
Abstract
Dravet syndrome (DS) is a severe and catastrophic epilepsy with childhood onset. The incidence and prevalence of sudden unexpected death in epilepsy (SUDEP) are significantly higher in DS patients than in general epileptic populations. Although extensive research conducted, the underlying mechanisms of SUDEP occurring in DS patients remain unclear. This review focuses on the link between DS and SUDEP and analyzes the potential pathogenesis. We summarize the genetic basis of DS and SUDEP and elucidate the pathophysiological mechanisms of SUDEP in DS. Furthermore, given the drug-resistant nature of this disorder, the pharmacological approach has limited efficacy and often causes side effects, therefore, the non-pharmacological approaches and precise treatment can reduce the risk of SUDEP in this condition, open a new window to cure this disease, and provide a widened landscape of treatment options for patients.

Highlights
-
DS is a severe, refractory childhood-onset epilepsy with extremely high premature mortality, with SUDEP being the leading cause of death.
-
Inherent genetic traits could be a nexus between DS and SUDEP, and DS patients exposed to various pathogenic genes or genetic modifiers may have different susceptibilities to SUDEP.
-
A confluence of different pathophysiological mechanisms synergistically triggers SUDEP in DS patients, with seizures-induced cardiopulmonary dysfunction representing a critical contributing factor.
-
Combined pharmacotherapy and non-pharmacologic interventions can improve therapeutic efficacy, while gene therapy has ushered in a new era with great potential.
Similar content being viewed by others
Introduction
Dravet syndrome (DS), also known as severe myoclonic epilepsy of infancy (SMEI), is a rare, severe and disastrous children-onset epilepsy [1, 2]. The estimated incidence ranges from 1 in 15,700 to 1 in 40,900 [3, 4]. In 70–80% of cases, DS is primarily attributed to de novo heterozygous loss-of-function mutations in the SCN1A gene that encodes the α-subunit of the voltage-gated sodium channel Nav1.1, leading to haploinsufficiency of the channel [5,6,7,8]. Missense and truncation (e.g., nonsense, frameshift) mutations are the most common types and significantly influence the severity of epilepsy phenotypes [9, 10]. Dysfunction of Nav1.1 decreases sodium currents in different types of GABAergic interneurons, especially the parvalbumin (PV)-positive interneurons where Nav1.1 is predominantly expressed [11, 12]. The reduced sodium currents lead to an imbalance between excitation and inhibition, which may contribute to hyperexcitability and disruptions in neural network function [13], potentially increasing the risk of epilepsy-related mortality. While multiple factors contribute to mortality, sudden unexpected death in epilepsy (SUDEP) is considered one of the primary causes.
SUDEP is defined as sudden, unexpected, witnessed or unwitnessed death in epilepsy patients without warning or other clear causes of death [14]. The public health burden of SUDEP ranks second only to stroke in terms of years of potential life lost, compared to other common neurological disorders. The incidence rate of SUDEP varies across different study cohorts, with the highest frequency observed in refractory and drug-resistant epilepsy [15]. The mechanism of SUDEP remains elusive, primarily due to its complex and multifactorial pathology, as well as the difficulty of obtaining real-time monitoring data in epileptic patients.
DS, characterized by high epilepsy-related premature mortality, is devastating and life-threatening. Death can occur at any age, but is more common during childhood. Compared to the general epileptic demographic, individuals with DS have an elevated risk of SUDEP. Several studies have shown that nearly half of all deaths in DS are attributed to SUDEP, which has now surpassed status epilepticus (SE) as the foremost cause of mortality [16, 17]. Despite some advancements in characterizing DS, the mechanisms underlying how DS leads to SUDEP remain unclear. Our group has long been dedicated to studying the pathogenesis of SUDEP. Since DS can cause detrimental comorbidities and is strongly associated with SUDEP, we believe a thorough exploration of the relationship between DS and SUDEP is imperative.
In this review, we integrate data from clinical and animal studies to evaluate the connection between DS and SUDEP and analyze the possible mechanisms. In addition, we discuss promising therapeutic approaches that could offer new insights for developing effective strategies to manage DS and reduce the incidence of SUDEP in DS patients.
Clinical manifestations of DS
The clinical manifestations of DS are primarily divided into two categories: epilepsy-related symptoms (e.g., seizures, epilepsy-induced death) and long-term comorbidities. DS is mainly characterized by hyperthermia sensitivity and typically manifests within the first year of life in an otherwise normal infant, usually between 4 and 8 months of age [4]. The most common initial symptom is a prolonged seizure triggered by hyperthermia that can manifest as recurrent focal clonic (hemiclonic) seizures or generalized clonic seizures [18, 19]. This stage is often diagnosed as febrile convulsions. By 1.5 to 5 years of age, additional seizure types may occur (but are not always present), such as myoclonic seizures, atypical absence seizures, atonic seizures, etc [20]. Some children with DS also develop acute encephalopathy. Notably, a history of multiple seizures is a significant risk factor for SUDEP. Mortality rates in DS patients reported in different studies vary from 3.7–20.8% [2, 21, 22], which are significantly higher than those in other pediatric epilepsy cohorts. Premature death is a major problem in DS, and the main causes of death have been classified into three types: SUDEP, status epilepticus (SE), and drowning. In a previous survey of 623 patients with DS from Japan, a total of 63 individuals died and data from 59 of these people were analyzed [23]. In this study, SUDEP was the cause of mortality in 53% of patients in this group and its incidence peaked at 1–3 and 18 years of age. In conjunction with other studies, it is evident that SUDEP most often attacks children, especially those under 10 years of age. Most dead patients were found in bed early in the morning or during sleep in the afternoon. The prevalence of SUDEP ranged from 49–61% in different studies and it is responsible for the vast majority of premature mortality in DS [16, 17].
In addition to epilepsy-related symptoms, long-term comorbidities, including developmental delay, hyperactivity, motor dysfunction, cognitive impairment, intellectual disability, and autistic like-behaviors, can also be present and become a tremendous burden [4, 24, 25]. Most living DS patients suffer from a lifelong disabling condition that limits daily activities and leads to a poor quality of life. Although less common, the incidence of SUDEP may increase with age.
Genetic features may be a link between DS and SUDEP
While mutations in the SCN1A gene are well-known as the primary cause of DS, it is increasingly recognized that DS can also occur in individuals with non-SCN1A mutations, leading to DS-like phenotypes [26]. These cases present with clinical characteristics that closely resemble classic DS but are driven by variations in other genes. In addition, different genetic modifiers are believed to significantly influence the severity of the phenotype of SCN1A‐induced DS, especially the risk of SUDEP. However, the underlying mechanisms leading to SUDEP remain unclear. Some evidence has emerged with respect to genetic susceptibility to SUDEP, suggesting that genetics is an additional important area for further understanding of SUDEP risk factors [27]. Therefore, researchers are intensifying their focus on the genetic characteristics of SUDEP. We found DS and SUDEP may have a common genetic basis, and there is an intersection of the causative genes of DS, genetic modifiers of DS, and the pathogenic gene of SUDEP (Fig. 1). The relationship between DS and SUDEP is complex, with genetic factors playing a central role in both conditions.

This figure summarizes the genetic features of DS and SUDEP. The genes that appear in the pink intersection are some DS-causing genes that are the main intrinsic genetic factors in the occurrence of SUDEP in DS patients.
Genes associated with DS
SCN1A is recognized as the most common genetic factor in DS. In addition to this, the PCDH19 gene is regarded as the secondary principal genetic cause of DS. PCDH19, expressed in the developing brains of humans, is the first member of the cadherin superfamily directly implicated in epilepsy. Depienne et al. detected that PCDH19 played an essential role in infantile epileptic encephalopathies, with a clinical spectrum overlapping that of SCN1A-DS [28]. PCDH19-related epilepsy is a special X-linked inherited disorder that mainly affects females [29]. In addition, several studies suggested that PCDH19 mutations could be found in some SCN1A-negative DS patients [30, 31].
Several genes encoding cation channels, such as SCN2A, SCN8A, SCN1B, KCNA2 and HCN1, have been proposed to be involved in DS or DS-like phenotypes. All of them control potential exchange between the extracellular and the intracellular spaces and may be tied to neuronal excitability. Mutations in these genes are linked to a spectrum of epilepsies and neurodevelopmental abnormalities. A de novo SCN2A mutation has been reported in one single case without coexisting SCN1A mutations, making it feasible to comprehend NaV1.2 dysfunction and its relationship to the etiology of DS [32]. SCN8A encodes NaV1.6, which, unlike NaV1.1, is primarily located in pyramidal neurons responsible for excitatory signals via glutamate release. Notably, the majority of SCN8A mutations result in the gain-of-function of NaV1.6, causing elevated excitability and firing rates of pyramidal cells. In 2015, some patients in the reported SCN8A cohort had previously been given a preliminary diagnosis of DS. Moreover, Patino et al. presented the first case of DS attributed to a homozygous mutation in SCN1B [33], and Gong et al. also discovered two SCN1B mutations in DS patients without SCN1A mutations [34]. It should be noted that no SCN1B mutations were identified in 54 patients with DS recruited from four centers in one report [35]. These results indicate that SCN1B is an etiologic candidate underlying DS, but is not a common cause. KV1.2 encoded by KCNA2 is typically concentrated along axons and axon terminals of neurons, as well as at presynaptic sites. In a clinical study, four different de novo KCNA2 variants have been described in six patients with epileptic encephalopathy, including one with clinical features that resemble DS [36]. In an animal study, it was discovered that Kcna2-null mice were more vulnerable to seizures and early death [37]. Furthermore, hyperpolarization-activated cyclic nucleotide-gated channel 1 is a nonselective cation channel that contributes to spontaneous rhythmic activity in neurons. HCN1 encoding this channel was found in patients with DS-like phenotypes [38]. In a word, these dysfunctional channel genes make hyperexcitability of the nervous system, leading to DS.
Mutations in the GABAA receptor genes (GABRA1, GABRB3 and GABRG2) are responsible for the pathogenesis of DS as the predominantly affected inhibitory neurons are GABAergic interneurons. For instance, GABRA1 is associated with early-onset epileptic encephalopathy as well as genetic generalized epilepsy and febrile seizures. Carvill et al. sequenced the genes of SCN1A-negative DS individuals and found that four of them carried GABRA1 mutation [39]. Two patients from another study with DS-like phenotype harbored a de novo variation in GABRA1 [40]. In 2017, Le et al. performed exome sequencing in six patients with SCN1A-negative DS to identify other genes related to this disorder. In one affected individual, they detected a de novo heterozygous missense variant in GABRB3 [41], and one case report showed a girl with DS presented novel GABRB3 mutations [42], implying that GABRB3 may be a putative candidate for DS. In addition, GABRG2 mutations have been reported to be associated with early-onset epileptic encephalopathy, with some cases showing core symptoms of typical DS [43]. Different phenotypes of DS are likely due to impaired axonal transport associated with the dominant-negative effects of GABRG2 [44]. All aforementioned mutations reduce GABA signaling by disrupting structural domains important for GABAA receptor function, thereby elucidating their role in the etiology of DS.
In addition, other causative genes encoding proteins, such as STXBP1, CHD2 and CPLX1, are involved in the etiology of DS. STXBP1 is pivotal in synaptic vesicle release. After excluding abnormalities in SCN1A, STXBP1 mutations contribute significantly to DS and are associated with the motor phenotype with parkinsonian symptoms in DS patients [39, 45]. Although loss-of-function mutations in CHD2 are rare, a previous study found that individuals carrying a CHD2 mutation displayed a spectrum of fever-sensitive generalized seizures similar to DS [46]. Abnormal expression of CPLX1 can be seen in several neurodegenerative and psychiatric disorders, but is rare in malignant epilepsy. In 2017, Redler et al. first described variants in CPLX1 in two families linked to severe infantile myoclonic epilepsy, developmental delay, and intellectual disability [47]. These genes are considered as emerging genetic contributors to DS and therefore warrant further investigation to elucidate their mechanisms in this disorder.
In summary, all the genes mentioned can be detected in SCN1A-positive DS and SCN1A-negative DS patients (Table 1). Although the clinical characteristics caused by other genes are slightly different from classical SCN1A-DS, they represent potential inherited pathogenic factors.
Genetic modifiers of DS
Several potential modifier genes have already been identified in human patients and Scn1a knock-out mice, including ion channel genes, GABA receptor subunit genes, and genes associated with seizures or hyperexcitability [48]. Variants in different genetic modifiers might alleviate or exacerbate clinical outcomes.
Modifiers that exacerbate the phenotype or increase DS severity include several genes. SCN2A could potentially underlie the genetic pathogenesis of DS without SCN1A mutations. Meanwhile, Scn2a mutants aggravate the phenotype and decrease the survival rate of DS mice [49]. In the same study, the combination of Scn1a and Kcnq2 mutations led to early-onset, generalized tonic-clonic seizures and juvenile lethality [49]. Similarly, variants in KCNQ2 exacerbate clinical presentation and severity in patients with DS [50]. GABRA2 is also a genetic modifier of DS. In an animal study, researchers found that a repaired Gabra2 allele increased Gabra2 expression, which dramatically lessened seizure burden and premature lethality (SUDEP-like) in the Scn1a+/− mice [51]. Furthermore, hepatic leukemia factor (Hlf) is a member of the PAR bZIP transcription factor family. Genetic deletion of Hlf increased premature mortality and exacerbated the phenotype of Scn1a+/− mice, thus also functioning as a genetic modifier of DS [52].
Similarly, SCN9A mutations have also been observed in certain patients with SCN1A mutations, correlating with a more severe phenotype [53]. In a case report, a missense mutation of CACNB4 (R468Q) was detected in one of 38 SCN1A mutation-positive DS patients, which causes greater Cav2.1 currents, thereby increasing the neurotransmitter release in excitatory neurons and enhancing seizure susceptibility [54]. Another human study has proposed that subjects with combinations of CACNA1A variants and SCN1A mutations showed a higher incidence of earlier onset, as well as longer and more severe seizures, compared to subjects with only SCN1A mutations [55]. Both CACNB4 and CACNA1A variants that alter Cav2.1 function worsen the epileptic phenotype of DS. Additionally, the rare heterozygous POLG variant impairs cortical metabolism during the high energy demands associated with status epilepticus, thereby increasing susceptibility to acute encephalopathy [56]. Although there is currently no clear evidence indicating that these genes increase the risk of SUDEP in DS patients, the more severe epilepsy phenotypes and complications they cause indirectly suggest that these genes may be associated with an increased risk of SUDEP.
Conversely, several genetic modifiers have been identified as ameliorating factors, reducing DS severity or improving survival. SCN8A mutants with reduced SCN8A expression exhibit increased resistance to induced seizures, improve survival and prolong the lifespan of DS mice [57]. In addition, CACNA1G was also recently nominated as a genetic modifier of survival in a mouse model of DS [58]. Scn1a+/− mice with decreased CACNA1G expression showed partial amelioration of the disease with improved survival and reduced spontaneous seizure frequency.
All genes can be demonstrated to be present in humans. They have been proposed as genetic modifiers leading to the variable expressivity of DS, which could provide a more accurate assessment of disease severity and alter the premature lethality of DS (Table 2). Thus, genetic modifiers may be novel risk genes associated with seizures and SUDEP, enhance our comprehension of the etiology of SUDEP in DS patients and emerge as potential targets for therapeutic intervention in patients.
Genetic basis of SUDEP
Several pathogenic modifications in various genes have been identified as factors that increase SUDEP risk through different pathophysiologic mechanisms, which has prompted a growing number of researchers to focus on studying the genetic underpinnings of SUDEP [59]. The genes associated with SUDEP can be broadly categorized into three major groups: cardiac arrhythmia genes, respiratory genes and genes associated with genetic epilepsies.
Genes associated with genetic epilepsies
Pathogenic variations associated with several genetic epilepsies that carry a high risk of SUDEP may serve as useful biomarkers. SCN1A-induced DS patients face a significantly higher risk of SUDEP, while SCN1B loss-of-function mutations also contribute to an increased risk of SUDEP [60]. Previous work suggested that Scn1b deletion can increase tetrodotoxin-sensitive sodium currents, alter intracellular calcium homeostasis, and induce arrhythmias in murine hearts. These factors underlie epileptogenesis and SUDEP [61]. In addition, SCN2A and SCN8A, primarily involved in developmental and epileptic encephalopathies, are also deleterious factors contributing to SUDEP [62]. Several studies have identified DEP domain containing 5 (DEPDC5) as a pathogenic gene for SUDEP [63, 64]. Mutations in DEPDC5 are increasingly recognized as a common cause of familial focal epilepsy with variable foci [65]. NPRL2 and NPRL3, together with DEPDC5, form the GATOR1 complex, a repressor of the mechanistic target of rapamycin complex 1 (mTORC1) pathway [66]. GATOR1 complex gene mutations that lead to mTORC1 pathway upregulation are a primary cause of SUDEP [67]. KCNA1, which encodes the voltage-gated potassium channel Kv1.1 α-subunit, is linked to episodic ataxia and myokymia syndrome [68]. In animal studies, Kcna1 gene deletion mice, frequently utilized in SUDEP research, displayed malignant interictal cardiac abnormalities, including a fivefold increase in atrioventricular conduction blocks, bradycardia, and premature ventricular contractions [69]. These mice exhibited an increased susceptibility to respiratory failure during severe seizures, potentially leading to sudden death [70]. Although direct links between KCNA1 gene variants and SUDEP in humans have yet to be established, these findings suggest that this gene could be a potential risk factor. Fibroblast growth factor homologous factors (FHFs) are brain and cardiac sodium channel binding proteins that modulate channel density and inactivation gating. According to a recent study, a de novo gain-of-function missense mutation in the FHF1 gene is associated with early infantile epileptic encephalopathy. This mutation enhances sodium channel Nav1.6 currents and induces SUDEP with bradycardia in mice [71]. Furthermore, variants in the PRRT2 gene, which encodes proline-rich transmembrane protein 2, are emerging as potential biomarkers for SUDEP. These variants are associated with benign familial infantile seizures. A previous case report has shown that a boy with a PRRT2 variant who had afebrile focal seizures was found dead in a supine position after having fallen asleep at the age of 14 due to probable SUDEP [72]. The potential link between mutations in the PRRT2 gene and susceptibility to SUDEP requires further investigation. In 2017, Coll et al. identified four missense variants in epilepsy-related genes—CDKL5(associated with X-linked infantile spasm syndrome), CNTNAP2(associated with cortical dysplasia focal epilepsy syndrome), GRIN2A (associated with focal epilepsies) and ADGRV1(associated with recurrent febrile seizures and afebrile seizures). These variants may contribute to SUDEP and were proposed as novel candidate [73].
Cardiac arrhythmia genes
Cardiac arrhythmias are widely recognized as a potential pathogenic mechanism in SUDEP. Genetic variants associated with arrhythmias are found in up to 15% of SUDEP cases, most of which involve ion channel genes that disrupt normal cardiac function and may increase the risk of SUDEP.
Long QT syndrome (LQTS) is an inherited cardiac channelopathy characterized by prolonged ventricular repolarization (prolonged QT interval) and a predisposition to torsade de pointes, ventricular fibrillation, and sudden cardiac death. Most congenital LQTS cases result from mutations in KCNQ1, KCNH2, and SCN5A [74]. Notably, SCN5A variants can also cause Brugada syndrome, a fatal cardiac disease [75]. Variants in all three genes have been identified in SUDEP cases, suggesting their potential role in the genetic basis of SUDEP [76, 77]. HCN channels are involved in generating spontaneous rhythmic activity in both cardiac pacemakers and neuronal cells, which can regulate neuronal excitability. Dysfunction in these channels may result in epileptogenesis. In 2011, Tu et al. analyzed the genes in 48 SUDEP cases and identified three non-synonymous variants in HCN2 and HCN4 [78]. A systematic review also identified all four HCN genes (HCN1–4) in some SUDEP cases [79]. Hence, HCN channels are strong candidates for genetic screening in SUDEP because of their association with human diseases. Other genetic variants associated with epilepsy and encoding cardiac ion channels could explain the SUDEP phenotype. One study identified additional potential candidate genes for SUDEP, including FBN1, SCN4A, EFHC1, CACNA1A, SCN11A, and SCN10A [80]. In conclusion, these findings support the hypothesis that genetic risk factors predisposing to cardiac arrhythmia may contribute to the risk of SUDEP in certain epilepsy patients.
Respiratory genes
Evidence for the connection between SUDEP and genes related to neuronal regulation of respiratory function is mainly from animal studies. DBA (DBA/1 and DBA/2) mice are useful animal models to study SUDEP because they exhibit generalized tonic-clonic seizures followed by seizure-induced respiratory arrest (S-IRA) and SUDEP. DBA mice exhibit impaired expression of serotonin(5-HT) receptors in the brainstem, and TPH2, the rate-limiting enzyme for 5-HT synthesis in the CNS, is also reduced [81]. Our previous studies revealed that 5-hydroxytryptophan, the precursor for 5-HT synthesis, can reduce the incidence of S-IRA and SUDEP in DBA/1. Additionally, enhancing 5-HT function in DBA/1 mice can reduce S-IRA and improve survival, indicating that the 5-HT system plays a key role in S-IRA and SUDEP [82, 83]. An animal study found that a mouse model with 5-HT2C receptor gene(htr2c) mutants is extremely susceptible to seizures, S-IRA and SUDEP [84] Meanwhile, a recent study also reported the first analysis of HTR2C variants found in human SUDEP [85].
Congenital central hypoventilation syndrome (CCHS) is a potentially fatal autonomic nervous system disorder characterized by hypoventilation and impaired ventilatory response to hypercapnia and hypoxemia during sleep, which is primarily caused by expansion of the alanine repeat in the homeobox gene PHOX2B. Although one human study has shown that PHOX2B mutations are not a common risk factor for SUDEP, this gene may still be a potential target for SUDEP [86]. Furthermore, in another study, no variants were identified in five genes with plausible roles in the central control of ventilation (ASCL1, BDNF, EDN3, GDNF, and RET) among our 61 SUDEP cases [59].
Emerging evidence suggests that the genetic landscape of DS and SUDEP may overlap. The identification of common genetic pathways between DS and SUDEP highlights the importance of further genetic studies of them.
Bioinformatics studies play a crucial role in understanding the complex interaction between DS and SUDEP, particularly in uncovering the genetic features underlying these conditions. Bioinformatics tools, such as genome-wide association studies (GWAS), next-generation sequencing (NGS), and RNA sequencing, enable researchers to analyze large-scale genomic data and identify novel genetic variants that may contribute to the increased risk of SUDEP in DS patients. Furthermore, bioinformatics approaches allow for the integration of genetic data with clinical and phenotypic information, providing deeper insights into how specific genetic variations interact with environmental factors.
In the future, bioinformatics will help to elucidate the genetic basis of DS and SUDEP, paving the way for personalized treatment strategies, and will be instrumental in understanding the mechanisms underlying the risk of SUDEP in DS patients, leading to more effective interventions and risk assessments for patients.
Pathophysiological mechanisms underlying the occurrence of SUDEP in DS
Central neuronal alteration
The “interneuron hypothesis” is the currently most well-recognized pathophysiological mechanism of DS [12]. Mutations in the main causative gene SCN1A, lead to the loss of NaV1.1 channels. In the brain, Nav1.1 is predominantly expressed in forebrain GABAergic neurons, especially in the axon initial segment and somata of PV-positive interneurons, whereas Nav1.1 is slightly expressed in excitatory pyramidal neurons [11, 12]. The mutant SCN1A results in severely impaired sodium current and action potential firing in forebrain GABAergic inhibitory interneurons without detectable changes in excitatory pyramidal neurons, leading to hypofunction of inhibitory circuits [12, 87, 88]. This leads to hyperexcitability in the neuronal network and seizures. Interneurons in the cortex and hippocampus are likely to be most affected. In contrast, Nav1.1 haploinsufficiency in excitatory neurons can ameliorate seizure-associated sudden death in a mouse model of DS [89]. Furthermore, both somatostatin-positive (SST + ) and vasoactive intestinal peptide-positive (VIP + ) interneurons contribute to the pathogenesis of DS. The excitability of SST+ and VIP+ interneurons is impaired in DS mice, leading to disrupted synaptic transmission [90]. The three major subtypes of GABAergic inhibitory interneurons (PV + , SST + , and VIP+ interneurons) synergistically affect the excitability of neural networks in the brain. It’s worth noting that Mistry et al. showed that elevated sodium current density in excitatory pyramidal neurons correlates with lethality in DS mice [91]. In addition, an animal study highlighted a novel perspective that the decline in acetylcholine and the excitability of pyramidal cells, along with inhibitory interneurons, are responsible for network hyperexcitability [92]. Decreased acetylcholine can lead to hippocampal excitation, as this neurotransmitter typically interacts with muscarinic receptors on GABAergic interneurons. This aligns with previous findings showing the protective effect of acetylcholine esterase inhibitors on triggered seizures in DS mice [93, 94]. Another group of GABAergic neurons expressing SCN1A is located in the nucleus reticularis thalami (nRT), which is thought to regulate information transmission between the thalamus and cortex. Alterations in the function of the corticothalamic circuit may underlie the mechanism of DS [95].
The origin of DS could also be explained by another mechanism. Different variants can alter the structure of the SCN1A-encoded protein in different ways, resulting in diverse functional outcomes [96]. Some SCN1A mutations have a gain-of-function effect and lead to overactivation of glutamatergic neurons. For example, electrophysiological analysis of iPSC-derived glutamatergic neurons from patients revealed a hyperexcitable state of enlarged and persistent sodium channel activation as well as typical epileptic spontaneous action potentials. This study suggested that at least a proportion of DS patients exhibit excessive activation of excitatory neurons [96]. Additionally, one study reported that enhanced NMDA receptor-mediated glutamatergic transmission facilitates abnormal neuronal excitability [97].
The neural network becomes overactive due to the hyperexcitability of glutamatergic neurons and/or the hypoexcitability of GABAergic neurons. This imbalance between excitation and inhibition is thought to be the primary cause of seizures [98]. SUDEP is closely associated with seizures. The most important risk factor for SUDEP is frequent generalized tonic-clonic seizures (GTCS) [99, 100], and SUDEP is more prevalent in patients with GTCS. Both poor management of GTCS and long-term GTCS duration are strongly linked to SUDEP. Similarly, focal seizures also can increase the incidence of SUDEP. Therefore, alterations in the neuronal network involved in epileptic seizures could be a potential etiology of SUDEP in DS patients (Fig. 2).

DS is caused by the deletion of Nav1.1 channels in GABAergic inhibitory interneurons (primarily PV + , and a few SST+ and VIP+ interneurons) due to a loss-of-function effect of SCN1A mutations, or by the excessive sodium channel activity in glutamate neurons due to a gain-of-function effect of few SCN1A mutations, both of which lead to excitation of downstream neural pathways as well as hyperactivity of neural network. Among them, the “inhibitory interneuron hypothesis” caused by GABAergic interneurons draws more popularity. Abbreviations: PV + : parvalbumin-positive; SST + : somatostatin-positive; VIP + : vasoactive intestinal peptide-positive; GABA, gamma-aminobutyric acid.
Cardiac dysfunctions
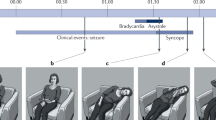
Cardiac arrhythmias are considered common causes of sudden death and potential risks for SUDEP. These arrhythmias are diverse and can occur during different types of seizures. Patients with DS often exhibit an imbalance in cardiac autonomic function, which may increase their risk of cardiac arrhythmias. A previous study suggested that parasympathetic nervous system hyperactivity, which disrupts cardiac regulation, can lead to severe bradycardia and electrical dysfunction of the ventricle [101]. This suggests that bradyarrhythmias pose a serious threat of death and may lead to cardiac arrests. Several case-control studies have investigated the alternation of heart-rate variability (HRV) in DS patients, consistently showing reduced HRV [102,103,104]. HPV is used to measure the balance between the sympathetic and parasympathetic systems, with decreased HRV values indicating increased adrenergic tone or sympathetic upregulation [105, 106]. In addition to HRV, increased QT interval and P wave dispersion can occur due to excessive sympathetic activity [105]. These are important indicators of cardiac autonomic dysfunction and may serve as potential biomarkers for SUDEP in DS. When the sympathetic system is overactivated and dominant, tachyarrhythmias occur, increasing the risk of life-threatening ventricular arrhythmias. Thus, patients have different types of arrhythmias due to parasympathetic or sympathetic dominance, which can progress to ventricular fibrillation, asystole, and death (Fig. 3A). However, bradyarrhythmias account for the vast majority of DS patients, distinguishing them from arrhythmias observed in other epilepsy disorders.

A DS patients, during seizures, produce abnormal impulsivity from cortical regions, which alters the direct input of DMV and RVLM from the upstream nuclei and results in sympathetic or parasympathetic hyperexcitability. Among them, bradyarrhythmia due to parasympathetic dominance accounts for the vast majority of DS patients. B Respiratory circuits and neural nuclei previously validated in DS studies cause respiratory dysfunction, and abnormalities in these circuits and neural nuclei lead to SUDEP. C We outline conceivable respiratory pathways that may lead to respiratory dysfunction during DS followed by SUDEP. There may be a circuit among these that explains the mechanism of breathing disorders in DS and SUDEP. Abbreviations: CeA: central amygdala; CVLM: caudal ventrolateral medulla; DMV: dorsal motor nucleus of the vagus; LC: locus coeruleus; NTS: nucleus tractus solitarius; OVLT: organum vasculosum of the lamina terminalis; PVN: paraventricular nucleus; RVLM: rostral ventrolateral medulla; SFO: subfornical organ; AMY: amygdala; BC: Bötzinger complex; BLA: basolateral amygdala; BNST: bed nucleus of the stria terminalis; CING: cingulate gyrus; DRN: dorsal raphe nucleus; KF: Kölliker-Fuse; MV: minute ventilation; PAG: periaqueductal gray; PBC: preBötzinger complex; PBN: parabrachial nucleus; pF: parafacial nuclei; PVN: paraventricular nucleus of the hypothalamus; RTN: retrotrapezoid nucleus; TP: temporal pole; TV: tidal volume; XIIn: hypoglossal nucleus.
The pathogenic SCN1A gene is expressed in both the heart and brain. As a neuronal gene, it can cause a CNS phenotype. Its product, NaV1.1, is also present and functionally important in the cardiomyocytes and the sinoatrial node [8]. Therefore, maintaining normal cardiac rhythm is crucial. SCN1A gene mutation leads to altered sodium current and is responsible for cardiac channelopathies. Some studies revealed DS mice and iPSC-derived cardiac myocytes exhibit increased sodium current, which contradicts the loss of NaV1.1 channels [107, 108]. This may be due to alterations in the expression of other ion channels, such as NaV1.5.
Overall, the cardiac autonomic imbalance and altered cardiac electrophysiology in DS patients are associated with an increased incidence of SUDEP, suggesting that cardiac dysfunctions may be a potential cause of death in a portion of DS patients.
Respiratory dysfunction
Respiratory dysfunction during seizures is considered the primary mechanism of SUDEP [109]. Some clinical case reports suggested that DS patients exhibited altered respiratory rhythms, central and obstructive apnea, as well as respiratory failure [70, 110, 111]. Seizures can cause laryngospasm, leading to complete or partial airway occlusion [112]. These respiratory dysfunctions result in reduced oxygen saturation followed by SUDEP. Seizure-induced respiratory arrest (S-IRA) is the leading cause of different respiratory dysfunctions. Our previous studies have shown that deficits in 5-HT and norepinephrine (NE) neurotransmission play an important role in the pathogenesis of S-IRA in the DBA/1 mouse model. We found that atomoxetine-mediated reduction of S-IRA evoked by acoustic stimulation or PTZ, can be significantly reversed by prazosin, a selective NE α-1 receptor antagonist. Additionally, the incidence of S-IRA can be reduced by administering 5-hydroxytryptophan, a precursor for serotonin synthesis [82, 113]. DS mice, like DBA/1 mice, also exhibit respiratory arrest prior to SUDEP. DS mice present respiratory phenotypes, such as hypoventilation and central apnea, which rapidly cause hypoxemia within seconds [114]. Hypoxemia then directly affects the heart, leading to bradycardia following respiratory arrest. Consistent with these findings in animal models, clinical studies have shown that DS patients initially experience peri-ictal respiratory dysfunction and apnea, followed by cardiac arrest and SUDEP. It was reported a child with acute and persistent postictal hypoventilation later died of SUDEP. These results suggest that immediate and fatal respiratory arrest may be a terminal event in DS patients and could serve as a biomarker for those at high risk. Other DS mice exhibited an increased respiratory rate and rapid, shallow breathing postictally [101, 115]. This respiratory abnormality is accompanied by tachycardia rather than the bradycardia previously mentioned.
Evidence shows that different brain regions are involved in respiratory dysfunctions in DS. In DS patients, hippocampal firing is increased, indicating hyperexcitability. Stimulation of the hippocampus induces consistent apnea via subcortical projections to brainstem respiratory centers [116]. Similarly, activation of the central amygdala (CeA) also produces fatal apnea in DS mice, possibly due to the spread of seizures to the CeA [117]. There is a bidirectional projection between the CeA and the bed nucleus of the stria terminalis (BNST), both of which project to the parabrachial nucleus (PBN). Research shows neurons projecting from the BNST to PBN are hypoexcitable in DS mice [118]. The BNST receives inputs from cortical and hippocampal regions and integrates these signals to influence downstream respiratory pathways. Further, the retrotrapezoid nucleus (RTN) is an important respiratory control center and functions as a central respiratory chemoreceptor to regulate respiratory depth and frequency. In DS mice, inactivation of inhibitory neurons in the RTN caused by loss of SCN1A function leads to enhanced sensitivity of glutamatergic chemosensitive RTN neurons [119]. In addition, SST+ parafacial neurons likely contribute to RTN chemoreception and regulate respiratory activity [120]. The RTN detects signals related to carbon dioxide and/or pH and transmits them to the preBötzinger complex (PBC) and other brainstem regions. GABAergic interneurons are located in the PBC, a brainstem region essential for respiratory rhythm generation and regulation. Loss of NaV 1.1 channels in DS could impair PBC function and cause respiratory dysfunctions that may contribute to the mechanisms of SUDEP in DS [121]. These cerebral nuclei are critical components of the neural pathway responsible for respiratory dysfunction in DS, leading to SUDEP. In summary, closely monitoring fatal respiratory dysfunction in DS patients may be conducive to preventing SUDEP. In Fig. 3, we illustrate the tested and possible respiratory circuits in DS (Fig. 3B, C).
Other possible mechanisms
The interaction between sleep and DS may have long-term effects, increasing susceptibility to SUDEP. SUDEP in DS patients typically occurs during sleep, highlighting its close relationship with sleep. DS patients frequently experience disrupted sleep due to seizures, which can, in turn, lead to more frequent and severe sleep disorders. Sleep disorders create a positive feedback loop, promoting seizures and increasing the risk of poorly controlled seizures and SUDEP [122]. Thus, sleep disturbances, including disorders of excessive somnolence and sleep-wake transition disorders, are important issues that cannot be ignored in DS [123]. The interactions between the brainstem arousal system and sleep control during seizures, as well as the circadian disruptions, significantly impact sleep disorders and SUDEP. Impaired circadian phenotype and sleep-wake regulation are present in DS patients. The SCN1A-encoded NaV1.1 channel is expressed in brain regions important for circadian rhythms and sleep-wake regulation, including the GABAergic neurons in the suprachiasmatic nucleus (SCN) of the hypothalamus, thalamic reticular nucleus (RTN), and cortex [124, 125]. The SCN contains a master circadian clock that controls circadian rhythms and regulates various circadian activities in the body. GABA, a neurotransmitter found in most SCN neurons, acts as a crucial signal to mediate the properties of the SCN neuronal network [126]. Decreased excitability of GABAergic interneurons in SCN due to dysfunctional NaV1.1 channels is a primary contributor to circadian defects, which may increase the risk of SUDEP. In addition, altered clock gene expression acts in conjunction with the impairments caused by SCN1A deficiency to result in aberrant circadian rhythmicity [126].
Tau, a microtubule-associated protein, is implicated in a number of neurodegenerative diseases, such as Alzheimer’s disease [127]. Intraneuronal accumulation of abnormal tau can explain their pathogenesis. However, no evidence shows that DS mice have microscopically visible accumulations of abnormal tau aggregates in neurons. In DS models, global genetic reduction of tau alleviates seizures and network hyperexcitability and improves survival [128]. Selective tau ablation in excitatory neurons also reduces SUDEP, epileptiform activity, and autism-like behaviors in DS mice [129]. These results suggest that nonaggregated tau has a pathogenic role in DS and may be a potential factor for SUDEP.
Therapeutic applications
Despite our increased knowledge of DS, the fact that DS pathogenesis remains poorly understood accounts for the limited therapeutic development of this disease to date. DS is drug-resistant and complete seizure control is generally not achievable with current therapies [130]. For the management of this disorder, the crucial aim is to significantly reduce seizure frequency and minimize the adverse effects of antiepileptic therapies [131].In addition to seizure control, treatment must also focus on the comorbidities that afflict patients with DS, which may have an even greater negative impact on the quality of life of the patient and their families. Many treatment options are being investigated to increase the longevity of patients, including pharmacotherapy, ketogenic diet, neuromodulation therapy, and gene therapy.
Pharmacological approach
A wide variety of antiepileptic drugs (AED) can be utilized to treat seizures in DS. Accepted first-line agents include broad-spectrum valproic acid (VPA) and clobazam (CLB). VPA, with multiple mechanisms of action, is effective for diverse types of seizures. Evidence of its efficacy in DS patients is derived from some retrospective studies, suggesting VPA is beneficial in DS [132, 133]. Like VPA, due to its good tolerability, safety profile, and broad-spectrum antiepileptic activity, CLB is commonly used to treat developmental and epileptic encephalopathy, including DS [134]. However, in most cases, seizures in DS cannot be absolutely controlled with VPA and CLB, and additional medications must be added if the seizures continue. That is why polypharmacy is common in DS. Due to the pathogenesis of most DS, it is indeed important to enhance GABA signaling, which may ameliorate seizures and comorbidities. Topiramate is regarded as a second-line drug for DS, and it is also a broad-spectrum AED, with several mechanisms of action, including enhancement of GABAergic activity [131]. Topiramate is effective and safe in both generalized and localized epilepsies in children. Several observational studies have evaluated the efficacy of topiramate in patients with DS and found that it can lead to a decrease in seizure frequency [135, 136]. Moreover, clonazepam is a positive allosteric modulator of GABAA receptors that protects against myoclonic and generalized tonic-clonic seizures. Given the synergistic GABA-enhancing effect of clonazepam and tiagabine gave increased seizure protection and decreased toxicity, the combination of these two drugs may be well tolerated and effective for controlling seizures in DS [137]. However, there are no clinical trials of clonazepam and tiagabine in DS patients. Other antiepileptic drugs, such as levetiracetam, zonisamide ethosuximide, and bromide, are later options for DS patients as a complementary therapy. Since the adverse effects of zonisamide may be related to the inhibition of carbonic anhydrase, it is best not to coadminister it with other carbonic anhydrase inhibitors (eg, topiramate). These four drugs may be capable of suppressing seizures, but their exact efficacy and safety require further evaluation [131, 138].
The results of new drugs for DS, including stiripentol and cannabidiol, are very promising. There is a consensus on the use of stiripentol as a first- or second-line treatment option. Stiripentol in combination with VPA and CLB can achieve better curative efficacy [139]. Therefore, Stiripentol is recommended as an adjuvant treatment for refractory generalized tonic-clonic seizures in DS patients together with VPA and CLB. However, stiripentol, when used with VPA and CLB, increases somnolence and gastrointestinal adverse events. Additionally, in animal studies, it is shown that cannabidiol was beneficial in seizures and behavioral comorbidities, as well as survival [140, 141]. In line with these observations in animal models, a randomized controlled trial reported that cannabidiol significantly reduced convulsive-seizure frequency and thus was effective in treating DS patients [142]. However, it should be noted that cannabidiol is associated with higher rates of adverse events, such as pyrexia and elevated transaminases. Thus, when treating patients with cannabidiol, it is important to provide patients with medical guidelines that describe crucial details about the usage and risks of this drug. Cannabidiol has a bidirectional interaction with clobazam potentially leading to increased somnolence and sedation, thus requiring dose reduction [134]. Some medications that modulate 5-HT signaling pathways, including fenfluramine, clemizole, lorcaserin, and trazodone, have recently emerged as potential treatments for DS. Previous studies have shown that 5-HT is significantly reduced in the head of DS zebrafish and 5-HT receptor subtypes can be targeted to trigger the antiseizure effects in DS, especially 5‐HT1D, 5‐HT2A, and 5‐HT2C receptors [143, 144]. For example, fenfluramine, which was recently approved to suppress seizures, is considered a second-line drug for DS and acts as an agonist of these three receptor subtypes. Clemizole, a recognized antihistamine, is an H1-receptor antagonist. However, the antiepileptic effect of clemizole is based on its affinity for the 5-HT2A receptor, and clemizole is under development as a therapeutic agent for DS [145]. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in DS mice and is currently under clinical investigation for the treatment of DS [146]. The long-term efficacy and safety of soticlestat are being assessed and it is found that the adverse events of soticlestat are noticeably less severe. In addition, its unique mechanism of action increases the possibility of combining it with other drugs. Another possible pharmacological agent is the modulator of calcium channels, and the administration of verapamil influences epileptic activity [147]. Add-on treatment with verapamil appears to have an effect on controlling seizures in DS patients, but further studies are needed to confirm its function. Importantly, one point to note is the influence of contraindicated medications. Seizures are typically worsened with sodium-channel agents, including lamotrigine, carbamazepine, oxcarbazepine and phenytoin [148]. Hence, these drugs must be avoided. Overall, although pharmacotherapy is the most important treatment, its efficacy is inadequate and even causes some side effects, necessitating non-pharmacological intervention.
In Table 3, we summarize the multiple drugs that may be effective against DS (Table 3).
Non-pharmacological approach
The common non-pharmacological approaches include ketogenic diet (KD) and neuromodulation techniques. KD is a high-fat, adequate-protein, and low-carbohydrate diet that uses fat as a replaceable fuel for glucose to alter energy metabolism [149]. The combination of high-fat intake and lowering glucose levels is thought to be the key to the effectiveness of this diet. Current studies have shown that KD can be used to rescue many different conditions, including obesity [150], type 2 diabetes [151], neurodegenerative disorders(e.g., Alzheimer’s disease, Parkinson’s disease) [152], and autism spectrum disorders [153]. Furthermore, the antiepileptic effects of KD have long been known, and it is a recognized, effective nonpharmacologic management option for refractory epilepsy in children, such as DS patients [154]. The production of ketones, which provides a more efficient source of energy than glucose for brain cells and leads to beneficial downstream metabolic changes, such as increasing adenosine levels, which may have an impact on seizure control, is considered to be the fundamental mechanism of this treatment. Evidence suggests that the gut microbiota can regulate brain GABA levels, alter inflammatory signaling and mediate the anti-seizure effect of the KD. KD-induced microbial changes correlated with a lower seizure density in Dravet mice and microbiota-gut-brain axis may be a novel potential target of KD for epilepsy patients [155, 156]. In addition to seizure control, KD treatment could improve survival rates and decrease behavior disturbances. KD can reduce premature mortality by reducing the incidence of S-IRA, a major cause of SUDEP [157]. Prior consensus papers from North America consider KD a second-line treatment for DS [158]. Complete seizure control is typically not achieved with pharmacotherapy alone in DS patients. The KD has fewer neurotoxic side effects than drug treatments, making it a relatively safe and effective therapeutic strategy.
While KD benefits seizure control, its metabolic shifts may negatively impact cardiac health. β-hydroxybutyric acid produced by KD may impair mitochondrial biogenesis, depleting energy in cardiomyocytes and triggering apoptosis and cardiac fibrosis [159]. KD can lead to electrolyte imbalances and sodium channel alterations, increasing depolarization and cardiac excitability [160], thereby exacerbating arrhythmia risks and compromising cardiac function, which is closely tied to cardiac events and SUDEP in DS patients. SCN1A mutations may further influence susceptibility to cardiac effects, underscoring the need for monitoring cardiac health in DS patients undergoing KD therapy, particularly in those at risk [161]. KD remains a powerful therapeutic option for seizure control, but its cardiac risks and side effects require careful assessment to ensure patient safety and optimize therapeutic benefits.
Another nonpharmacological approach is neuromodulation techniques, mainly including vagus nerve stimulation (VNS) and deep brain stimulation (DBS). In some DS patients, reduced seizure frequency and shorter recovery time could be observed after the application of VNS, suggesting VNS may be beneficial in the treatment of DS [162]. However, it does not guarantee improvement of the disease and carries the risk of infection and subsequent device failure [163]. With regard to DBS, there is no strong evidence for its curative effect on DS, and the role of DBS in DS needs further study [164]. Nevertheless, neuromodulation therapy is an emerging addition to the DS treatment landscape with significant unexplored potential for further optimization.
Novel genetic therapy
In the last few years, there have been significant advances in the field of gene therapy, which has expanded the potential treatment options for DS. Currently, the dominant gene therapies are antisense oligonucleotides (ASOs), viral vectors-mediated gene modulation, and gene editing strategies, and some of these are in transition to clinical trials [165, 166]. Since DS is mainly caused by mutations in one of the copies of the SCN1A gene, gene therapies aim to reduce seizures and severe comorbidities by utilizing the non-mutant copy of the SCN1A gene to upregulate Nav1.1 protein and restore normal Nav1.1 levels. STK-001, developed by Targeted Augmentation of Nuclear Gene Output (TANGO) technology, is an investigational new medicine for DS and is currently being evaluated in ongoing clinical trials [167]. STK-001 is an ASO designed to bind to pre-mRNA to enhance the level of productive SCN1A mRNA and then improve the production of the Nav1.1 protein. Preclinical studies suggested a positive impact of Scn1a-ASO on seizures and the incidence of SUDEP in DS mice [168]. Similarly, data from clinical trials indicated that STK-001 was well tolerated, with 70.6% of individuals treated with it experiencing a decrease in seizure frequency. Thus, this medicine could represent a major advance in gene therapy for DS patients. In addition, several different types of viral vectors have recently come up as emerging avenues for gene therapy carrying the potential to treat DS, yet adeno-associated virus (AAV) is by far the most widely used one. Transcriptional activators and CRISPR-Cas9-mediated gene editing techniques can be applied to facilitate AAV-based SCN1A gene upregulation [169]. EXT is a GABAergic interneuron selective AAV containing an engineered transcription factor for the upregulation of SCN1A expression. The efficacy of ETX101 has also been validated in preclinical studies: ETX101 administration reduced seizures and the risk of sudden death in DS mice and was well tolerated in nonhuman primates, and this medicine will probably start clinical trials soon [170]. Further, the CRISPR-Cas9 technique has shown promising results in several studies. This approach could restore inhibitory interneuron excitability and ameliorate epileptic and behavioral phenotypes in DS mice, suggesting that it may be an effective treatment for seizures and behavioral deficits [171, 172]. However, additional investigation is needed to further elucidate its role in human patients. The clinical utility of these mentioned therapies is limited to DS patients with SCN1A variants that result in Nav1.1 haploinsufficiency and therapies targeting other gene variants are supposed to emerge in the future as well. In conclusion, gene therapy, as a form of precision medicine and targeted therapeutics, holds great promise to provide sustained seizure freedom, reduce the risk of SUDEP risk, and even completely cure devastating DS.
Conclusion and perspectives
In this review, we focus on DS, a severe form of intractable pediatric epilepsy with a notably high incidence of SUDEP. The pathogenesis of SUDEP in DS is complex, and progress in understanding it has been hindered by the unpredictable and irreversible nature of the disease. Here, we review the currently identified pathogenesis of DS and consider the association between DS and SUDEP, proposing potential explanations for the occurrence of SUDEP in DS patients. Genetic factors may provide a link between DS and SUDEP. Different genetic mutations underlying DS may affect the susceptibility to SUDEP. Genetic modifiers may exacerbate or alleviate this risk. Additionally, pathophysiological factors are equally important. Proposed mechanisms, such as fatal arrhythmias and respiratory dysfunction, offer significant insights into the pathogenesis of SUDEP in DS patients. Moreover, the increased frequency and severity of seizures, coupled with sleep disorders, create a positive feedback loop that may ultimately lead to SUDEP.
DS remains a serious public health issue, complicated by drug resistance and a lack of effective preventive and therapeutic measures. Combining pharmacological treatments with non-pharmacologic therapies holds promise for improving therapeutic outcomes. Future therapeutic strategies are likely to focus on precision medicine. For example, recent advancements in gene therapy highlight its potential to revolutionize the treatment of epilepsy. Gene therapy may play a critical role in developing novel therapies for epilepsy, though further research is necessary to understand the interactions between gene therapy and conventional treatments, as well as their respective advantages and limitations in DS.
References
Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52:3–9. https://doi.org/10.1111/j.1528-1167.2011.02994.x
Dravet C, Oguni H. Dravet syndrome (severe myoclonic epilepsy in infancy). Handb Clin Neurol. 2013;111:627–33. https://doi.org/10.1016/b978-0-444-52891-9.00065-8
Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, et al. Incidence of Dravet Syndrome in a US Population. Pediatrics. 2015;136:e1310–1315,. https://doi.org/10.1542/peds.2015-1807
Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135:2329–36. https://doi.org/10.1093/brain/aws151
Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–32. https://doi.org/10.1086/320609
Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. 2010;51:1650–8. https://doi.org/10.1111/j.1528-1167.2010.02640.x
Parihar R, Ganesh S. The SCN1A gene variants and epileptic encephalopathies. J Hum Genet. 2013;58:573–80. https://doi.org/10.1038/jhg.2013.77
Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–59. https://doi.org/10.1113/jphysiol.2010.187484
Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. 2011;76:594–600. https://doi.org/10.1212/WNL.0b013e31820c309b
Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia. 2012;53:87–100. https://doi.org/10.1111/j.1528-1167.2011.03346.x
Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–14. https://doi.org/10.1523/jneurosci.5270-06.2007
Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–9. https://doi.org/10.1038/nn1754
Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini PP. SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. 2012;23:177–86. https://doi.org/10.1016/j.yebeh.2011.11.022
Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53:227–33. https://doi.org/10.1111/j.1528-1167.2011.03358.x
Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–85. https://doi.org/10.1111/epi.12666
Cooper MS, McIntosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–47. https://doi.org/10.1016/j.eplepsyres.2016.10.006
Shmuely S, Sisodiya SM, Gunning WB, Sander JW, Thijs RD. Mortality in Dravet syndrome: a review. Epilepsy Behav. 2016;64:69–74. https://doi.org/10.1016/j.yebeh.2016.09.007
Fisher RS, Cross JH, D’Souza C, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. 2017;58:531–42. https://doi.org/10.1111/epi.13671
Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE task force on nosology and definitions. Epilepsia. 2022;63:1349–97. https://doi.org/10.1111/epi.17239
Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure. 2017;44:58–64. https://doi.org/10.1016/j.seizure.2016.10.014
Skluzacek JV, Watts KP, Parsy O, Wical B, Camfield P. Dravet syndrome and parent associations: the IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia. 2011;52:95–101. https://doi.org/10.1111/j.1528-1167.2011.03012.x.
Cheah CS, Westenbroek RE, Roden WH, Kalume F, Oakley JC, Jansen LA, et al. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels (Austin, Tex.). 2013;7:468–72. https://doi.org/10.4161/chan.26023
Sakauchi M, Oguni H, Kato I, Osawa M, Hirose S, Kaneko S, et al. Mortality in Dravet syndrome: search for risk factors in Japanese patients. Epilepsia. 2011;52:50–54. https://doi.org/10.1111/j.1528-1167.2011.03002.x
Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52:44–9. https://doi.org/10.1111/j.1528-1167.2011.03001.x
Selvarajah A, Gorodetsky C, Marques P, Zulfiqar Ali Q, Berg AT, Fasano A, et al. Progressive worsening of gait and motor abnormalities in older adults with Dravet syndrome. Neurology. 2022;98:e2204–e2210. https://doi.org/10.1212/wnl.0000000000200341
Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: Beyond SCN1A. Epilepsia. 2017;58:1807–16. https://doi.org/10.1111/epi.13889
Leu C, Balestrini S, Maher B, Hernández-Hernández L, Gormley P, Hämäläinen E, et al. Genome-wide polygenic burden of rare deleterious variants in sudden unexpected death in epilepsy. EBioMedicine. 2015;2:1063–70. https://doi.org/10.1016/j.ebiom.2015.07.005
Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009;5:e1000381 https://doi.org/10.1371/journal.pgen.1000381
Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40:776–81. https://doi.org/10.1038/ng.149
Trivisano M, Pietrafusa N, Ciommo V, Cappelletti S, Palma L, Terracciano A, et al. PCDH19-related epilepsy and Dravet syndrome: face-off between two early-onset epilepsies with fever sensitivity. Epilepsy Res. 2016;125:32–6. https://doi.org/10.1016/j.eplepsyres.2016.05.015
Liu YH, Cheng YT, Tsai MH, Chou IJ, Hung PC, Hsieh MY, et al. Genetics and clinical correlation of Dravet syndrome and its mimics - experience of a tertiary center in Taiwan. Pediatr Neonatol. 2021;62:550–8. https://doi.org/10.1016/j.pedneo.2021.05.022
Shi X, Yasumoto S, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. 2009;31:758–62. https://doi.org/10.1016/j.braindev.2009.08.009
Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. 2009;29:10764–78. https://doi.org/10.1523/jneurosci.2475-09.2009
Gong JE, Liao HM, Long HY, Li XM, Long LL, Zhou L, et al. SCN1B and SCN2B gene variants analysis in dravet syndrome patients: analysis of 22 cases. Medicine. 2019;98:e14974 https://doi.org/10.1097/md.0000000000014974
Kim YO, Dibbens L, Marini C, Suls A, Chemaly N, Mei D, et al. Do mutations in SCN1B cause Dravet syndrome? Epilepsy Res. 2013;103:97–100. https://doi.org/10.1016/j.eplepsyres.2012.10.009
Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet. 2015;47:393–9. https://doi.org/10.1038/ng.3239
Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, et al. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol. 2007;98:1501–25. https://doi.org/10.1152/jn.00640.2006
Nava C, Dalle C, Rastetter A, Striano P, de Kovel CG, Nabbout R, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet. 2014;46:640–5. https://doi.org/10.1038/ng.2952
Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Møller RS, Hjalgrim H, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 2014;82:1245–53. https://doi.org/10.1212/wnl.0000000000000291
Johannesen K, Marini C, Pfeffer S, Møller RS, Dorn T, Niturad CE, et al. Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology. 2016;87:1140–51. https://doi.org/10.1212/wnl.0000000000003087
Le SV, Le PHT, Le TKV, Kieu Huynh TT, Hang Do TT. A mutation in GABRB3 associated with Dravet syndrome. Am J Med Genet A. 2017;173:2126–31. https://doi.org/10.1002/ajmg.a.38282
Pavone P, Pappalardo XG, Marino SD, Sciuto L, Corsello G, Ruggieri M, et al. A novel GABRB3 variant in Dravet syndrome: Case report and literature review. Mol Genet Genomic Med. 2020;8:e1461 https://doi.org/10.1002/mgg3.1461
Shen D, Hernandez CC, Shen W, Hu N, Poduri A, Shiedley B, et al. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain. 2017;140:49–67. https://doi.org/10.1093/brain/aww272
Ishii A, Kanaumi T, Sohda M, Misumi Y, Zhang B, Kakinuma N, et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. 2014;108:420–32. https://doi.org/10.1016/j.eplepsyres.2013.12.005
Álvarez Bravo G, Yusta Izquierdo A. The adult motor phenotype of Dravet syndrome is associated with mutation of the STXBP1 gene and responds well to cannabidiol treatment. Seizure. 2018;60:68–70. https://doi.org/10.1016/j.seizure.2018.06.010
Suls A, Jaehn JA, Kecskés A, Weber Y, Weckhuysen S, Craiu DC, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. 2013;93:967–75. https://doi.org/10.1016/j.ajhg.2013.09.017
Redler S, Strom TM, Wieland T, Cremer K, Engels H, Distelmaier F, et al. Variants in CPLX1 in two families with autosomal-recessive severe infantile myoclonic epilepsy and ID. Eur J Hum Genet. 2017;25:889–93. https://doi.org/10.1038/ejhg.2017.52
Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2014;13:163–72. https://doi.org/10.1111/gbb.12099
Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41:655–60. https://doi.org/10.1016/j.nbd.2010.11.016
Hammer MF, Ishii A, Johnstone L, Tchourbanov A, Lau B, Sprissler R, et al. Rare variants of small effect size in neuronal excitability genes influence clinical outcome in Japanese cases of SCN1A truncation-positive Dravet syndrome. PLoS ONE. 2017;12:e0180485 https://doi.org/10.1371/journal.pone.0180485
Hawkins NA, Nomura T, Duarte S, Barse L, Williams RW, Homanics GE, et al. Gabra2 is a genetic modifier of Dravet syndrome in mice. Mamm Genome. 2021;32:350–63. https://doi.org/10.1007/s00335-021-09877-1
Hawkins NA, Kearney JA. Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res. 2016;119:20–3. https://doi.org/10.1016/j.eplepsyres.2015.11.016
Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649 https://doi.org/10.1371/journal.pgen.1000649
Ohmori I, Ouchida M, Miki T, Mimaki N, Kiyonaka S, Nishiki T, et al. A CACNB4 mutation shows that altered Ca(v)2.1 function may be a genetic modifier of severe myoclonic epilepsy in infancy. Neurobiol Dis. 2008;32:349–54. https://doi.org/10.1016/j.nbd.2008.07.017
Ohmori I, Ouchida M, Kobayashi K, Jitsumori Y, Mori A, Michiue H, et al. CACNA1A variants may modify the epileptic phenotype of Dravet syndrome. Neurobiol Dis. 2013;50:209–17. https://doi.org/10.1016/j.nbd.2012.10.016
Gaily E, Anttonen AK, Valanne L, Liukkonen E, Träskelin AL, Polvi A, et al. Dravet syndrome: new potential genetic modifiers, imaging abnormalities, and ictal findings. Epilepsia. 2013;54:1577–85. https://doi.org/10.1111/epi.12256
Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–9. https://doi.org/10.1093/hmg/ddm248
Calhoun JD, Hawkins NA, Zachwieja NJ, Kearney JA. Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia. 2017;58:e111–e115. https://doi.org/10.1111/epi.13811
Bagnall RD, Ingles J, Yeates L, Berkovic SF, Semsarian C. Exome sequencing-based molecular autopsy of formalin-fixed paraffin-embedded tissue after sudden death. Genet Med. 2017;19:1127–33. https://doi.org/10.1038/gim.2017.15
O’Malley HA, Hull JM, Clawson BC, Chen C, Owens-Fiestan G, Jameson MB, et al. Scn1b deletion in adult mice results in seizures and SUDEP. Ann Clin Transl Neurol. 2019;6:1121–6. https://doi.org/10.1002/acn3.785
Lin X, O’Malley H, Chen C, Auerbach D, Foster M, Shekhar A, et al. Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol. 2015;593:1389–407. https://doi.org/10.1113/jphysiol.2014.277699
Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–10. https://doi.org/10.1016/j.ajhg.2012.01.006
Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI, et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. 2016;79:522–34. https://doi.org/10.1002/ana.24596
Nascimento FA, Borlot F, Cossette P, Minassian BA, Andrade DM. Two definite cases of sudden unexpected death in epilepsy in a family with a DEPDC5 mutation. Neurol Genet. 2015;1:e28 https://doi.org/10.1212/nxg.0000000000000028
Klofas LK, Short BP, Zhou C, Carson RP. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum Mol Genet. 2020;29:1365–77. https://doi.org/10.1093/hmg/ddaa068
Baldassari S, Picard F, Verbeek NE, van Kempen M, Brilstra EH, Lesca G, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019;21:398–408. https://doi.org/10.1038/s41436-018-0060-2
Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia. 2016;57:994–1003. https://doi.org/10.1111/epi.13391
Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–40. https://doi.org/10.1038/ng1094-136
Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–75. https://doi.org/10.1523/jneurosci.5591-09.2010
Simeone KA, Hallgren J, Bockman CS, Aggarwal A, Kansal V, Netzel L, et al. Respiratory dysfunction progresses with age in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia. 2018;59:345–57. https://doi.org/10.1111/epi.13971
Velíšková J, Marra C, Liu Y, Shekhar A, Park DS, Iatckova V, et al. Early onset epilepsy and sudden unexpected death in epilepsy with cardiac arrhythmia in mice carrying the early infantile epileptic encephalopathy 47 gain-of-function FHF1(FGF12) missense mutation. Epilepsia. 2021;62:1546–58. https://doi.org/10.1111/epi.16916
Labate A, Tarantino P, Palamara G, Gagliardi M, Cavalcanti F, Ferlazzo E, et al. Mutations in PRRT2 result in familial infantile seizures with heterogeneous phenotypes including febrile convulsions and probable SUDEP. Epilepsy Res. 2013;104:280–4. https://doi.org/10.1016/j.eplepsyres.2012.10.014
Coll M, Striano P, Ferrer-Costa C, Campuzano O, Matés J, Del Olmo B, et al. Targeted next-generation sequencing provides novel clues for associated epilepsy and cardiac conduction disorder/SUDEP. PLoS ONE. 2017;12:e0189618 https://doi.org/10.1371/journal.pone.0189618
Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77. https://doi.org/10.1161/circep.111.962019
Wilde AAM, Amin AS. Clinical spectrum of SCN5A mutations: long QT syndrome, brugada syndrome, and cardiomyopathy. JACC Clin Electrophysiol. 2018;4:569–79. https://doi.org/10.1016/j.jacep.2018.03.006
Tu E, Bagnall RD, Duflou J, Semsarian C. Post-mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol. 2011;21:201–8. https://doi.org/10.1111/j.1750-3639.2010.00438.x
Rosamilia MB, Lu IM, Landstrom AP. Pathogenicity assignment of variants in genes associated with cardiac channelopathies evolve toward diagnostic uncertainty. Circ Genom Precis Med. 2022;15:e003491 https://doi.org/10.1161/circgen.121.003491
Tu E, Waterhouse L, Duflou J, Bagnall RD, Semsarian C. Genetic analysis of hyperpolarization-activated cyclic nucleotide-gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol. 2011;21:692–8. https://doi.org/10.1111/j.1750-3639.2011.00500.x
Kessi M, Peng J, Duan H, He H, Chen B, Xiong J, et al. The contribution of HCN channelopathies in different epileptic syndromes, mechanisms, modulators, and potential treatment targets: a systematic review. Front Mol Neurosci. 2022;15:807202 https://doi.org/10.3389/fnmol.2022.807202
Coll M, Allegue C, Partemi S, Mates J, Del Olmo B, Campuzano O, et al. Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int J Legal Med. 2016;130:331–9. https://doi.org/10.1007/s00414-015-1269-0
Feng HJ, Faingold CL. Abnormalities of serotonergic neurotransmission in animal models of SUDEP. Epilepsy Behav. 2017;71:174–80. https://doi.org/10.1016/j.yebeh.2015.06.008
Zhang H, Zhao H, Yang X, Xue Q, Cotten JF, Feng HJ. 5-Hydroxytryptophan, a precursor for serotonin synthesis, reduces seizure-induced respiratory arrest. Epilepsia. 2016;57:1228–35. https://doi.org/10.1111/epi.13430
Zhang H, Zhao H, Zeng C, Van Dort C, Faingold CL, Taylor NE, et al. Optogenetic activation of 5-HT neurons in the dorsal raphe suppresses seizure-induced respiratory arrest and produces anticonvulsant effect in the DBA/1 mouse SUDEP model. Neurobiol Dis. 2018;110:47–58. https://doi.org/10.1016/j.nbd.2017.11.003
Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–90. https://doi.org/10.1038/ng0897-387
Massey CA, Thompson SJ, Ostrom RW, Drabek J, Sveinsson OA, Tomson T, et al. X-linked serotonin 2C receptor is associated with a non-canonical pathway for sudden unexpected death in epilepsy. Brain Commun. 2021;3:fcab149 https://doi.org/10.1093/braincomms/fcab149
Bagnall RD, Crompton DE, Cutmore C, Regan BM, Berkovic SF, Scheffer IE, et al. Genetic analysis of PHOX2B in sudden unexpected death in epilepsy cases. Neurology. 2014;83:1018–21. https://doi.org/10.1212/wnl.0000000000000781
Sun Y, Paşca SP, Portmann T, Goold C, Worringer KA, Guan W, et al. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. eLife. 2016;5:e13073 https://doi.org/10.7554/eLife.13073
Oakley JC, Kalume F, Catterall WA. Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia. 2011;52:59–61. https://doi.org/10.1111/j.1528-1167.2011.03004.x
Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–804. https://doi.org/10.1093/hmg/ddt331
Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2014;111:E3139–3148. https://doi.org/10.1073/pnas.1411131111
Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL Jr., Kearney JA. Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis. 2014;65:1–11. https://doi.org/10.1016/j.nbd.2014.01.006
Dyment DA, Schock SC, Deloughery K, Tran MH, Ure K, Nutter LMJ, et al. Electrophysiological alterations of pyramidal cells and interneurons of the CA1 region of the hippocampus in a novel mouse model of Dravet syndrome. Genetics. 2020;215:1055–66. https://doi.org/10.1534/genetics.120.303399
Wong JC, Dutton SB, Collins SD, Schachter S, Escayg A. Huperzine a provides robust and sustained protection against induced seizures in Scn1a mutant mice. Front Pharmacol. 2016;7:357 https://doi.org/10.3389/fphar.2016.00357
Wong JC, Thelin JT, Escayg A. Donepezil increases resistance to induced seizures in a mouse model of Dravet syndrome. Ann Clin Transl Neurol. 2019;6:1566–71. https://doi.org/10.1002/acn3.50848
Studtmann C, Ladislav M, Topolski MA, Safari M, Swanger SA. Na(V)1.1 haploinsufficiency impairs glutamatergic and GABAergic neuron function in the thalamus. Neurobiol Dis. 2022;167:105672 https://doi.org/10.1016/j.nbd.2022.105672
Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan Y, et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet. 2013;22:4241–52. https://doi.org/10.1093/hmg/ddt275
Hunt RF, Hortopan GA, Gillespie A, Baraban SC. A novel zebrafish model of hyperthermia-induced seizures reveals a role for TRPV4 channels and NMDA-type glutamate receptors. Exp Neurol. 2012;237:199–206. https://doi.org/10.1016/j.expneurol.2012.06.013
Smith PE. Introduction: the causes of epilepsy. Epilepsia. 2012;53:1–2. https://doi.org/10.1111/j.1528-1167.2012.03607.x
Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol. 2016;15:1075–88. https://doi.org/10.1016/s1474-4422(16)30158-2
Sveinsson O, Andersson T, Mattsson P, Carlsson S, Tomson T. Clinical risk factors in SUDEP: a nationwide population-based case-control study. Neurology. 2020;94:e419–e429. https://doi.org/10.1212/wnl.0000000000008741
Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–808. https://doi.org/10.1172/jci66220
Myers KA, Bello-Espinosa LE, Symonds JD, Zuberi SM, Clegg R, Sadleir LG, et al. Heart rate variability in epilepsy: a potential biomarker of sudden unexpected death in epilepsy risk. Epilepsia. 2018;59:1372–80. https://doi.org/10.1111/epi.14438
Lyu SY, Nam SO, Lee YJ, Kim G, Kim YA, Kong J, et al. Longitudinal change of cardiac electrical and autonomic function and potential risk factors in children with dravet syndrome. Epilepsy Res. 2019;152:11–17. https://doi.org/10.1016/j.eplepsyres.2019.02.018
Perulli M, Battista A, Sivo S, Turrini I, Musto E, Quintiliani M, et al. Heart rate variability alterations in Dravet syndrome: the role of status epilepticus and a possible association with mortality risk. Seizure. 2022;94:129–35. https://doi.org/10.1016/j.seizure.2021.11.023
Ergul Y, Ekici B, Tatli B, Nisli K, Ozmen M. QT and P wave dispersion and heart rate variability in patients with Dravet syndrome. Acta Neurol Belg. 2013;113:161–6. https://doi.org/10.1007/s13760-012-0140-z
Delogu AB, Spinelli A, Battaglia D, Dravet C, De Nisco A, Saracino A, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia. 2011;52:55–58. https://doi.org/10.1111/j.1528-1167.2011.03003.x
Auerbach DS, Jones J, Clawson BC, Offord J, Lenk GM, Ogiwara I, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS ONE. 2013;8:e77843 https://doi.org/10.1371/journal.pone.0077843
Frasier CR, Zhang H, Offord J, Dang LT, Auerbach DS, Shi H, et al. Channelopathy as a SUDEP Biomarker in Dravet Syndrome Patient-Derived Cardiac Myocytes. Stem Cell Reports. 2018;11:626–34. https://doi.org/10.1016/j.stemcr.2018.07.012
Surges R, Thijs RD, Tan HL, Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5:492–504. https://doi.org/10.1038/nrneurol.2009.118
Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–77. https://doi.org/10.1016/s1474-4422(13)70214-x
Jefferys JGR, Arafat MA, Irazoqui PP, Lovick TA. Brainstem activity, apnea, and death during seizures induced by intrahippocampal kainic acid in anaesthetized rats. Epilepsia. 2019;60:2346–58. https://doi.org/10.1111/epi.16374
Nakase K, Kollmar R, Lazar J, Arjomandi H, Sundaram K, Silverman J, et al. Laryngospasm, central and obstructive apnea during seizures: defining pathophysiology for sudden death in a rat model. Epilepsy Res. 2016;128:126–39. https://doi.org/10.1016/j.eplepsyres.2016.08.004
Shen Y, Ma HX, Lu H, Zhao HT, Sun JL, Cheng Y, et al. Central deficiency of norepinephrine synthesis and norepinephrinergic neurotransmission contributes to seizure-induced respiratory arrest. Biomed Pharmacother. 2021;133:111024 https://doi.org/10.1016/j.biopha.2020.111024
Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest. 2018;128:1141–53. https://doi.org/10.1172/jci94999
Sahai N, Bard AM, Devinsky O, Kalume F. Disordered autonomic function during exposure to moderate heat or exercise in a mouse model of Dravet syndrome. Neurobiol Dis. 2021;147:105154 https://doi.org/10.1016/j.nbd.2020.105154
Lacuey N, Zonjy B, Londono L, Lhatoo SD. Amygdala and hippocampus are symptomatogenic zones for central apneic seizures. Neurology. 2017;88:701–5. https://doi.org/10.1212/wnl.0000000000003613
Dlouhy BJ, Gehlbach BK, Kreple CJ, Kawasaki H, Oya H, Buzza C, et al. Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci. 2015;35:10281–9. https://doi.org/10.1523/jneurosci.0888-15.2015
Yan WW, Xia M, Chiang J, Levitt A, Hawkins N, Kearney J et al. Enhanced synaptic transmission in the extended amygdala and altered excitability in an extended amygdala to brainstem circuit in a dravet syndrome mouse model. eNeuro 8, https://doi.org/10.1523/eneuro.0306-20.2021 (2021).
Kuo FS, Cleary CM, LoTurco JJ, Chen X, Mulkey DK. Disordered breathing in a mouse model of Dravet syndrome. eLife 2019;8 https://doi.org/10.7554/eLife.43387.
Cleary CM, Milla BM, Kuo FS, James S, Flynn WF, Robson P et al. Somatostatin-expressing parafacial neurons are CO(2)/H(+) sensitive and regulate baseline breathing. eLife 10, https://doi.org/10.7554/eLife.60317 (2021).
Kalume F. Sudden unexpected death in Dravet syndrome: respiratory and other physiological dysfunctions. Respir Physiol Neurobiol. 2013;189:324–8. https://doi.org/10.1016/j.resp.2013.06.026
Sanchez REA, Bussi IL, Ben-Hamo M, Caldart CS, Catterall WA, De La Iglesia HO. Circadian regulation of sleep in a pre-clinical model of Dravet syndrome: dynamics of sleep stage and siesta re-entrainment. Sleep 2019;42 https://doi.org/10.1093/sleep/zsz173.
Licheni SH, McMahon JM, Schneider AL, Davey MJ, Scheffer IE. Sleep problems in Dravet syndrome: a modifiable comorbidity. Dev Med Child Neurol. 2018;60:192–8. https://doi.org/10.1111/dmcn.13601
Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, et al. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol Dis. 2015;77:141–54. https://doi.org/10.1016/j.nbd.2015.02.016
Dhamija R, Erickson MK, St Louis EK, Wirrell E, Kotagal S. Sleep abnormalities in children with Dravet syndrome. Pediatr Neurol. 2014;50:474–8. https://doi.org/10.1016/j.pediatrneurol.2014.01.017
Han S, Yu FH, Schwartz MD, Linton JD, Bosma MM, Hurley JB, et al. Na(V)1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci USA. 2012;109:E368–377. https://doi.org/10.1073/pnas.1115729109
Chang CW, Shao E, Mucke L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science. 2021;371:eabb8255 https://doi.org/10.1126/science.abb8255
Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, et al. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol. 2014;76:443–56. https://doi.org/10.1002/ana.24230
Shao E, Chang CW, Li Z, Yu X, Ho K, Zhang M, et al. TAU ablation in excitatory neurons and postnatal TAU knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model. Sci Transl Med. 2022;14:eabm5527 https://doi.org/10.1126/scitranslmed.abm5527
Ceulemans B. Overall management of patients with Dravet syndrome. Dev Med Child Neurol. 2011;53:19–23. https://doi.org/10.1111/j.1469-8749.2011.03968.x.
Knupp KG, Wirrell EC. Treatment strategies for Dravet syndrome. CNS Drugs. 2018;32:335–50. https://doi.org/10.1007/s40263-018-0511-y
Dressler A, Trimmel-Schwahofer P, Reithofer E, Mühlebner A, Gröppel G, Reiter-Fink E, et al. Efficacy and tolerability of the ketogenic diet in Dravet syndrome - Comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015;109:81–89. https://doi.org/10.1016/j.eplepsyres.2014.10.014
Shi XY, Tomonoh Y, Wang WZ, Ishii A, Higurashi N, Kurahashi H, et al. Efficacy of antiepileptic drugs for the treatment of Dravet syndrome with different genotypes. Brain Dev. 2016;38:40–46. https://doi.org/10.1016/j.braindev.2015.06.008
Strzelczyk A, Schubert-Bast S. A practical guide to the treatment of Dravet syndrome with anti-seizure medication. CNS Drugs. 2022;36:217–37. https://doi.org/10.1007/s40263-022-00898-1
Coppola G, Capovilla G, Montagnini A, Romeo A, Spanò M, Tortorella G, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res. 2002;49:45–48. https://doi.org/10.1016/s0920-1211(02)00010-4
Kröll-Seger J, Portilla P, Dulac O, Chiron C. Topiramate in the treatment of highly refractory patients with Dravet syndrome. Neuropediatrics. 2006;37:325–9. https://doi.org/10.1055/s-2007-964867
Oakley JC, Cho AR, Cheah CS, Scheuer T, Catterall WA. Synergistic GABA-enhancing therapy against seizures in a mouse model of Dravet syndrome. J Pharmacol Exp Ther. 2013;345:215–24. https://doi.org/10.1124/jpet.113.203331
Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia. 2011;52:72–75. https://doi.org/10.1111/j.1528-1167.2011.03007.x
Peigné S, Chhun S, Tod M, Rey E, Rodrigues C, Chiron C, et al. Population pharmacokinetics of stiripentol in paediatric patients with Dravet syndrome treated with stiripentol, valproate and clobazam combination therapy. Clin Pharmacokinet. 2018;57:739–48. https://doi.org/10.1007/s40262-017-0592-7
Kaplan JS, Stella N, Catterall WA, Westenbroek RE. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2017;114:11229–34. https://doi.org/10.1073/pnas.1711351114
Patra PH, Serafeimidou-Pouliou E, Bazelot M, Whalley BJ, Williams CM, McNeish AJ. Cannabidiol improves survival and behavioural co-morbidities of Dravet syndrome in mice. Br J Pharmacol. 2020;177:2779–92. https://doi.org/10.1111/bph.15003
Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Eng. J. Med. 2017;376:2011–20. https://doi.org/10.1056/NEJMoa1611618
Sourbron J, Lagae L. Serotonin receptors in epilepsy: novel treatment targets? Epilepsia Open. 2022;7:231–46. https://doi.org/10.1002/epi4.12580
Sourbron J, Schneider H, Kecskés A, Liu Y, Buening EM, Lagae L, et al. Serotonergic Modulation as Effective Treatment for Dravet Syndrome in a Zebrafish Mutant Model. ACS Chem Neurosci. 2016;7:588–98. https://doi.org/10.1021/acschemneuro.5b00342
Griffin A, Hamling KR, Knupp K, Hong S, Lee LP, Baraban SC. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain. 2017;140:669–83. https://doi.org/10.1093/brain/aww342
Hawkins NA, Jurado M, Thaxton TT, Duarte SE, Barse L, Tatsukawa T, et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia. 2021;62:2845–57. https://doi.org/10.1111/epi.17062
Nicita F, Spalice A, Papetti L, Nikanorova M, Iannetti P, Parisi P. Efficacy of verapamil as an adjunctive treatment in children with drug-resistant epilepsy: a pilot study. Seizure. 2014;23:36–40. https://doi.org/10.1016/j.seizure.2013.09.009
Wirrell EC, Hood V, Knupp KG, Meskis MA, Nabbout R, Scheffer IE, et al. International consensus on diagnosis and management of Dravet syndrome. Epilepsia. 2022. https://doi.org/10.1111/epi.17274
Kraeuter AK, Guest PC, Sarnyai Z. The therapeutic potential of ketogenic diet throughout life: focus on metabolic, neurodevelopmental and neurodegenerative disorders. Adv Exp Med Biol. 2019;1178:77–101. https://doi.org/10.1007/978-3-030-25650-0_5
Paoli A. Ketogenic diet for obesity: friend or foe? Int J Environ Res Public Health. 2014;11:2092–107. https://doi.org/10.3390/ijerph110202092
Dashti HM, Mathew TC, Al-Zaid NS. Efficacy of low-carbohydrate ketogenic diet in the treatment of type 2 diabetes. Med Princ Pract. 2021;30:223–35. https://doi.org/10.1159/000512142
Włodarek D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019;11 https://doi.org/10.3390/nu11010169.
Karhu E, Zukerman R, Eshraghi RS, Mittal J, Deth RC, Castejon AM, et al. Nutritional interventions for autism spectrum disorder. Nutr Rev. 2020;78:515–31. https://doi.org/10.1093/nutrit/nuz092
van der Louw E, van den Hurk D, Neal E, Leiendecker B, Fitzsimmon G, Dority L, et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur J Paediatr Neurol. 2016;20:798–809. https://doi.org/10.1016/j.ejpn.2016.07.009
Miljanovic N, Potschka H. The impact of Scn1a deficiency and ketogenic diet on the intestinal microbiome: a study in a genetic Dravet mouse model. Epilepsy Res. 2021;178:106826 https://doi.org/10.1016/j.eplepsyres.2021.106826
Tang Y, Wang Q, Liu J. Microbiota-gut-brain axis: A novel potential target of ketogenic diet for epilepsy. Curr Opin Pharmacol. 2021;61:36–41. https://doi.org/10.1016/j.coph.2021.08.018
Crotts MS, Kim Y, Bravo E, Richerson GB, Teran FA. A ketogenic diet protects DBA/1 and Scn1a(R1407X/+) mice against seizure-induced respiratory arrest independent of ketosis. Epilepsy Behav. 2021;124:108334 https://doi.org/10.1016/j.yebeh.2021.108334
Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American consensus panel. Pediatr Neurol. 2017;68:18–34.e13. https://doi.org/10.1016/j.pediatrneurol.2017.01.025
Xu S, Tao H, Cao W, Cao L, Lin Y, Zhao S-M, et al. Ketogenic diets inhibit mitochondrial biogenesis and induce cardiac fibrosis. Signal Transduct Target Ther. 2021;6:54 https://doi.org/10.1038/s41392-020-00411-4
Hegyi B, Ko CY, Bossuyt J, Bers DM. Two-hit mechanism of cardiac arrhythmias in diabetic hyperglycaemia: reduced repolarization reserve, neurohormonal stimulation, and heart failure exacerbate susceptibility. Cardiovasc Res. 2021;117:2781–93. https://doi.org/10.1093/cvr/cvab006
van Hugte EJH, Lewerissa EI, Wu KM, Scheefhals N, Parodi G, van Voorst TW, et al. SCN1A-deficient excitatory neuronal networks display mutation-specific phenotypes. Brain. 2023;146:5153–67. https://doi.org/10.1093/brain/awad245
Orosz I, McCormick D, Zamponi N, Varadkar S, Feucht M, Parain D, et al. Vagus nerve stimulation for drug-resistant epilepsy: a European long-term study up to 24 months in 347 children. Epilepsia. 2014;55:1576–84. https://doi.org/10.1111/epi.12762
Ding J, Wang L, Li W, Wang Y, Jiang S, Xiao L, et al. Up to what extent does Dravet syndrome benefit from neurostimulation techniques? Front Neurol. 2022;13:843975 https://doi.org/10.3389/fneur.2022.843975
Andrade DM, Hamani C, Lozano AM, Wennberg RA. Dravet syndrome and deep brain stimulation: seizure control after 10 years of treatment. Epilepsia. 2010;51:1314–6. https://doi.org/10.1111/j.1528-1167.2009.02408.x
Chilcott E, Díaz JA, Bertram C, Berti M, Karda R. Genetic therapeutic advancements for Dravet syndrome. Epilepsy Behav. 2022;132:108741 https://doi.org/10.1016/j.yebeh.2022.108741
Isom LL, Knupp KG. Dravet syndrome: novel approaches for the most common genetic epilepsy. Neurotherapeutics. 2021;18:1524–34. https://doi.org/10.1007/s13311-021-01095-6
Wengert ER, Wagley PK, Strohm SM, Reza N, Wenker IC, Gaykema RP, et al. Targeted Augmentation of Nuclear Gene Output (TANGO) of Scn1a rescues parvalbumin interneuron excitability and reduces seizures in a mouse model of Dravet Syndrome. Brain Res. 2022;1775:147743 https://doi.org/10.1016/j.brainres.2021.147743
Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12:eaaz6100 https://doi.org/10.1126/scitranslmed.aaz6100
Lersch R, Jannadi R, Grosse L, Wagner M, Schneider MF, von Stülpnagel C et al. Targeted molecular strategies for genetic neurodevelopmental disorders: emerging lessons from Dravet syndrome. The Neuroscientist 2022;10738584221088244 https://doi.org/10.1177/10738584221088244.
Tanenhaus A, Stowe T, Young A, McLaughlin J, Aeran R, Lin IW, et al. Cell-Selective adeno-associated virus-mediated SCN1A gene regulation therapy rescues mortality and seizure phenotypes in a Dravet syndrome mouse model and is well tolerated in nonhuman primates. Hum Gene Ther. 2022;33:579–97. https://doi.org/10.1089/hum.2022.037
Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, et al. dCas9-Based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in dravet syndrome mice. Mol Ther. 2020;28:235–53. https://doi.org/10.1016/j.ymthe.2019.08.018
Yamagata T, Raveau M, Kobayashi K, Miyamoto H, Tatsukawa T, Ogiwara I, et al. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis. 2020;141:104954 https://doi.org/10.1016/j.nbd.2020.104954
Lossin C, Shi X, Rogawski MA, Hirose S. Compromised function in the Na(v)1.2 Dravet syndrome mutation R1312T. Neurobiol Dis. 2012;47:378–84. https://doi.org/10.1016/j.nbd.2012.05.017
Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84:480–9. https://doi.org/10.1212/wnl.0000000000001211
Ogiwara I, Nakayama T, Yamagata T, Ohtani H, Mazaki E, Tsuchiya S, et al. A homozygous mutation of voltage-gated sodium channel β(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia. 2012;53:e200–203. https://doi.org/10.1111/epi.12040
Shi YW, Zhang Q, Cai K, Poliquin S, Shen W, Winters N, et al. Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain. 2019;142:3028–44. https://doi.org/10.1093/brain/awz250
Kang JQ, Macdonald RL. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 2016;73:1009–16,. https://doi.org/10.1001/jamaneurol.2016.0449
Huang X, Tian M, Hernandez CC, Hu N, Macdonald RL. The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol Dis. 2012;48:115–23. https://doi.org/10.1016/j.nbd.2012.06.013
Hernandez CC, Kong W, Hu N, Zhang Y, Shen W, Jackson L et al. Altered channel conductance states and gating of GABA(A) receptors by a pore mutation linked to Dravet syndrome. eNeuro 2017;4. https://doi.org/10.1523/eneuro.0251-16.2017.
Ohmori I, Kobayashi K, Ouchida M. Scn1a and Cacna1a mutations mutually alter their original phenotypes in rats. Neurochem Int. 2020;141:104859 https://doi.org/10.1016/j.neuint.2020.104859
Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: Treatment options and management of prolonged seizures. Epilepsia. 2019;60:S39–s48. https://doi.org/10.1111/epi.16334
Striano P, Coppola A, Pezzella M, Ciampa C, Specchio N, Ragona F, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology. 2007;69:250–4. https://doi.org/10.1212/01.wnl.0000265222.24102.db
Tanabe T, Awaya Y, Matsuishi T, Iyoda K, Nagai T, Kurihara M, et al. Management of and prophylaxis against status epilepticus in children with severe myoclonic epilepsy in infancy (SMEI; Dravet syndrome)–a nationwide questionnaire survey in Japan. Brain Dev. 2008;30:629–35. https://doi.org/10.1016/j.braindev.2008.03.002
Lotte J, Haberlandt E, Neubauer B, Staudt M, Kluger GJ. Bromide in patients with SCN1A-mutations manifesting as Dravet syndrome. Neuropediatrics. 2012;43:17–21. https://doi.org/10.1055/s-0032-1307454
Chiron C. Stiripentol for the treatment of seizures associated with Dravet syndrome. Expert Rev Neurother. 2019;19:301–10. https://doi.org/10.1080/14737175.2019.1593142
Devinsky O, Patel AD, Thiele EA, Wong MH, Appleton R, Harden CL, et al. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90:e1204–e1211. https://doi.org/10.1212/wnl.0000000000005254
Cross JH, Galer BS, Gil-Nagel A, Devinsky O, Ceulemans B, Lagae L, et al. Impact of fenfluramine on the expected SUDEP mortality rates in patients with Dravet syndrome. Seizure. 2021;93:154–9. https://doi.org/10.1016/j.seizure.2021.10.024
Sourbron J, Smolders I, de Witte P, Lagae L. Pharmacological Analysis of the Anti-epileptic Mechanisms of Fenfluramine in scn1a Mutant Zebrafish. Front Pharmacol. 2017;8:191 https://doi.org/10.3389/fphar.2017.00191
Odi R, Invernizzi RW, Gallily T, Bialer M, Perucca E. Fenfluramine repurposing from weight loss to epilepsy: what we do and do not know. Pharmacol Ther. 2021;226:107866. https://doi.org/10.1016/j.pharmthera.2021.107866
Bialer M, Perucca E. Lorcaserin for Dravet syndrome: a potential advance over fenfluramine? CNS Drugs. 2022;36:113–22. https://doi.org/10.1007/s40263-022-00896-3
Koike T, Yoshikawa M, Ando HK, Farnaby W, Nishi T, Watanabe E, et al. Discovery of soticlestat, a potent and selective inhibitor for cholesterol 24-hydroxylase (CH24H). J Med Chem. 2021;64:12228–44. https://doi.org/10.1021/acs.jmedchem.1c00864
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81974205 and 81771403) to HHZ; by the Natural Science Foundation of Zhejiang Province (LZ25H090003 to HHZ; LQN25H090016 to Yuling Wang); by the Program of New Century 131 Outstanding Young Talent Plan Top-level of Hang Zhou to HHZ; and by the Construction Fund of Key Medical Disciplines of Hangzhou.
Author information
Authors and Affiliations
Contributions
HHZ and WHS designed and conceptualized this review. WHS wrote the original and revised drafts, drew the figures, and reviewed and edited the original and revised manuscripts. LL and JXG drew the figures and reviewed and edited the original and revised manuscripts. YY, YXW and ZYZ reviewed and edited the original and revised manuscripts. QX, YLW, YS, LYG, and YC reviewed the manuscript and participated in the revisions. All authors contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shao, W., Liu, L., Gu, J. et al. Spotlight on mechanism of sudden unexpected death in epilepsy in Dravet syndrome. Transl Psychiatry 15, 84 (2025). https://doi.org/10.1038/s41398-025-03304-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-025-03304-8
This article is cited by
-
Electrocardiographic abnormalities in epilepsy: analysis of cardiac conduction patterns and SUDEP Risk
Neurological Sciences (2025)
