramidal cells via positive allostatic modulation of metabotropic glutamate receptors (mGlu2/3). Finally oxidative stress can lead to dysfunction and loss of PVI, therefore antioxidants and anti-inflammatory agents have been investigated in SSD.
GABAergic mechanisms
Enhancing postsynaptic GABA transmission is one strategy to compensate for the loss of PVI function. There are ionotropic GABAA and metabotropic GABAB receptors, each with diverse subunits and uneven tissue distribution [143].
Broad modulators like benzodiazepines act on multiple GABAA subunits, which can lead to sedation, tolerance and dependence. An observational cohort study found that benzodiazepine exposure in CHR-P individuals was not associated with a significant change in psychosis transition risk after controlling for confounding by indication [144]. A single dose of diazepam significantly reduced hippocampal blood flow in CHR-P individuals [145], aligning with pre-clinical evidence suggesting that enhancing GABA signalling can reduce hippocampal/amygdala hyperactivity, by attenuating hippocampal parvalbumin loss [146, 147]. Clinical application in SSD requires further investigation and will benefit from the use of more selective medications.
GABAA-α5 subunits are abundant in limbic areas and regulate inhibitory input to pyramidal neurons without impacting sedation [148]. PET studies have demonstrated lower α5 subtype GABAA receptor availability in the hippocampus of antipsychotic-free patients with SSD [149]. In animal models, selective GABAA-α5 positive allosteric modulators reversed ventral hippocampal hyperactivity [150]. An α2/3 selective GABAA medication (MK-0777/TPA023) showed promise in enhancing frontal gamma activity and improving cognitive performance in psychosis [151], but there was no significant cognitive benefit in a larger trial [152].
Potassium channels are extensively expressed on PVI, allowing them to fire at the high frequencies needed to synchronise pyramidal cells. Kv3-type voltage-gated potassium channels are highly expressed in corticolimbic brain circuits [153]; they have been shown to improve PVI firing [154] and rescue hippocampal network synchrony in vitro [155]. In animal models, they improved PVI firing and reversed behavioural impairments [156]. In healthy human participants, a Kv3-modulator (AUT00206) attenuated ketamine effects on fMRI BOLD activation in the ACC, precuneus and thalamus [157]. A PET study found that Kv3-modulation had no overall impact on dopamine synthesis capacity in patients with SSD [158]. However, there was a significant positive correlation between dopamine synthesis capacity reduction and positive symptom reduction in the group that received the Kv3-modulator.
Glutamatergic mechanisms
Hypofunction of NMDAR on GABAergic neurons may lead to loss of PVI activity in the hippocampus and PFC. Medications that increase the activity of NMDAR without leading to excitotoxicity have therefore been investigated.
NMDAR co-agonists targeting the glycine binding-site (glycine, d-serine, d-cycloserine and d-alanine) showed promise in small early trials [159, 160], but failed to meet clinical endpoints in phase III trials [161]. A meta-analysis of adjunctive treatment in patients with persistent negative symptoms found no benefit for d-cycloserine, while the combination of glycine or d-serine with d-cycloserine showed a small improvement in negative symptoms [162]. A large multicentre trial showed no significant improvement in negative or cognitive symptoms with glycine or d-cycloserine [161]. In a small sample of CHR-P individuals, d-serine resulted in a significant reduction in negative symptoms [163], which requires confirmation in larger samples. Small EEG studies have found improvements in mismatch negativity frequency with acute administration of glycine [164] and 6 weeks of d-serine [165] in SSD, but pharmaco-neuroimaging evidence is limited for these compounds.
Selective glycine transporter inhibitors have also been evaluated. Biopertin resulted in a significant reduction in negative symptoms in a phase II trial [166], but later trials found no significant improvement in patients with prominent negative symptoms [167]. Iclepertin showed significant improvements in cognitive symptoms in a phase II study [168], but the relationship between the treatment response and EEG measures, including mismatch negativity and resting gamma power, were modest and inconsistent [169]. There are phase III trials ongoing [170]. Sarcosine was also promising in initial trials [171, 172], but in larger trials, there were no symptom improvements [173]. However, there were improvements in cognitive symptoms when sarcosine was combined with benzoate [173], which inhibits the enzyme that metabolises D-serine. In small studies, benzoate reduced negative and cognitive symptoms in chronic [174] and clozapine-resistant SSD [175]. A recent meta-analysis found that adjunctive benzoate improved positive symptoms but had no effect on negative symptoms or cognition [176]. There was a significantly higher occurrence of extrapyramidal symptoms with benzoate, which raised safety concerns [176]. Imaging studies with benzoate have been limited; a small study with individuals with mild cognitive impairment reported decreased fMRI regional homogeneity in the right orbitofrontal cortex after benzoate treatment [177].
Meta-analyses with memantine, a low-affinity non-selective NMDAR antagonist, have had mixed results (reviewed [178]), with the most recent reporting improved negative and cognitive symptoms in chronic SSD [179]. Due to strong voltage-dependency and rapid channel-blocking, memantine can block pathological NMDA activity while maintaining signalling related to cognitive functioning [178]. EEG studies found that a single dose of memantine normalised gamma oscillatory power and phase synchrony in SSD [180, 181]. An ongoing study in antipsychotic-naïve first-episode psychosis will investigate its effects on regional glutamatergic metabolites and structural brain changes [182]. Memantine has generally been reported to be well-tolerated in clinical practice, with low rates of central nervous system side effects [178]. This is thought to be due to its low affinity, fast off-rate and preferential inhibition of extra-synaptic receptors [178]. Further research is needed to establish whether there is a role for memantine in the treatment of SSD, possibly in combination with other medications acting on the NMDAR.
By depolarising neurons, allosteric modulators of AMPAR can remove the magnesium block of NMDAR, therefore increasing the availability for glutamatergic signalling. CX-516 showed some promise as an adjunctive treatment with clozapine for cognitive symptoms [183], but in a larger study, there were no significant improvements when added to clozapine, olanzapine or risperidone [184]. Similar compounds (piracetam and diazoxide) resulted in positive symptom improvements in small trials [185, 186]. Overall, there is limited evidence for these compounds and a lack of pharmaco-imaging studies.
Another approach to decrease the excitability of pyramidal neurons is reducing pre-synaptic glutamate release via positive allostatic modulation of metabotropic receptors. mGluR2/3 is highly expressed in limbic areas and regulates excessive glutamate release [187]. mGluR2/3 agonists reduced hippocampal activity and atrophy in pre-clinical studies [25], and reduced ketamine associated BOLD fMRI increases in healthy individuals [188, 189]. Pomaglumetad methionil reduced positive symptoms in initial studies but failed in phase II/III trials [190]. Re-analyses suggested that pomaglumetad may be effective in earlier psychosis stages [191]. Other mGlu2-specific compounds (e.g. AZD8529 and LY2979165) showed promise in phase I trials and pharmaco-fMRI studies [189, 192], but had negative results in other trials [193].
Riluzole prevents glutamate release by inhibiting voltage-gated calcium channels and increasing the uptake of extracellular glutamate by astrocytes [194]. It also increases the depolarising effect of GABA and interacts with dopamine and acetylcholine [194]. A small clinical trial in SSD found that adjunctive riluzole reduced negative symptoms [195]. In treatment-resistant SSD, a 2-day riluzole challenge led to a reduction in glutamate + glutamine concentration and increased ACC-anterior PFC connectivity [196]. Higher baseline glutamate + glutamine concentrations were associated with more severe negative symptoms. A more recent review with treatment-resistant participants also found increased frontal connectivity after riluzole treatment [197].
Anticonvulsants
Several anticonvulsant medications have been investigated for their potential role in treating SSD. These medications have complex actions on multiple targets, but several act on voltage-gated sodium channels and reduce synaptic release of glutamate (e.g. lamotrigine, carbamazepine, topiramate). A meta-analysis of lamotrigine in SSD found small improvements in negative, cognitive and positive symptoms, but these are unlikely to be clinically significant, and the evidence was not robust [198]. Topiramate has a broad range of actions, including AMPA/kainite receptors, calcium/sodium channels and blocking GABA reuptake [199]. Several meta-analyses indicate topiramate improved all symptom domains, but these findings are considered preliminary, and there are safety concerns regarding possible psychiatric side effects [200, 201]; larger studies are thus needed to further investigate these findings. Imaging studies with topiramate have been limited to healthy participants and individuals with epilepsy or migraine; they have reported a pattern of reduced activation across the inferior and middle frontal gyri, and a reduced ability to deactivate task-negative networks [202, 203]. In healthy individuals, a study combining transcranial magnetic stimulation with sodium valproate/lamotrigine showed that lamotrigine increased connectivity between the dlPFC and the ACC when transcranial magnetic stimulation was applied to the PFC [204]. Meta-analytic evidence indicates that sodium valproate augmentation in SSD significantly improved total symptoms when open-labelled studies were included, but this was not significant when only RCTs were pooled [205].
Levetiracetam targets synaptic vesicle protein 2A and reduces pre-synaptic glutamate by regulating the expression/trafficking of synaptic vesicle sensor proteins and blocking voltage-gated calcium channels [206]. It also increases GABA receptor activation [206]. Levetiracetam was found to have significant effects on global symptoms, particularly negative symptoms, in a small RCT with individuals with SSD [207]. A recent neuroimaging study found increased resting-state fractional amplitude of low-frequency fluctuations (fALFF) in the hippocampus of patients with SSD, reflecting increased spontaneous neural activity in this region. Levetiracetam reduced hippocampal fALFF to a level that did not differ from controls [208]. Another study in medication naïve SSD reported that levetiracetam reduced hippocampal blood flow, but this non-crossover study was limited by very small sample sizes [209]. There are studies ongoing investigating the effect of levetiracetam on symptom domains and/or hippocampal hyperactivity in SSD (NCT04317807, NCT02647437 and NCT03034356) and CHR-P (NCT06224530). It will also be important to evaluate the safety profile of levetiracetam in SSD, given concerns regarding potential psychiatric side effects primarily from case reports and retrospective studies in epilepsy [210].
Targeting oxidative stress and neuroinflammation
Given the evidence for increased oxidative stress and its impact on PVI function, there have been trials of antioxidants (e.g. N-acetyl-cysteine (NAC), vitamins C/E, allopurinol, omega-3-polyunsaturated fatty acids and ginkgo biloba [211]) and anti-inflammatory agents (e.g. minocycline, NSAIDs, monoclonal antibodies [212]) in SSD. While large meta-analyses suggested an overall improvement in psychopathology with some of these compounds, many studies were small, and the mechanisms likely extend beyond the direct anti-inflammatory/antioxidant effects [213].
NAC has been extensively investigated in patients with SSD. NAC has multiple protective mechanisms, including: increasing glutamate transport into glial cells, increasing extra-synaptic glutamate release via activation of mGlu2/3, and antioxidant effects as the precursor to glutathione [214]. Meta-analyses indicate superiority to placebo in all symptom domains, although the RCTs were small and heterogeneous [215, 216]. The effect size was larger for patients in the acute phase of illness and at longer treatment durations (>24 weeks). Despite this, another meta-analysis reported no significant difference in any symptom domain in both early (<24 weeks) and late (>24 weeks) timepoints [217]. Pharmaco-imaging studies in patients with SSD found that NAC modulated EEG synchronisation over the left parietal-temporal, right temporal and bilateral PFC regions [218] and reduced fMRI resting-state connectivity in medial frontal areas [219]. Changes in structural connectivity have also been demonstrated after 6 months of NAC, with an 11% increase in fornix white matter integrity [220]. NAC increased peripheral and MRS mPFC measures of glutathione in early psychosis [221] and in chronic SSD [222], therefore, peripheral redox markers could be used to stratify patients into subgroups that will benefit more from NAC supplementation [221].
Targeting the mitochondria more directly with mitoquinone mesylate (MitoQ) can rescue oxidative stress and mitochondrial damage in animal models [223]. Alterations in oxidative stress blood markers have been identified in early psychosis, and these marker differences correlated with reductions in auditory response gamma oscillations in EEG [223]. As a subgroup of SSD with high mitochondrial dysfunction and more severe symptoms can be identified using these markers [223], a clinical trial is ongoing to test if these mechanism-based biomarkers could be used to stratify patients most likely to respond to MitoQ (NCT06191965).
Targeting the other interacting systems
Although we have focused on the role of excitation/inhibition balance in the corticolimbic system we acknowledge that there are complex interactions with other systems, including serotonin, acetylcholine and endocannabinoid systems. A comprehensive review of compounds targeting these systems is beyond the scope of this review, but we briefly outline results that are relevant to the corticolimbic system in Table 1.
Non-pharmacological approaches
Non-invasive brain stimulation has potential benefits in improving corticolimbic system function. In one of the few studies investigating neural changes in SSD with transcranial direct current stimulation, left dlPFC stimulation led to significant increases in gamma synchronisation [224]. Reviews suggest that transcranial magnetic stimulation and transcranial direct current stimulation may have promise in managing negative and cognitive symptoms in psychosis, but the evidence is mixed, and the studies are heterogeneous [225, 226].
Cognitive training involves performing cognitive tasks which are intended to improve function by activating relevant neuronal circuits and altering plasticity. A meta-analysis of neuroimaging studies with cognitive training showed that increased left PFC activation was most frequently observed, but there were effects on many regions, possibly due to heterogeneous training programmes [227]. Gamma oscillatory power has been shown to predict cognitive improvement after a course of cognitive remediation [228]. Combining cognitive training with either pharmacological interventions [229] or non-invasive brain stimulation may enhance the pro-cognitive effects [230].
Challenges and opportunities
Despite progress in understanding the role of corticolimbic circuit disruption in SSD and its potential therapeutic manipulation, promising results from pre-clinical and early clinical studies have largely not translated in later-phase clinical trials. There are multiple factors contributing to translation barriers, including intervention timing, target population heterogeneity, lack of biomarkers and lack of medication selectivity.
Many of the studies discussed were conducted with individuals with established SSD who have been treated with antipsychotics for many years, sometimes with a brief washout period. Antipsychotics exert long-term effects on neurotransmitter systems and brain circuits, which may be sufficient in making novel agents non-effective [231]. For instance, compensatory brain changes due to D2 antagonists can lead to increased sensitivity to dopamine receptor stimulation, known as dopamine receptor supersensitivity [232]. Pre-clinical studies have shown that haloperidol pretreatment-induced D2 supersensitivity which masked the efficacy of a GABAA-α5 compound [231]. There is evidence that active psychosis is also harmful to prognosis, although evidence of global neurobiological changes with untreated psychosis is more mixed [233]. Addressing these challenges could involve testing novel drugs in patients who have been on medications which are less likely to produce supersensitivity, such as partial dopamine agonists, and early interventions or preventative methods in at-risk/early psychosis populations.
Adolescence/early adulthood is a critical period due to multiple neurobiological and environmental factors. Heightened plasticity and perineuronal net development during this time makes these structures susceptible to oxidative stress, which may lead to PVI dysfunction [234]. Animal models with peripubertal administration of pharmacological treatments, including antioxidants, glutamate or GABA modulating agents, have been shown to prevent perineuronal net damage, PVI loss and increased dopaminergic activity [25, 33]. This suggests that interventions targeting excitation/inhibition imbalances and oxidative stress may have preventive potential in SSD, although the long-term broad administration of some of these medications would not be feasible in at-risk human populations due to poor tolerability. Longitudinal research tracking the pathological changes in corticolimbic circuits could help to identify the optimal timing for different interventions.
Another major obstacle to translation is the continued reliance on diagnostic categories, disregarding the considerable pathophysiological and phenomenological heterogeneity in SSD. Meta-analyses typically examine the entire patient population, potentially overlooking treatment response in specific subgroups. Patient stratification based on relevant biomarkers would enhance group homogeneity, but this relies on identifying appropriate predictive markers of treatment response. Neuroimaging methodologies provide a promising route for identifying biomarkers, but scalability issues may hinder clinical application [235]. Some progress has been made in identifying blood makers, such as for oxidative stress and mitochondrial function [221, 223]. Promising new evidence also suggests that increased peripheral blood levels of endothelial glycocalyx, which indicates blood–brain barrier dysfunction, can differentiate first-episode psychosis from healthy controls and is associated with increased symptom severity [236]. Despite these advances, targeted treatment approaches remain in the early stages of development in SSD research.
Many of the novel pharmacological approaches outlined above interact with several neurotransmitter and receptor subtypes, both within and beyond the corticolimbic system. This lack of selectivity makes it challenging to distinguish which mechanism is beneficial and why. The development of compounds with greater regional and subunit specificity would help untangle this complexity. A better understanding of the neurobiological mechanisms that underlie the considerable heterogeneity in psychosis will also help identify the most effective interventions for the individual. Neuroimaging combined with pharmacological and non-pharmacological interventions can enhance our understanding of how molecular targets impact wider neurocircuits. It is also important to integrate different research modalities to test hypotheses across computational, cellular, animal and human scales.
Conclusions
The progress that has been made in understanding the role of the corticolimbic system in SSD holds great promise for future successful translation of treatments. Further research to better understand why many of the novel compounds have failed and the role of illness stage and prior treatment exposure will be essential to progressing drug discovery. Combining multiple modalities to examine the same hypotheses can help to increase our understanding of the neuropathological processes within the corticolimbic system. For instance, utilising neuroimaging across species can explore homology between circuits and validate theories via back translation. The combination of neuroimaging measures with experimental interventions can substantially improve our understanding of how molecular modulation influences wider corticolimbic function and behaviour/symptomatology. Large-scale consortia afford the power needed to use multivariate data-driven methods and normative modelling to make inferences at the individual level. Increasing our knowledge of biomarkers and their use to stratify patient samples will also be integral to moving towards a precision medicine approach in SSD.
References
Benes FM. Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacology. 2010;35:239–57.
Kovner R, Oler JA, Kalin NH. Cortico-limbic interactions mediate adaptive and maladaptive responses relevant to psychopathology. AJP. 2019;176:987–99.
Lavigne KM, Kanagasabai K, Palaniyappan L. Ultra-high field neuroimaging in psychosis: a narrative review. Front Psychiatry. 2022;13:994372.
Sydnor VJ, Roalf DR. A meta-analysis of ultra-high field glutamate, glutamine, GABA and glutathione in psychosis: implications for studies of psychosis risk. Schizophr Res. 2020;226:61–9.
Zhang Y-D, Dong Z, Wang S-H, Yu X, Yao X, Zhou Q, et al. Advances in multimodal data fusion in neuroimaging: overview, challenges, and novel orientation. Inf Fusion. 2020;64:149–87.
Georgiadis F, Larivière S, Glahn D, Hong LE, Kochunov P, Mowry B, et al. Connectome architecture shapes large-scale cortical alterations in schizophrenia: a worldwide ENIGMA study. Mol Psychiatry. 2024;29:1869–81.
Demro C, Mueller BA, Kent JS, Burton PC, Olman CA, Schallmo M-P, et al. The psychosis human connectome project: an overview. Neuroimage. 2021;241:118439.
Dwyer DB, Chand GB, Pigoni A, Khuntia A, Wen J, Antoniades M, et al. Psychosis brain subtypes validated in first-episode cohorts and related to illness remission: results from the PHENOM consortium. Mol Psychiatry. 2023;28:2008–17.
Smucny J, Davidson I, Carter CS. Comparing machine and deep learning-based algorithms for prediction of clinical improvement in psychosis with functional magnetic resonance imaging. Hum Brain Mapp. 2021;42:1197–205.
Rutherford S, Kia SM, Wolfers T, Fraza C, Zabihi M, Dinga R, et al. The normative modeling framework for computational psychiatry. Nat Protoc. 2022;17:1711–34.
Aryutova K, Stoyanov D. Pharmaco-magnetic resonance as a tool for monitoring the medication-related effects in the brain may provide potential biomarkers for psychotic disorders. Int J Mol Sci. 2021;22:9309.
Phelps EA. Human emotion and memory: interactions of the amygdala and hippocampal complex. Curr Opin Neurobiol. 2004;14:198–202.
Lieberman J, Girgis R, Brucato G, Moore H, Provenzano F, Kegeles L, et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018;23:1764–72.
Knight S, McCutcheon R, Dwir D, Grace AA, O'Daly O, McGuire P, et al. Hippocampal circuit dysfunction in psychosis. Transl Psychiatry. 2022;12:1–13.
Adriano F, Caltagirone C, Spalletta G. Hippocampal volume reduction in first-episode and chronic schizophrenia: a review and meta-analysis. Neuroscientist. 2012;18:180–200.
Talati P, Rane S, Kose S, Blackford JU, Gore J, Donahue MJ, et al. Increased hippocampal CA1 cerebral blood volume in schizophrenia. Neuroimage Clin. 2014;5:359–64.
McHugo M, Talati P, Armstrong K, Vandekar SN, Blackford JU, Woodward ND, et al. Hyperactivity and reduced activation of anterior hippocampus in early psychosis. Am J Psychiatry. 2019;176:1030–8.
Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11:543–50.
Allen P, Chaddock CA, Egerton A, Howes OD, Bonoldi I, Zelaya F, et al. Resting hyperperfusion of the hippocampus, midbrain, and basal ganglia in people at high risk for psychosis. Am J Psychiatry. 2016;173:392–9.
Allen P, Azis M, Modinos G, Bossong MG, Bonoldi I, Samson C, et al. Increased resting hippocampal and basal ganglia perfusion in people at ultra high risk for psychosis: replication in a second cohort. Schizophr Bull. 2018;44:1323–31.
Modinos G, Şimşek F, Azis M, Bossong M, Bonoldi I, Samson C, et al. Prefrontal GABA levels, hippocampal resting perfusion and the risk of psychosis. Neuropsychopharmacol. 2018;43:2652–9.
Modinos G, Egerton A, McMullen K, McLaughlin A, Kumari V, Barker GJ, et al. Increased resting perfusion of the hippocampus in high positive schizotypy: a pseudocontinuous arterial spin labeling study. Hum Brain Mapp. 2018;39:4055–64.
Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, et al. Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. AJP. 2014;171:549–56.
Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–46.
Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a pathogenic driver. Neuron. 2013;78:81–93.
Samudra N, Ivleva EI, Hubbard NA, Rypma B, Sweeney JA, Clementz BA, et al. Alterations in hippocampal connectivity across the psychosis dimension. Psychiatry Res Neuroimaging. 2015;233:148–57.
Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. 2019;24:1248–57.
DeFelipe J, López-Cruz PL, Benavides-Piccione R, Bielza C, Larrañaga P, Anderson S, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci. 2013;14:202–16.
Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–20.
Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H, et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res. 2011;131:165–73.
Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55:1–10.
Chung DW, Fish KN, Lewis DA. Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am J Psychiatry. 2016;173:1131–9.
Phensy A, Driskill C, Lindquist K, Guo L, Jeevakumar V, Fowler B, et al. Antioxidant treatment in male mice prevents mitochondrial and synaptic changes in an NMDA receptor dysfunction model of schizophrenia. eNeuro. 2017;4:ENEURO.0081-17.2017.
Abdul-Monim Z, Neill JC, Reynolds GP. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol. 2007;21:198–205.
Zaitsev AV, Povysheva NV, Lewis DA, Krimer LS. P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex. J Neurophysiol. 2007;97:3567–73.
Cabungcal J-H, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci USA. 2013;110:9130–5.
Cuenod M, Steullet P, Cabungcal J-H, Dwir D, Khadimallah I, Klauser P, et al. Caught in vicious circles: a perspective on dynamic feed-forward loops driving oxidative stress in schizophrenia. Mol Psychiatry. 2022;27:1886–97.
Perkins DO, Jeffries CD, Do KQ. Potential roles of redox dysregulation in the development of schizophrenia. Biol Psychiatry. 2020;88:326–36.
Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38:950–7.
Do KQ, Trabesinger AH, Kirsten-Krüger M, Lauer CJ, Dydak U, Hell D, et al. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci. 2000;12:3721–8.
Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. 2011;29:311–24.
Comer AL, Carrier M, Tremblay M-È, Cruz-Martín A. The inflamed brain in schizophrenia: the convergence of genetic and environmental risk factors that lead to uncontrolled neuroinflammation. Front Cell Neurosci. 2020;14:274.
Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–32.
Grace AA, Gomes FV. The circuitry of dopamine system regulation and its disruption in schizophrenia: insights into treatment and prevention. Schizophr Bull. 2019;45:148–57.
Kaar SJ, Natesan S, McCutcheon R, Howes OD. Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology. 2020;172:107704.
Charara A, Grace A. Dopamine receptor subtypes selectively modulate excitatory afferents from the hippocampus and amygdala to rat nucleus accumbens neurons. Neuropsychopharmacology. 2003;28:1412–21.
Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006;31:1356–61.
Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharm Sci. 2011;32:507–13.
Grace AA. Dopamine system dysregulation by the ventral subiculum as the common pathophysiological basis for schizophrenia psychosis, psychostimulant abuse, and stress. Neurotox Res. 2010;18:367–76.
Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–73.
Floresco SB, Todd CL, Grace AA. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci. 2001;21:4915–22.
Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–76.
Kesby JP, Murray GK, Knolle F. Neural circuitry of salience and reward processing in psychosis. Biol Psychiatry Glob Open Sci. 2023;3:33–46.
Knolle F, Ermakova AO, Justicia A, Fletcher PC, Bunzeck N, Düzel E, et al. Brain responses to different types of salience in antipsychotic naïve first episode psychosis: an fMRI study. Transl Psychiatry. 2018;8:196.
Modinos G, Allen P, Zugman A, Dima D, Azis M, Samson C, et al. Neural circuitry of novelty salience processing in psychosis risk: association with clinical outcome. Schizophr Bull. 2020;46:670–9.
Taylor SF, Phan KL, Britton JC, Liberzon I. Neural response to emotional salience in schizophrenia. Neuropsychopharmacology. 2005;30:984–95.
Morris RW, Vercammen A, Lenroot R, Moore L, Langton JM, Short B, et al. Disambiguating ventral striatum fMRI-related bold signal during reward prediction in schizophrenia. Mol Psychiatry. 2012;17:280–9.
Murray GK, Corlett PR, Clark L, Pessiglione M, Blackwell AD, Honey G, et al. Substantia nigra/ventral tegmental reward prediction error disruption in psychosis. Mol Psychiatry. 2008;13:267–76.
Millman ZB, Gallagher K, Demro C, Schiffman J, Reeves GM, Gold JM, et al. Evidence of reward system dysfunction in youth at clinical high-risk for psychosis from two event-related fMRI paradigms. Schizophr Res. 2020;226:111–9.
Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry. 2011;168:1311–7.
McCutcheon RA, Merritt K, Howes OD. Dopamine and glutamate in individuals at high risk for psychosis: a meta‐analysis of in vivo imaging findings and their variability compared to controls. World Psychiatry. 2021;20:405–16.
McCutcheon R, Beck K, Jauhar S, Howes OD. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophr Bull. 2018;44:1301–11.
Sorg C, Manoliu A, Neufang S, Myers N, Peters H, Schwerthöffer D, et al. Increased intrinsic brain activity in the striatum reflects symptom dimensions in schizophrenia. Schizophr Bull. 2013;39:387–95.
Stone JM, Howes OD, Egerton A, Kambeitz J, Allen P, Lythgoe DJ, et al. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry. 2010;68:599–602.
McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci. 2019;42:205–20.
Pani SM, Sabaroedin K, Tiego J, Bellgrove MA, Fornito A. A multivariate analysis of the association between corticostriatal functional connectivity and psychosis-like experiences in the general community. Psychiatry Res Neuroimaging. 2021;307:111202.
Dandash O, Fornito A, Lee J, Keefe RSE, Chee MWL, Adcock RA, et al. Altered striatal functional connectivity in subjects with an at-risk mental state for psychosis. Schizophr Bull. 2014;40:904–13.
Chechko N, Cieslik EC, Müller VI, Nickl-Jockschat T, Derntl B, Kogler L, et al. Differential resting-state connectivity patterns of the right anterior and posterior dorsolateral prefrontal cortices (DLPFC) in schizophrenia. Front Psychiatry. 2018;9:211.
Sarpal DK, Robinson DG, Lencz T, Argyelan M, Ikuta T, Karlsgodt K, et al. Antipsychotic treatment and functional connectivity of the striatum in first-episode schizophrenia. JAMA Psychiatry. 2015;72:5–13.
Simpson EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron. 2010;65:585–96.
Morris RW, Quail S, Griffiths KR, Green MJ, Balleine BW. Corticostriatal control of goal-directed action is impaired in schizophrenia. Biol Psychiatry. 2015;77:187–95.
Strauss GP, Frank MJ, Waltz JA, Kasanova Z, Herbener ES, Gold JM. Deficits in positive reinforcement learning and uncertainty-driven exploration are associated with distinct aspects of negative symptoms in schizophrenia. Biol Psychiatry. 2011;69:424–31.
Blanchard JJ, Cohen AS. The structure of negative symptoms within schizophrenia: implications for assessment. Schizophr Bull. 2006;32:238–45.
Carbon M, Correll CU. Thinking and acting beyond the positive: the role of the cognitive and negative symptoms in schizophrenia. CNS Spectr. 2014;19:35–53.
Galderisi S, Merlotti E, Mucci A. Neurobiological background of negative symptoms. Eur Arch Psychiatry Clin Neurosci. 2015;265:543–58.
Metzak PD, Devoe DJ, Iwaschuk A, Braun A, Addington J. Brain changes associated with negative symptoms in clinical high risk for psychosis: a systematic review. Neurosci Biobehav Rev. 2020;118:367–83.
Anderson AK, Phelps EA. Lesions of the human amygdala impair enhanced perception of emotionally salient events. Nature. 2001;411:305–9.
Yaniv D, Desmedt A, Jaffard R, Richter-Levin G. The amygdala and appraisal processes: Stimulus and response complexity as an organizing factor. Brain Res Brain Res Rev. 2004;44:179–86.
Banks SJ, Eddy KT, Angstadt M, Nathan PJ, Phan KL. Amygdala–frontal connectivity during emotion regulation. Soc Cogn Affect Neurosci. 2007;2:303–12.
Cunningham WA, Brosch T. Motivational salience: amygdala tuning from traits, needs, values, and goals. Curr Dir Psychol Sci. 2012;21:54–9.
Lawrie SM, Abukmeil SS. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br J Psychiatry. 1998;172:110–20.
Wright IC, Rabe-Hesketh S, Woodruff PWR, David AS, Murray RM, Bullmore ET. Meta-analysis of regional brain volumes in schizophrenia. AJP. 2000;157:16–25.
Okada N, Fukunaga M, Yamashita F, Koshiyama D, Yamamori H, Ohi K, et al. Abnormal asymmetries in subcortical brain volume in schizophrenia. Mol Psychiatry. 2016;21:1460–6.
van Erp TGM, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. 2016;21:547–53.
Keshavan MS, Dick E, Mankowski I, Harenski K, Montrose DM, Diwadkar V, et al. Decreased left amygdala and hippocampal volumes in young offspring at risk for schizophrenia. Schizophr Res. 2002;58:173–83.
Lawrie SM, Whalley HC, Job DE, Johnstone EC. Structural and functional abnormalities of the amygdala in schizophrenia. Ann N Y Acad Sci. 2003;985:445–60.
Ho NF, Chong PLH, Lee DR, Chew QH, Chen G, Sim K. The amygdala in schizophrenia and bipolar disorder: a synthesis of structural MRI, diffusion tensor imaging, and resting-state functional connectivity findings. Harv Rev Psychiatry. 2019;27:150.
Kitis O, Ozalay O, Zengin EB, Haznedaroglu D, Eker MC, Yalvac D, et al. Reduced left uncinate fasciculus fractional anisotropy in deficit schizophrenia but not in non-deficit schizophrenia. Psychiatry Clin Neurosci. 2012;66:34–43.
Jalbrzikowski M, Murty VP, Tervo-Clemmens B, Foran W, Luna B. Age associated deviations of amygdala functional connectivity in psychosis spectrum youth: relevance to psychotic symptoms. Am J Psychiatry. 2019;176:196–207.
Anticevic A, Tang Y, Cho YT, Repovs G, Cole MW, Savic A, et al. Amygdala connectivity differs among chronic, early course, and individuals at risk for developing schizophrenia. Schizophr Bull. 2014;40:1105–16.
Walton E, Hibar DP, Erp van TGM, Potkin SG, Roiz-Santiañez R, Crespo-Facorro, et al. Prefrontal cortical thinning links to negative symptoms in schizophrenia via the ENIGMA consortium. Psychol Med. 2018;48:82–94.
Barkl SJ, Lah S, Harris AWF, Williams LM. Facial emotion identification in early-onset and first-episode psychosis: A systematic review with meta-analysis. Schizophr Res. 2014;159:62–9.
Gao Z, Zhao W, Liu S, Liu Z, Yang C, Xu Y. Facial emotion recognition in schizophrenia. Front Psychiatry. 2021;12:633717.
Martin D, Croft J, Pitt A, Strelchuk D, Sullivan S, Zammit S. Systematic review and meta-analysis of the relationship between genetic risk for schizophrenia and facial emotion recognition. Schizophr Res. 2020;218:7–13.
Addington J, Penn D, Woods SW, Addington D, Perkins DO. Facial affect recognition in individuals at clinical high risk for psychosis. Br J Psychiatry. 2008;192:67–8.
Amminger GP, Schäfer MR, Papageorgiou K, Klier CM, Schlögelhofer M, Mossaheb N, et al. Emotion recognition in individuals at clinical high-risk for schizophrenia. Schizophr Bull. 2012;38:1030–9.
Lukow PB, Kiemes A, Kempton MJ, Turkheimer FE, McGuire P, Modinos G. Neural correlates of emotional processing in psychosis risk and onset – A systematic review and meta-analysis of fMRI studies. Neurosci Biobehav Rev. 2021;128:780–8.
Dong D, Wang Y, Jia X, Li Y, Chang X, Vandekerckhove M, et al. Abnormal brain activation during threatening face processing in schizophrenia: a meta-analysis of functional neuroimaging studies. Schizophr Res. 2018;197:200–8.
Taylor SF, Kang J, Brege IS, Tso IF, Hosanagar A, Johnson TD. Meta-analysis of functional neuroimaging studies of emotion perception and experience in schizophrenia. Biol Psychiatry. 2012;71:136–45.
Modinos G, Ormel J, Aleman A. Altered activation and functional connectivity of neural systems supporting cognitive control of emotion in psychosis proneness. Schizophr Res. 2010;118:88–97.
Morris RW, Sparks A, Mitchell PB, Weickert CS, Green MJ. Lack of cortico-limbic coupling in bipolar disorder and schizophrenia during emotion regulation. Transl Psychiatry. 2012;2:e90.
Dichter GS, Bellion C, Casp M, Belger A. Impaired modulation of attention and emotion in schizophrenia. Schizophr Bull. 2010;36:595–606.
Aghevli MA, Blanchard JJ, Horan WP. The expression and experience of emotion in schizophrenia: a study of social interactions. Psychiatry Res. 2003;119:261–70.
Myin-Germeys I, Delespaul PAEG, deVries MW. Schizophrenia patients are more emotionally active than is assumed based on their behavior. Schizophr Bull. 2000;26:847–54.
Frost KH, Strauss GP. A review of anticipatory pleasure in schizophrenia. Curr Behav Neurosci Rep. 2016;3:232–47.
Knutson B, Adams CM, Fong GW, Hommer D. Anticipation of increasing monetary reward selectively recruits nucleus accumbens. J Neurosci. 2001;21:RC159.
Arrondo G, Segarra N, Metastasio A, Ziauddeen H, Spencer J, Reinders NR, et al. Reduction in ventral striatal activity when anticipating a reward in depression and schizophrenia: a replicated cross-diagnostic finding. Front Psychol. 2015;6:1280.
Juckel G, Schlagenhauf F, Koslowski M, Filonov D, Wüstenberg T, Villringer A, et al. Dysfunction of ventral striatal reward prediction in schizophrenic patients treated with typical, not atypical, neuroleptics. Psychopharmacology. 2006;187:222–8.
Simon JJ, Biller A, Walther S, Roesch-Ely D, Stippich C, Weisbrod M, et al. Neural correlates of reward processing in schizophrenia — Relationship to apathy and depression. Schizophr Res. 2010;118:154–61.
Kirschner M, Hager OM, Bischof M, Hartmann MN, Kluge A, Seifritz E, et al. Ventral striatal hypoactivation is associated with apathy but not diminished expression in patients with schizophrenia. J Psychiatry Neurosci. 2016;41:152–61.
Grimm O, Heinz A, Walter H, Kirsch P, Erk S, Haddad L, et al. Striatal response to reward anticipation: evidence for a systems-level intermediate phenotype for schizophrenia. JAMA Psychiatry. 2014;71:531–9.
Zeng J, Yan J, Cao H, Su Y, Song Y, Luo Y, et al. Neural substrates of reward anticipation and outcome in schizophrenia: a meta-analysis of fMRI findings in the monetary incentive delay task. Transl Psychiatry. 2022;12:448.
Barch DM, Dowd EC. Goal representations and motivational drive in schizophrenia: the role of prefrontal–striatal interactions. Schizophr Bull. 2010;36:919–34.
McCleery A, Nuechterlein KH. Cognitive impairment in psychotic illness: prevalence, profile of impairment, developmental course, and treatment considerations. Dialogues Clin Neurosci. 2019;21:239–48.
Cowman M, Holleran L, Lonergan E, O’Connor K, Birchwood M, Donohoe G. Cognitive predictors of social and occupational functioning in early psychosis: a systematic review and meta-analysis of cross-sectional and longitudinal data. Schizophr Bull. 2021;47:1243–53.
Jay TM, Thierry A-M, Wiklund L, Glowinski J. Excitatory amino acid pathway from the hippocampus to the prefrontal cortex. Contribution of AMPA receptors in hippocampo-prefrontal cortex transmission. Eur J Neurosci. 1992;4:1285–95.
Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73.
Berdenis van Berlekom A, Muflihah CH, Snijders GJLJ, MacGillavry HD, Middeldorp J, Hol EM, et al. Synapse pathology in schizophrenia: a meta-analysis of postsynaptic elements in postmortem brain Studies. Schizophr Bull. 2020;46:374–86.
Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry. 2019;24:549–61.
Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25.
Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–13.
Wang X-J. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. 2010;90:1195–268.
Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci USA. 2006;103:19878–83.
Minzenberg MJ, Firl AJ, Yoon JH, Gomes GC, Reinking C, Carter CS. Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology. 2010;35:2590–9.
Chen C-MA, Stanford AD, Mao X, Abi-Dargham A, Shungu DC, Lisanby SH, et al. GABA level, gamma oscillation, and working memory performance in schizophrenia. Neuroimage Clin. 2014;4:531–9.
Haenschel C, Bittner RA, Waltz J, Haertling F, Wibral M, Singer W, et al. Cortical oscillatory activity is critical for working memory as revealed by deficits in early-onset schizophrenia. J Neurosci. 2009;29:9481–9.
Sohal VS. Transforming discoveries about cortical microcircuits and gamma oscillations into new treatments for cognitive deficits in schizophrenia. AJP. 2022;179:267–76.
Tukker JJ, Fuentealba P, Hartwich K, Somogyi P, Klausberger T. Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J Neurosci. 2007;27:8184–9.
Deleuze C, Bhumbra GS, Pazienti A, Lourenço J, Mailhes C, Aguirre A, et al. Strong preference for autaptic self-connectivity of neocortical PV interneurons facilitates their tuning to γ-oscillations. PLoS Biol. 2019;17:e3000419.
Cho YK, Li D. Optogenetics: methods and protocols. In: Kianianmomeni A, editor. Springer, 2016.
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702.
Cho KKA, Hoch R, Lee AT, Patel T, Rubenstein JLR, Sohal VS. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/-) mice. Neuron. 2015;85:1332–43.
Cho KKA, Davidson TJ, Bouvier G, Marshall JD, Schnitzer MJ, Sohal VS. Cross-hemispheric gamma synchrony between prefrontal parvalbumin interneurons supports behavioral adaptation during rule shift learning. Nat Neurosci. 2020;23:892–902.
Boudewyn MA, Scangos K, Ranganath C, Carter CS. Using prefrontal transcranial direct current stimulation (tDCS) to enhance proactive cognitive control in schizophrenia. Neuropsychopharmacology. 2020;45:1877–83.
Merritt K, McGuire PK, Egerton A. 1H-MRS in schizophrenia investigators, Aleman A, Block W, et al. Association of age, antipsychotic medication, and symptom severity in schizophrenia with proton magnetic resonance spectroscopy brain glutamate level: a mega-analysis of individual participant-level data. JAMA Psychiatry. 2021;78:667–81.
Smucny J, Carter CS, Maddock RJ. Medial prefrontal cortex glutamate is reduced in schizophrenia and moderated by measurement quality: a meta-analysis of proton magnetic resonance spectroscopy studies. Biol Psychiatry. 2021;90:643–51.
Kumar V, Vajawat B, Rao NP. Frontal GABA in schizophrenia: a meta-analysis of 1H-MRS studies. World J Biol Psychiatry. 2021;22:1–13.
Yoon JH, Grandelis A, Maddock RJ. Dorsolateral prefrontal cortex GABA concentration in humans ppredicts working memory load processing capacity. J Neurosci. 2016;36:11788–94.
Ragland JD, Maddock RJ, Hurtado MY, Tanase C, Lesh TA, Niendam TA, et al. Disrupted GABAergic facilitation of working memory performance in people with schizophrenia. Neuroimage Clin. 2020;25:102127.
Frankle WG, Cho RY, Mason NS, Chen C-M, Himes M, Walker C, et al. [11C]flumazenil Binding is increased in a dose-dependent manner with tiagabine-induced elevations in GABA levels. PLoS ONE. 2012;7:e32443.
Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML, et al. In vivo measurement of GABA transmission in healthy subjects and schizophrenia patients. Am J Psychiatry. 2015;172:1148–59.
Uliana DL, Lisboa JRF, Gomes FV, Grace AA. The excitatory-inhibitory balance as a target for the development of novel drugs to treat schizophrenia. Biochem Pharmacol. 2024;228:116298.
Enna SJ. The GABA receptors. In: Enna SJ, Möhler H, editors. Humana Press:Totowa. 2007. p. 1–21.
Livingston NR, De Micheli A, McCutcheon RA, Butler E, Hamdan M, Grace AA, et al. Effects of benzodiazepine exposure on real-world clinical outcomes in individuals at clinical high risk for psychosis. Schizophr Bull. 2024. https://doi.org/10.1093/schbul/sbae036.
Livingston NR, Kiemes A, Devenyi GA, Knight S, Lukow PB, Jelen LA, et al. Effects of diazepam on hippocampal blood flow in people at clinical high risk for psychosis. Neuropsychopharmacol. 202;49:1448–58.
Du Y, Grace AA. Peripubertal diazepam administration prevents the emergence of dopamine system hyperresponsivity in the MAM developmental disruption model of schizophrenia. Neuropsychopharmacology. 2013;38:1881–8.
Du Y, Grace AA. Loss of parvalbumin in the hippocampus of MAM schizophrenia model rats is attenuated by peripubertal diazepam. Int J Neuropsychopharmacol. 2016;19:pyw065.
Gill KM, Grace AA. The role of α5 GABAA receptor agonists in the treatment of cognitive deficits in schizophrenia. Curr Pharm Des. 2014;20:5069–76.
Marques TR, Ashok AH, Angelescu I, Borgan F, Myers J, Lingford-Hughes A, et al. GABA-A receptor differences in schizophrenia: a positron emission tomography study using [11C]Ro154513. Mol Psychiatry. 2021;26:2616–25.
Gill KM, Lodge DJ, Cook JM, Aras S, Grace AA. A novel α5GABAAR-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacol. 2011;36:1903–11.
Lewis DA, Cho RY, Carter CS, Eklund K, Forster S, Kelly MA, et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry. 2008;165:1585–93.
Buchanan RW, Keefe RSE, Lieberman JA, Barch DM, Csernansky JG, Goff DC, et al. A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol Psychiatry. 2011;69:442–9.
Yanagi M, Joho RH, Southcott SA, Shukla AA, Ghose S, Tamminga CA. Kv3.1-containing K(+) channels are reduced in untreated schizophrenia and normalized with antipsychotic drugs. Mol Psychiatry. 2014;19:573–9.
Boddum K, Hougaard C, Xiao-Ying Lin J, von Schoubye NL, Jensen HS, Grunnet M, et al. Kv3.1/Kv3.2 channel positive modulators enable faster activating kinetics and increase firing frequency in fast-spiking GABAergic interneurons. Neuropharmacology. 2017;118:102–12.
Large C, Neill J, Harte M, Cadinu D, Cunningham M, LeBeau F, et al. Entraining neural networks through parvalbumin-positive interneurons: can this offer a better way to treat schizophrenia? Schizophr Bull. 2017;43:S3.
Leger M, Alvaro G, Large C, Harte M, Neill JC. Efficacy of AUT6, a novel and selective Kv3 channel modulator, to alleviate cognitive and neurobiological dysfunction in the sub-chronic PCP rat model of schizophrenia symptomatology. J Psychopharmacol Suppl. 2015;29.
Deakin B, Perini F, Nazimek J, McKie S, Hutchison J, Alvaro G, et al. AUT00206, a novel Kv3 channel modulator, reduces ketamine induced BOLD signalling in healthy male volunteers: a randomised placebo-controlled crossover trial. Schizophr Bull. 2019;45:S245–6.
Angelescu I, Kaar SJ, Marques TR, Borgan F, Veronesse M, Sharman A, et al. The effect of AUT00206, a Kv3 potassium channel modulator, on dopamine synthesis capacity and the reliability of [18F]-FDOPA imaging in schizophrenia. J Psychopharmacol. 2022;36:1061–9.
Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry. 1999;56:29–36.
Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 1998;44:1081–9.
Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, et al. The cognitive and negative symptoms in schizophrenia trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. AJP. 2007;164:1593–602.
Tiihonen J, Wahlbeck K. Glutamatergic drugs for schizophrenia. Cochrane Database Syst Rev. 2006. https://doi.org/10.1002/14651858.CD003730.pub2.
Kantrowitz JT, Woods SW, Petkova E, Cornblatt B, Corcoran CM, Chen H, et al. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: a pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry. 2015;2:403–12.
Greenwood L-M, Leung S, Michie PT, Green A, Nathan PJ, Fitzgerald P, et al. The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophr Res. 2018;191:61–9.
Kantrowitz JT, Epstein ML, Lee M, Lehrfeld N, Nolan KA, Shope C, et al. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: correlation with symptoms. Schizophr Res. 2018;191:70–9.
Umbricht D, Alberati D, Martin-Facklam M, Borroni E, Youssef EA, Ostland M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71:637–46.
Blaettler T, Bugarski-Kirola D, Fleischhacker WW, Bressan R, Arango C, Abi-Saab D, et al. Efficacy and safety of adjunctive bitopertin (10 and 20mg) versus placebo in subjects with persistent predominant negative symptoms of schizophrenia treated with antipsychotics — Results from the phase III FlashLyte study. Schizophr Res. 2014;158:e2–e3.
Fleischhacker WW, Podhorna J, Gröschl M, Hake S, Zhao Y, Huang S, et al. Efficacy and safety of the novel glycine transporter inhibitor BI 425809 once daily in patients with schizophrenia: a double-blind, randomised, placebo-controlled phase 2 study. Lancet Psychiatry. 2021;8:191–201.
Schultheis C, Rosenbrock H, Mack SR, Vinisko R, Schuelert N, Plano A, et al. Quantitative electroencephalography parameters as neurophysiological biomarkers of schizophrenia-related deficits: a Phase II substudy of patients treated with iclepertin (BI 425809). Transl Psychiatry. 2022;12:329.
Rosenbrock H, Desch M, Wunderlich G. Development of the novel GlyT1 inhibitor, iclepertin (BI 425809), for the treatment of cognitive impairment associated with schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2023;273:1557–66.
Lane H-Y, Liu Y-C, Huang C-L, Chang Y-C, Liau C-H, Perng C-H, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63:9–12.
Lane H-Y, Lin C-H, Huang Y-J, Liao C-H, Chang Y-C, Tsai GE. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and d-serine add-on treatment for schizophrenia. Int J Neuropsychopharmacol. 2010;13:451–60.
Lin C-Y, Liang S-Y, Chang Y-C, Ting S-Y, Kao C-L, Wu Y-H, et al. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: a randomised, double-blind, placebo-controlled trial. World J Biol Psychiatry. 2017;18:357–68.
Lane H-Y, Lin C-H, Green MF, Hellemann G, Huang C-C, Chen P-W, et al. Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of d -amino acid oxidase inhibitor. JAMA Psychiatry. 2013;70:1267.
Lin C-H, Lin C-H, Chang Y-C, Huang Y-J, Chen P-W, Yang H-T, et al. Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry. 2018;84:422–32.
Seetharam JC, Maiti R, Mishra A, Mishra BR. Efficacy and safety of add-on sodium benzoate, a D-amino acid oxidase inhibitor, in treatment of schizophrenia: a systematic review and meta-analysis. Asian J Psychiatry. 2022;68:102947.
Lane H-Y, Tu C-H, Lin W-C, Lin C-H. Brain activity of benzoate, a D-amino acid oxidase inhibitor, in patients with mild cognitive impairment in a randomized, double-blind, placebo controlled clinical trial. Int J Neuropsychopharmacol. 2021;24:392–9.
Kikuchi T. Is memantine effective as an NMDA receptor antagonist in adjunctive therapy for schizophrenia? Biomolecules. 2020;10:1134.
Zheng W, Zhu X-M, Zhang Q-E, Cai D-B, Yang X-H, Zhou Y-L, et al. Adjunctive memantine for major mental disorders: a systematic review and meta-analysis of randomized double-blind controlled trials. Schizophr Res. 2019;209:12–21.
Light GA, Zhang W, Joshi YB, Bhakta S, Talledo JA, Swerdlow NR. Single-dose memantine improves cortical oscillatory response dynamics in patients with schizophrenia. Neuropsychopharmacol. 2017;42:2633–9.
Molina JL, Voytek B, Thomas ML, Joshi YB, Bhakta SG, Talledo JA, et al. Memantine effects on electroencephalographic measures of putative excitatory/inhibitory balance in schizophrenia. Biol Psychiatry Cogn Neurosci Neuroimaging. 2020;5:562–8.
Sandström KO, Baltzersen OB, Marsman A, Lemvigh CK, Boer VO, Bojesen KB, et al. Add-on MEmaNtine to dopamine antagonism to improve negative symptoms at first psychosis- the AMEND trial protocol. Front Psychiatry. 2022;13:889572.
Goff DC, Leahy L, Berman I, Posever T, Herz L, Leon AC, et al. A placebo-controlled pilot study of the ampakine CX516 added to clozapine in schizophrenia. J Clin Psychopharmacol. 2001;21:484–7.
Goff DC, Lamberti JS, Leon AC, Green MF, Miller AL, Patel J, et al. A placebo-controlled add-on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology. 2008;33:465–72.
Akhondzadeh S, Mojtahedzadeh V, Mirsepassi G-R, Moin M, Amini-Nooshabadi H, Kamalipour A. Diazoxide in the treatment of schizophrenia: novel application of potassium channel openers in the treatment of schizophrenia. J Clin Pharm Ther. 2002;27:453–9.
Noorbala AA, Akhondzadeh S, Davari-Ashtiani R, Amini-Nooshabadi H. Piracetam in the treatment of schizophrenia: implications for the glutamate hypothesis of schizophrenia. J Clin Pharm Ther. 1999;24:369–74.
Milczarek MM, Perry JC, Amin E, Haniffa S, Hathaway T, Vann SD. Impairments in the early consolidation of spatial memories via group II mGluR agonism in the mammillary bodies. Sci Rep. 2024;14:5977.
Kantrowitz JT, Grinband J, Goff DC, Lahti AC, Marder SR, Kegeles LS, et al. Proof of mechanism and target engagement of glutamatergic drugs for the treatment of schizophrenia: RCTs of pomaglumetad and TS-134 on ketamine-induced psychotic symptoms and pharmacoBOLD in healthy volunteers. Neuropsychopharmacol. 2020;45:1842–50.
Mehta MA, Schmechtig A, Kotoula V, McColm J, Jackson K, Brittain C, et al. Group II metabotropic glutamate receptor agonist prodrugs LY2979165 and LY2140023 attenuate the functional imaging response to ketamine in healthy subjects. Psychopharmacology. 2018;235:1875–86.
Marek GJ. When is a proof-of-concept (POC) not a POC? Pomaglumetad (LY2140023) as a case study for antipsychotic efficacy. Curr Pharm Des. 2015;21:3788–96.
Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry. 2015;78:754–62.
McColm J, Brittain C, Suriyapperuma S, Swanson S, Tauscher-Wisniewski S, Foster J, et al. Evaluation of single and multiple doses of a novel mGlu2 agonist, a potential antipsychotic therapy, in healthy subjects. Br J Clin Pharmacol. 2017;83:1654–67.
Litman RE, Smith MA, Doherty JJ, Cross A, Raines S, Gertsik L, et al. AZD8529, a positive allosteric modulator at the mGluR2 receptor, does not improve symptoms in schizophrenia: a proof of principle study. Schizophr Res. 2016;172:152–7.
de Boer JN, Vingerhoets C, Hirdes M, McAlonan GM, Amelsvoort TV, Zinkstok JR. Efficacy and tolerability of riluzole in psychiatric disorders: a systematic review and preliminary meta-analysis. Psychiatry Res. 2019;278:294–302.
Farokhnia M, Sabzabadi M, Pourmahmoud H, Khodaie-Ardakani M-R, Hosseini S-M-R, Yekehtaz H, et al. A double-blind, placebo controlled, randomized trial of riluzole as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia. Psychopharmacology. 2014;231:533–42.
Pillinger T, Rogdaki M, McCutcheon RA, Hathway P, Egerton A, Howes OD. Altered glutamatergic response and functional connectivity in treatment resistant schizophrenia: the effect of riluzole and therapeutic implications. Psychopharmacology. 2019;236:1985–97.
Tuovinen N, Hofer A. Resting-state functional MRI in treatment-resistant schizophrenia. Front Neuroimaging. 2023;2:1127508.
Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006. https://doi.org/10.1002/14651858.CD005962.pub2.
Sánchez-Villalobos JM, Aledo-Serrano Á, Villegas-Martínez I, Shaikh MF, Alcaraz M, Epilepsy treatment in neuro-oncology: a rationale for drug choice in common clinical scenarios. Front Pharmacol. 2022;13:991244.
Correll CU, Maayan L, Kane J, Hert MD, Cohen D. Efficacy for psychopathology and body weight and safety of topiramate-antipsychotic cotreatment in patients with schizophrenia spectrum disorders: results from a meta-analysis of randomized controlled trials. J Clin Psychiatry. 2016;77:21061.
Okuyama Y, Oya K, Matsunaga S, Kishi T, Iwata N. Efficacy and tolerability of topiramate-augmentation therapy for schizophrenia: a systematic review and meta-analysis of randomized controlled trials. Neuropsychiatr Dis Treat. 2016;12:3221–36.
De Ciantis A, Muti M, Piccolini C, Principi M, Di Renzo A, De Ciantis R, et al. A functional MRI study of language disturbances in subjects with migraine headache during treatment with topiramate. Neurol Sci. 2008;29:141–3.
Tang Y, Xia W, Yu X, Zhou B, Wu X, Lui S, et al. Altered cerebral activity associated with topiramate and its withdrawal in patients with epilepsy with language impairment: An fMRI study using the verb generation task. Epilepsy Behav. 2016;59:98–104.
Li X, Large CH, Ricci R, Taylor JJ, Nahas Z, Bohning DE, et al. Using interleaved transcranial magnetic stimulation/functional magnetic resonance imaging (fMRI) and dynamic causal modeling to understand the discrete circuit specific changes of medications: lamotrigine and valproic acid changes in motor or prefrontal effective connectivity. Psychiatry Res. 2011;194:141–8.
Tseng P-T, Chen Y-W, Chung W, Tu K-Y, Wang H-Y, Wu C-K, et al. Significant effect of valproate augmentation therapy in patients with schizophrenia: a meta-analysis study. Medicine. 2016;95:e2475.
Contreras-García IJ, Cárdenas-Rodríguez N, Romo-Mancillas A, Bandala C, Zamudio SR, Gómez-Manzo S, et al. Levetiracetam mechanisms of action: from molecules to systems. Pharmaceuticals. 2022;15:475.
Behdani F, Hassanzadeh B, Eslamzadeh M, Moradi M, Hebrani P, Dadgarmoghaddam M, et al. Can levetiracetam improve clinical symptoms in schizophrenic patients? A randomized placebo-controlled clinical trial. Int Clin Psychopharmacol. 2022;37:159–65.
Roeske MJ, McHugo M, Rogers B, Armstrong K, Avery S, Donahue M, et al. Modulation of hippocampal activity in schizophrenia with levetiracetam: a randomized, double-blind, cross-over, placebo-controlled trial. Neuropsychopharmacol. 2023;49:1–9.
Goff DC, Santacatterina M, Capichioni G, Ando F, Hart K, Convit A, et al. Levetiracetam effects on hippocampal blood flow and symptoms in medication-free individuals with nonaffective first episode psychosis (letter). Schizophr Res. 2023;260:140–2.
de Toffol B. Epilepsy and psychosis. Rev Neurologique. 2024;180:298–307.
De Simone G, Mazza B, Vellucci L, Barone A, Ciccarelli M, de Bartolomeis A. Schizophrenia synaptic pathology and antipsychotic treatment in the framework of oxidative and mitochondrial dysfunction: translational highlights for the clinics and treatment. Antioxidants. 2023;12:975.
Hong J, Bang M. Anti-inflammatory strategies for schizophrenia: a review of evidence for therapeutic applications and drug repurposing. Clin Psychopharmacol Neurosci. 2020;18:10–24.
Jeppesen R, Christensen RHB, Pedersen EMJ, Nordentoft M, Hjorthøj C, Köhler-Forsberg O, et al. Efficacy and safety of anti-inflammatory agents in treatment of psychotic disorders - a comprehensive systematic review and meta-analysis. Brain Behav Immun. 2020;90:364–80.
Bradlow RCJ, Berk M, Kalivas PW, Back SE, Kanaan RA. The potential of N-acetyl-L-cysteine (NAC) in the treatment of psychiatric disorders. CNS Drugs. 2022;36:451–82.
Kishi T, Sakuma K, Hatano M, Iwata N. N-acetylcysteine for schizophrenia: a systematic review and meta-analysis. Psychiatry Clin Neurosci. 2023;77:119–21.
Yolland CO, Hanratty D, Neill E, Rossell SL, Berk M, Dean OM, et al. Meta-analysis of randomised controlled trials with N-acetylcysteine in the treatment of schizophrenia. Aust N Z J Psychiatry. 2020;54:453–66.
Zhang Q, Liu Z, Wang T, Yu M, Li X. Efficacy and acceptability of adjunctive n-acetylcysteine for psychotic disorders: Systematic review and meta-analysis. Hum Psychopharmacol. 2023;39:e2880.
Carmeli C, Knyazeva MG, Cuénod M, Do KQ. Glutathione precursor N-acetyl-cysteine modulates EEG synchronization in schizophrenia patients: a double-blind, randomized, placebo-controlled trial. PLoS ONE. 2012;7:e29341.
McQueen G, Lay A, Lally J, Gabay AS, Collier T, Lythgoe DJ, et al. Effect of single dose N-acetylcysteine administration on resting state functional connectivity in schizophrenia. Psychopharmacology. 2020;237:443–51.
Klauser P, Xin L, Fournier M, Griffa A, Cleusix M, Jenni R, et al. N-acetylcysteine add-on treatment leads to an improvement of fornix white matter integrity in early psychosis: a double-blind randomized placebo-controlled trial. Transl Psychiatry. 2018;8:1–8.
Conus P, Seidman LJ, Fournier M, Xin L, Cleusix M, Baumann PS, et al. N-acetylcysteine in a double-blind randomized placebo-controlled trial: toward biomarker-guided treatment in early psychosis. Schizophr Bull. 2018;44:317–27.
Yang YS, Maddock RJ, Zhang H, Lee J, Hellemann G, Marder SR, et al. N-acetylcysteine effects on glutathione and glutamate in schizophrenia: a preliminary MRS study. Psychiatry Res Neuroimaging. 2022;325:111515.
Khadimallah I, Jenni R, Cabungcal J-H, Cleusix M, Fournier M, Beard E, et al. Mitochondrial, exosomal miR137-COX6A2 and gamma synchrony as biomarkers of parvalbumin interneurons, psychopathology, and neurocognition in schizophrenia. Mol Psychiatry. 2022;27:1192–204.
Hoy KE, Bailey NW, Arnold SL, Fitzgerald PB. The effect of transcranial direct current stimulation on gamma activity and working memory in schizophrenia. Psychiatry Res. 2015;228:191–6.
Cheng PWC, Louie LLC, Wong YL, Wong SMC, Leung WY, Nitsche MA, et al. The effects of transcranial direct current stimulation (tDCS) on clinical symptoms in schizophrenia: a systematic review and meta-analysis. Asian J Psychiatry. 2020;53:102392.
Manfredi N, Zhang R, Seltzberg H, Johnson M, Ehrie J. Transcranial magnetic stimulation in the treatment of positive, negative, and cognitive symptoms of psychosis. Curr Behav Neurosci Rep. 2023;10:82–90.
Mothersill D, Donohoe G. Neural effects of cognitive training in schizophrenia: a systematic review and activation likelihood estimation meta-analysis. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:688–96.
Molina JL, Thomas ML, Joshi YB, Hochberger WC, Koshiyama D, Nungaray JA, et al. Gamma oscillations predict pro-cognitive and clinical response to auditory-based cognitive training in schizophrenia. Transl Psychiatry. 2020;10:405.
Guercio GD, Thomas ME, Cisneros-Franco JM, Voss P, Panizzutti R, de Villers-Sidani E. Improving cognitive training for schizophrenia using neuroplasticity enhancers: Lessons from decades of basic and clinical research. Schizophr Res. 2019;207:80–92.
Burton CZ, Garnett EO, Capellari E, Chang S-E, Tso IF, Hampstead BM, et al. Combined cognitive training and transcranial direct current stimulation in neuropsychiatric disorders: a systematic review and meta-analysis. Biol Psychiatry Cogn Neurosci Neuroimaging. 2023;8:151–61.
Gill KM, Cook JM, Poe MM, Grace AA. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophr Bull. 2014;40:341–50.
Servonnet A, Samaha A-N. Antipsychotic-evoked dopamine supersensitivity. Neuropharmacology. 2020;163:107630.
Anderson KK, Rodrigues M, Mann K, Voineskos A, Mulsant BH, George TP, et al. Minimal evidence that untreated psychosis damages brain structures: a systematic review. Schizophr Res. 2015;162:222–33.
Yang J-M, Zhang J, Yu Y-Q, Duan S, Li X-M. Postnatal development of 2 microcircuits involving fast-spiking interneurons in the mouse prefrontal cortex. Cereb Cortex. 2014;24:98–109.
Preller KH, Scholpp J, Wunder A, Rosenbrock H. Neuroimaging biomarkers for drug discovery and development in schizophrenia. Biol Psychiatry. 2024;96:666-73.
Andersen HG, DellaValle B, Bøgehave H, Mogensen PB, Hahn MK, Goth CK, et al. Glycocalyx shedding patterns identifies antipsychotic-naïve patients with first-episode psychosis. Psychiatry Res. 2024;339:116037.
Crippa JA, Zuardi AW, Garrido GE, Wichert-Ana L, Guarnieri R, et al. Effects of cannabidiol (CBD) on regional cerebral blood flow. Neuropsychopharmacology. 2004;29:417–26.
Fusar-Poli P, Allen P, Bhattacharyya S, Crippa JA, Mechelli A, Borgwardt S, et al. Modulation of effective connectivity during emotional processing by Delta 9-tetrahydrocannabinol and cannabidiol. Int J Neuropsychopharmacol. 2010;13:421–32.
O’Neill A, Wilson R, Blest-Hopley G, Annibale L, Colizzi M, Brammer M, et al. Normalization of mediotemporal and prefrontal activity, and mediotemporal-striatal connectivity, may underlie antipsychotic effects of cannabidiol in psychosis. Psychol Med. 2021;51:596–606.
Appiah-Kusi E, Petros N, Wilson R, Colizzi M, Bossong MG, Valmaggia L, et al. Effects of short-term cannabidiol treatment on response to social stress in subjects at clinical high risk of developing psychosis. Psychopharmacology. 2020;237:1121–30.
Bossong M, Wilson R, Appiah-Kusi E, Linsen F, Zelaya F, Allen P, et al. Treatment with cannabidiol reduces resting state perfusion in individuals at clinical high risk for psychosis. Schizophr Bull. 2019;45:S200.
McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am J Psychiatry. 2018;175:225–31.
Baker S, Chin C-L, Basso AM, Fox GB, Marek GJ, Day M. Xanomeline modulation of the blood oxygenation level-dependent signal in awake rats: development of pharmacological magnetic resonance imaging as a translatable pharmacodynamic biomarker for central activity and dose selection. J Pharm Exp Ther. 2012;341:263–73.
Montani C, Canella C, Schwarz AJ, Li J, Gilmour G, Galbusera A, et al. The M1/M4 preferring muscarinic agonist xanomeline modulates functional connectivity and NMDAR antagonist-induced changes in the mouse brain. Neuropsychopharmacol. 2021;46:1194–206.
Dean B, Scarr E. Muscarinic M1 and M4 receptors: hypothesis driven drug development for schizophrenia. Psychiatry Res. 2020;288:112989.
Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–9.
Brannan SK, Sawchak S, Miller AC, Lieberman JA, Paul SM, Breier A. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. N Engl J Med. 2021;384:717–26.
Kaul I, Sawchak S, Correll CU, Kakar R, Breier A, Zhu H, et al. Efficacy and safety of the muscarinic receptor agonist KarXT (xanomeline–trospium) in schizophrenia (EMERGENT-2) in the USA: results from a randomised, double-blind, placebo-controlled, flexible-dose phase 3 trial. Lancet. 2024;403:160–70.
Krystal JH, Kane JM, Correll CU, Walling DP, Leoni M, Duvvuri S, et al. Emraclidine, a novel positive allosteric modulator of cholinergic M4 receptors, for the treatment of schizophrenia: a two-part, randomised, double-blind, placebo-controlled, phase 1b trial. The Lancet. 2022;400:2210–2220.
Kantrowitz JT, Javitt DC, Freedman R, Sehatpour P, Kegeles LS, Carlson M, et al. Double blind, two dose, randomized, placebo-controlled, cross-over clinical trial of the positive allosteric modulator at the alpha7 nicotinic cholinergic receptor AVL-3288 in schizophrenia patients. Neuropsychopharmacol. 2020;45:1339–45.
Antonio-Tolentino K, Hopkins CR. Selective α7 nicotinic receptor agonists and positive allosteric modulators for the treatment of schizophrenia – a review. Expert Opin Invest Drugs. 2020;29:603–10.
Recio-Barbero M, Segarra R, Zabala A, González-Fraile E, González-Pinto A, Ballesteros J Cognitive enhancers in schizophrenia: a systematic review and meta-analysis of alpha-7 nicotinic acetylcholine receptor agonists for cognitive deficits and negative symptoms. Front Psychiatry. 2021;12:631589.
Li Z, Ichikawa J, Huang M, Prus AJ, Dai J, Meltzer HY. ACP-103, a 5-HT2A/2C inverse agonist, potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and nucleus accumbens. Psychopharmacology. 2005;183:144–53.
Meltzer HY, Elkis H, Vanover K, Weiner DM, van Kammen DP, Peters P, et al. Pimavanserin, a selective serotonin (5-HT)2A-inverse agonist, enhances the efficacy and safety of risperidone, 2mg/day, but does not enhance efficacy of haloperidol, 2mg/day: comparison with reference dose risperidone, 6mg/day. Schizophr Res. 2012;141:144–52.
Bugarski-Kirola D, Arango C, Fava M, Nasrallah H, Liu I-Y, Abbs B, et al. Pimavanserin for negative symptoms of schizophrenia: results from the ADVANCE phase 2 randomised, placebo-controlled trial in North America and Europe. Lancet Psychiatry. 2022;9:46–58.
Bugarski-Kirola D, Bitter I, Liu I-Y, Abbs B, Stankovic S. ENHANCE: Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Adjunctive Pimavanserin for Schizophrenia in Patients With an Inadequate Response to Antipsychotic Treatment. Schizophr Bull Open. 2022;3:sgac006.
Ebdrup BH, Rasmussen H, Arnt J, Glenthøj B. Serotonin 2A receptor antagonists for treatment of schizophrenia. Expert Opin Invest Drugs. 2011;20:1211–23.
Davidson M, Saoud J, Staner C, Noel N, Werner S, Luthringer E, et al. Efficacy and safety of roluperidone for the treatment of negative symptoms of schizophrenia. Schizophr Bull. 2022;48:609–19.
Acknowledgements
This research was partly funded by the Wellcome Trust [223486/Z/21/Z to AG] and the National Institute for Health and Care Research (NIHR) Maudsley Biomedical Research Centre (BRC). The views expressed are those of the authors and not necessarily those of the Welcome Trust, NIHR or the Department of Health and Social Care. For the purpose of open access, the author has applied a CC-BY public copyright licence to any author-accepted manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
GM, AG and PD conceived the project. AG wrote the first draft of the manuscript. AG obtained the funding. GM, PD and AAG provided critical input into this first draft and approved the final version of this article.
Corresponding author
Ethics declarations
Competing interests
GM has received consulting fees from Boehringer Ingelheim. AAG has received funds from Lundbeck, Pfizer, Lilly, Roche, Janssen, Alkermes, Newron, Takeda and Merck. PD has received speaker’s fees from Lundbeck and Janssen. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gee, A., Dazzan, P., Grace, A.A. et al. Corticolimbic circuitry as a druggable target in schizophrenia spectrum disorders: a narrative review. Transl Psychiatry 15, 21 (2025). https://doi.org/10.1038/s41398-024-03221-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-03221-2
