
Abstract
Inflammation is associated with a range of neuropsychiatric symptoms, but the issue of causality remains unclear. We used complementary non-genetic, genetic risk score (GRS), and Mendelian randomization (MR) analyses to examine whether inflammatory markers are associated with affect, depressive and anxiety disorders, and cognition. We tested in ≈55,098 (59% female) individuals from the Dutch Lifelines cohort the concurrent/prospective associations of C-reactive protein (CRP) with: depressive and anxiety disorders; positive/negative affect; and attention, psychomotor speed, episodic memory, and executive functioning at baseline and a follow-up assessment occurring 3.91 years later (SD = 1.21). Additionally, we examined the association between inflammatory GRSs (CRP, interleukin-6 [IL-6], IL-6 receptor [IL-6R and soluble IL-6R (sIL-6R)], glycoprotein acetyls [GlycA]) on these same outcomes (Nmin = 35,300; Nmax = 57,946), followed by MR analysis examining evidence of causality of CRP on outcomes (Nmin=22,154; Nmax = 23,268). In non-genetic analyses, higher CRP was associated with depressive disorder, lower positive/higher negative affect, and worse executive function, attention, and psychomotor speed after adjusting for potential confounders. In genetic analyses, CRPGRS was associated with any anxiety disorder (β = 0.002, p = 0.037) whereas GlycAGRS was associated with major depressive disorder (β = 0.001, p = 0.036). Both CRPGRS (β = 0.006, p = 0.035) and GlycAGRS (β = 0.006, p = 0.049) were associated with greater negative affect. Inflammatory GRSs were not associated with cognition, except sIL-6RGRS which was associated with poorer memory (β = −0.009, p = 0.018). There was a non-significant CRP-anxiety association using MR (β = 0.12; p = 0.054). Genetic and non-genetic analyses provide consistent evidence for an association between CRP and negative affect. These results suggest that inflammation may impact a broad range of trans-diagnostic affective symptoms.
Similar content being viewed by others


Introduction
Depression is the leading cause of mental health-related global disease burden [1, 2]. Persistent cognitive problems, such as poor memory and concentration, are reported in 11% of adults aged ≥45 years [3] and are frequently observed across physical [cancer (35%); COVID-19 (22%); HIV (43%); hepatitis C (50%)] [4,5,6,7] and mental health conditions [depression (30%); schizophrenia (50%)] [8, 9]. Existing treatments for depression are only modestly effective [10] and almost inexistent for cognitive dysfunction [11]. A mechanistic understanding of depression and cognitive dysfunction is urgently needed to inform the development of effective treatments and prevention approaches.
Chronic, low-grade systemic inflammation may represent one such mechanism. Indices of inflammation [e.g., circulating levels of cytokines (e.g., interleukin-6 (IL-6) and acute phase proteins (e.g., C-reactive protein (CRP)] are elevated in individuals with depression compared to controls [12] and inflammatory biomarkers have been linked to specific aspects of depression, such as anhedonia and negative affect [13, 14]. Further, longitudinal observational studies have found that higher levels of inflammatory biomarkers (e.g., IL-6, CRP) are prospectively associated with higher depressive symptoms [15]. Observational studies have linked inflammation with impaired cognition in population-based [16,17,18,19] and in physical [20,21,22] and mental health conditions [23,24,25,26]. Inflammation also impacts neural circuitry relevant to affective disorder and cognitive task performance [27, 28], particularly the hippocampus [29] and striatum [30,31,32]. To date, inflammation-cognition research has primarily relied upon observational data.
Inferring causality from observational studies is a challenge due to confounding (e.g., stress, poor sleep [22]) and reverse causality (i.e., whether inflammation impacts depression/cognition, or vice versa). Mendelian randomization (MR) is a genetic epidemiological method that can assess evidence of causality by using genetic variants associated with an exposure (e.g., inflammatory protein) as proxies for the exposure [33], given key assumptions are met (see Methods). As genetic variants are randomly inherited from parents to offspring and are fixed at conception, they are less likely to be associated with confounders and overcome issues of reverse causation [33]. MR is analogous to an RCT, providing stronger evidence suggestive of causality compared to standard observational analyses (see Supplementary Figure 1). Preliminary evidence, using MR, implicate IL-6 and its soluble IL-6 receptor (sIL-6R) in depression [34,35,36,37]. To date, most MR studies examining the effect of IL-6 on health have focused on circulating IL-6 levels. However, IL-6 signals via multiple pathways (trans-signaling, classical-signaling, and trans-presentation) and there is growing evidence that IL-6 trans-signaling is primarily responsible for the pathogenic inflammatory effects of IL-6 [38]. Here, we include variants related to circulating IL-6 levels, and sIL-6R levels (relevant for IL-6 trans-signaling). Causal evidence for CRP and other proinflammatory markers [i.e., Glycoprotein Acetyls (GlycA): a composite biomarker thought to provide a more stable marker of inflammation which reflects the glycosylation of multiple acute-phase proteins [39]] on depression are mixed [34, 37, 40,41,42,43]. Regarding cognition, few studies have examined potential causal relationships with inflammation. MR analyses using available genome-wide association studies (GWAS) report both null results of inflammatory biomarkers on emotion recognition, working memory, response inhibition [44] as well as effects of specific cytokines/chemokines (i.e., Eotaxin, IL-8) on fluid intelligence [45].
The current study used data from the Lifelines Cohort Study – a large population-based cohort in the Netherlands – to conduct complementary non-genetic and genetic analysis to investigate the potential causal effect of inflammation on negative affect, depressive/anxiety disorders, and cognitive task performance. We triangulate findings from different methods (non-genetic and genetic) which have distinct strengths and sources of potential bias [46]. If we observe consistent findings across these different methods (i.e., results point to the same conclusion), we can be more confident in the conclusions we draw as to the causal nature of the relationship between specific immune biomarkers and depression [46]. If we do not observe consistent findings, then we must be more cautious in our conclusions and discrepant findings may help us understand the sources of bias and how they may be influencing our results [46]. For instance, a significant association of CRP and depression based on observational data coupled with no significant link based on MR analyses would indicate that CRP is not causally linked to depression and is a by-product of the true causal process, whether immunological, biological, cognitive or behavioral in nature. Inconsistent findings across MR and non-genetic analyses would decrease confidence in conclusions. For example, evidence in MR analyses (but not non-genetic analyses) could indicate that a false negative had been observed for non-genetic analyses or a false positive had been observed for MR analyses (e.g., potentially due to violation of an MR assumption). Further investigation of potential discrepancies between MR and non-genetic analyses would be warranted (for example, MR focuses on lifetime effect versus non-genetic analysis focuses on specific time periods; there may be differences in populations examined in non-genetic versus MR analyses such as ancestry, age, sex). See Munafo et al. (2021) for more details on triangulation [46].
First, we used cross-sectional and longitudinal non-genetic analysis to examine the association between circulating levels of CRP and depression/cognitive performance. Second, we conducted genetic risk score (GRS) and MR analysis to test whether genetic variants regulating levels and activity of CRP, IL-6, and GlycA were causally related with depression/cognitive performance. We also conducted the above analyses on closely related constructs (e.g., anxiety, negative/positive affect), for which associations with inflammation have previously been observed [47,48,49,50,51,52] but for which considerably less empirical data has been published. We hypothesized that higher levels of both circulating CRP levels and genetically predicted inflammatory biomarkers (i.e., CRP, IL-6, sIL-6R, and GlycA) would be associated with more severe depressive and anxious symptoms, higher levels of negative affect, and worse cognitive task performance.
Methods and materials
Participants
Lifelines is a multi-disciplinary prospective population-based cohort study examining in a unique three-generation design the health and health-related behaviors of 167,729 persons living in the North of the Netherlands. It employs a broad range of investigative procedures in assessing the biomedical, socio-demographic, behavioral, physical and psychological factors which contribute to the health and disease of the general population, with a special focus on multi-morbidity and complex genetics [53]. This cohort has previously been described in detail [53] and further information on the types of data collected (https://data-catalogue.lifelines.nl/) and when assessment occurred (https://wiki.lifelines.nl/doku.php?id=general_assessments) are available online. In brief, both the baseline and follow-up visits were divided into two parts occurring two weeks apart. For both baseline and follow-up visits, diagnostic interviewing and cognitive assessment occurred in the first part and blood draw occurred in the second part two weeks later. The follow-up assessment occurred, on average, 3.91 years later (SD = 1.23).
In brief, participants were recruited via their general practitioner (49%), participating family members (38%), and self-registration on the Lifelines website (13%). Exclusion criteria for recruitment through the general practitioner included: insufficient knowledge of Dutch language, severe psychiatric or physical illness, limited life expectancy ( < 5 years). Baseline data included approximately: 140,000 adults (18–65 years), 15,000 children (0–17 years), 12,000 elderly individuals (65+ years). Following baseline assessment, participants are invited to complete an in-person study visit every 5 years. Phenotypic and genotypic data are collected by Lifelines to permit investigation on determinants of health. Data for the current study were drawn from 147,815 individuals who were aged 18+ years at baseline and who did not report a diagnosis that typically impairs cognitive function, specifically Alzheimer’s disease, other dementias, epilepsy, multiple sclerosis, Parkinson’s disease, and stroke. In the non-genetic analyses, the analytic sample is smaller as CRP was assessed in a sub-sample of individuals (N = 55,098) as was baseline cognitive performance on the Ruff Figural Fluency Test (N = 88,096). The analytic sample is smaller for non-genetic (N ≤ 55,098), GRS (N ≤ 57,946) and MR (N ≤ 23,268) analysis as only a subset of participants who met inclusion criteria had outcome data and CRP data (non-genetic analysis), genetic data (GRS), or genetic and CRP data (MR) due to time and cost constraints. Phenotypic data were drawn from both the baseline assessment and the first follow-up assessment; whether a specific measure was assessed at baseline, first follow-up or both assessments is noted for each measure.
Ethics approval and consent to participate
The Lifelines study was approved by the ethics committee of the University Medical Center Groningen, document number METC UMCG METc 2007/152. Informed consent was obtained from all individuals included in the study. All methods were performed in accordance with relevant guidelines and regulations.
Measures
Cognition
Ruff figural fluency test (baseline assessment)
The Ruff Figural Fluency Test (RFFT) is a reliable and valid measure of figural fluency, a dimension of executive function [54]. Participants were asked to draw as many unique designs as possible within 60 s by connecting dots in different patterns. The task is composed of five parts, with each part containing 35 identical five-dot patterns (with or without distractors). The total number of unique designs was used as the dependent variable in the analyses, consistent with previous studies [55]. In Lifelines, the RFFT was administered to all participants until April 2012, and subsequently in a random half of the sample. Data from participants who failed to generate a single unique design per trial (n = 181) were deemed invalid and removed.
Cogstate test battery (first follow-up assessment)
The Cogstate Test Battery took approximately 10–15 min to complete and consisted of four tasks: detection task (psychomotor speed), identification task (attention), one-back task (working memory), and one card learning task (episodic memory). For each task, outcomes recommended by Cogstate were selected, specifically: log10 transformed response time in millisecond (detection/identification tasks) and arcsine-transformed response accuracy (one-back/one card learning tasks). For the detection and identification tasks, higher values reflect poorer performance and for the one-back and one card learning tasks, higher values equal better performance. Data cleaning involved excluding participants with a high number of errors. The percentage of successful trials per Cogstate task was high, averaging 66% (n = 85,050; SD = 0.11) on the episodic memory task, 91% (n = 85,053; SD = 0.17) on the visual attention task, 92% (n = 85,053; SD = 0.20) on the psychomotor speed task, and 90% (n = 85,051; SD = 0.15) on the working memory task. A small number of participants exhibiting implausibly low accuracy rates indicative of poor effort, failure to comprehend task instructions, or technical errors were excluded from analyses. Specifically individuals with an accuracy rate less than: 25% on the episodic memory task (n = 231), 40% on the visual attention task (n = 2,878), 45% on the psychomotor speed task (n = 3,914), and 35% on the working memory task (n = 1,330). For more details on the Cogstate Test Battery, see Supplementary Methods 1.1.
Clinical assessments
Anxiety and depressive disorders (baseline/first follow-up assessments)
The Mini International Neuropsychiatric Interview – Simplified (MINI) is a reliable, valid, and brief structured interview that was designed to screen for psychiatric disorders [56,57,58]. Lifelines used an adapted version of a Dutch translation of the MINI that was administered by trained interviewers at baseline and self-administered on location at follow-up – details on the version used in Lifelines have previously been published [59]. Participants were considered to meet criteria for any depressive disorder if they met the Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria for Major Depressive Disorder (MDD) or dysthymia at the time of the interview. Impairment was assessed in the MINI for dysthymia but not depression and consequently, impairment was not used as a criterion for MDD. Any anxiety disorder refers to meeting current criteria for any one of the following conditions that was assessed using the MINI: panic disorder, agoraphobia, social phobia, or Generalized Anxiety Disorder (GAD). We used four diagnostic groups as outcome variables: MDD, any depressive disorder, GAD, and any anxiety disorder.
Positive and negative affect schedule (baseline assessment)
The Positive and Negative Affect Schedule (PANAS) assesses positive and negative affect [60] using two 10-item subscales (examples of items include ‘excited’ on positive subscale; ‘upset’ on negative subscale). Participants are asked to rate the extent that they experienced each item during the last four weeks on a five-point scale (ranging from ‘not at all’ to ‘extremely’). The outcome is the summed score on each subscale, which ranges from 10 to 50 (higher value reflects higher positive or negative affect, respectively). The PANAS has been shown to be reliable and valid [61]. Reliability, as estimated using Cronbach’s alpha, for the positive subscale of the PANAS at baseline was 0.79 and 0.84 for the negative subscale.
C-reactive protein (baseline assessment)
Participants gave blood samples before 10AM via venipuncture following an overnight fast. Complete details on blood specimen data collection have previously been reported [53, 55]. Due to assay costs, CRP was assessed in approximately 35% of Lifelines participants and data were available for 55,098 individuals in the analytic sample. CRP was quantified using three separate methods over the course of baseline assessment (Method 1: 12.90% of total CRP values assessed in serum; CardioPhase hsCRP; Method 2: 84.58% of total CRP values, assessed in plasma; CardioPhase high sensitivity (hs)CRP, Siemens Healthcare Diagnostics, Marburg, Germany; Method 3: 2.52% of total CRP values; assessed in plasma; CRPL3, Roche Diagnostics, Mannheim, Germany). Assay methods 2 and 3 were identical and only differed in terms of the manufacturer. A conversion formula (new = 0.92 x old - 0.01) was derived from an internal validation using 39 samples, according to the AMC (alternative method comparison, Deming Regression) protocol in order that Method 1 could be compared with Method 2 and 3 [55]. For CardioPhase hsCRP, the intra-assay coefficient of variability was 3.45% and the inter-assay coefficient of variability was 3.15%. For CRPL3, the intra-assay coefficient of variability was 4.15% and the inter-assay coefficient of variability was 5.8%.
Genetic data
Genotype data were available for a subgroup of participants in Lifelines. Genotyping was conducted using three chip arrays: Illumina CytoSNP-12 Bead Chip v2 array (N = 17,033), Infinium Global Screening Array (GSA) Beadchip-24 v1.0 (N = 38,030), FinnGen Thermo Fisher Axiom ® custom array (Affymetrix; N = 29,166). For details on quality checks (QC’s) and imputation conducted by Lifelines, see Supplementary Methods 1.2. Following Lifelines QC’s, the total sample size for participants who met criteria for this study: CytoSNP (N = 14,942), GSA (N = 31,810) and Affymetrix (N = 26,334). We applied additional QC’s which included removing: one of the duplicates (individuals who were genotyped on more than one chip) and first-degree relatives between chips, non-European individuals (identified by Lifelines), and genetic outliers (identified by Lifelines); see Supplementary Figure 2 for more details. This resulted in a total of 58,713 participants with genetics data included in this study (CytoSNP N = 7,632; GSA N = 24,975; Affymetrix N = 26,106). For more details on the genetic data in Lifelines, see Supplementary Methods 1.2.
Covariates
Covariates included age, sex, educational attainment, body mass index (BMI) and health status. Age, sex, and educational attainment were self-reported by participants. Educational attainment was determined using a single-item question and was categorized by Lifelines as: low [no education, primary education, lower/preparatory vocational education, lower general secondary education (leaving secondary school aged >16 years)], moderate (intermediate vocational education/apprenticeship, higher secondary education), and high (higher vocational education, university). We recoded educational attainment so that higher values represent lower educational attainment. To estimate body mass index (BMI), height was measured to the closest 0.1 cm and body weight was measured without shoes to 0.1 kg precision. For health status, a composite measure was created counting several self-reported chronic medical conditions related to increased levels of inflammation (i.e., arthritis, asthma, coeliac disease, Crohn’s disease, diabetes, and psoriasis); we then categorized participants into those with no relevant chronic medical condition, 1, 2 or 3+ conditions.
Analyses
Analyses were conducted in R version 4.1.1.
Non-genetic analyses
Multivariable linear and logistic regression models were estimated using base functions in R (i.e., ‘lm’, ‘glm’). Assumptions underpinning regression analyses were investigated using ‘broom’ and ‘ggfortify’ packages [62]. Models using CRP on its naturally occurring scale (mg/L in this study) demonstrated deviations in the assumptions of (i) a linear relationship between the predictor and outcome variables and (ii) homogeneity of variance of the residuals that were all substantially improved when CRP was log-transformed; thus, CRP was transformed by natural log for all analyses.
Genetic risk scores
Genetic risk scores (GRS) were calculated to determine whether GRS for inflammatory markers (CRP, IL-6, IL-6R, sIL-6R, GlycA) were associated with depression/anxiety, affect and cognitive outcomes. To create GRS for each inflammatory marker, we identified genetic variants (single nucleotide polymorphisms [SNPs]) associated with these proteins in large available GWAS or using SNP lists from previous publications, see Supplementary Table 1. For details on the GWAS used and accessing summary statistics, please see Supplementary Methods 1.3 and Supplementary Tables 2, 3. The following criteria were used to identify SNPs from GWAS for each inflammatory marker: p-value < 5 × 10−8, linkage disequilibrium clumping (r2 = 0.01, kb = 1000 based on the European-clustering individuals in the 1000 genomes reference panel) using ld_clump() [63] in the ieugwasr package, minor allele frequency >0.01. In the primary analysis, we restricted the SNP set to cis variants (SNPs +/−1Mb from protein coding gene based on Genome Reference Consortium Human Build used in the GWAS) [64,65,66]. The reason for restricting to cis variants in the primary analysis is because, due to their proximity to the protein coding gene, they are more likely to be valid instruments, as they are more likely to influence mRNA expression and protein levels (thus being less pleiotropic) [67]. For GlycA, which does not have a single coding gene due to its composite nature, we used the largest available GWAS in our primary analysis. In our secondary analyses, we used both cis and trans variants from GWAS (i.e., we did not restrict to cis variants). Each SNP list was used to create a weighted GRS for each Lifelines participant. Specifically, the risk alleles were weighted by the effect size (beta) reported in the GWAS/previous study and then summed to provide a risk score. Any SNP identified in GWAS/previous study that was not available in Lifelines was replaced with a proxy (where possible) that had r2 > 0.8 (using LDproxy_batch function in EUR population in R), rsID (SNP name) available, SNP available in full summary statistic GWAS, and in Lifelines [68, 69]. GRS were created in Plink v1.90 [70] and continuous phenotypes were standardized within each chip (z-scored) for direct comparison (CRP levels were log transformed but not standardized). To adjust for relatedness within each chip, two approaches were taken. The primary approach applied the GRAMMAR method [71] and the secondary approach involved re-running analyses removing close relatives (up to first-degree, up to second-degree, and up to third-degree), see Supplementary Materials 1.4 and 1.5 for details. We then ran regression models predicting each outcome using the standardized GRS, including top 10 genetic PCs (calculated on merged Lifelines genotype data), age, sex, and chip. Maximum sample size for analyses: no relatives within chips removed (N = 58,713), up to first-degree removed (N = 50,955), up to second-degree removed (N = 50,255), up to third-degree removed (N = 48,880). Unadjusted analyses are also reported in the Supplementary Tables 6 and 7.
Mendelian randomization
To conduct MR using individual level data and two-stage least squares regression, genetic data, exposure data, and outcome data are required. As only CRP is available within the Lifelines cohort (IL-6 and GlycA are not currently available), only this inflammatory marker could be assessed in the MR analysis. Where there was evidence of associations between CRP GRS and outcomes, we followed this up with MR to assess potential causality. Two-stage least squares regressions were conducted using the AER package [72]. Analyses were GRAMMAR adjusted for relatedness and all regression models adjusted for age, sex, and chip. Three assumptions are necessary for valid inferences from MR: (i) the genetic variants are robustly associated with the exposure, (ii) there is no confounding of the genetic variants and the outcome and (iii) the genetic variants are independent of the outcome given the exposure. Only the first assumption can be formally tested (typically using the F-statistic of the association between the genetic instrument and the exposure). Whilst the other two assumptions cannot be proven to be true, they can be disproven [73]. Please see for a primer on MR [73]. To test the first MR assumption, we ran linear regressions to check whether CRP GRS were associated with circulating CRP levels in participants with both genetic and CRP data available (n = 23,607) using the GRAMMAR method. We also checked whether any inflammatory marker GRS were associated with potential confounders (BMI, current smoking status, educational attainment; all models were adjusted for age, sex, and chip).
Results
The characteristics of the Lifelines cohort sample are described in Table 1 and Pearson correlations between study variables are presented in Table 2.
Association of CRP with affect, depressive and anxiety disorders, and cognition
The association of (log-transformed) CRP with: clinical outcomes (i.e., MDD, any depressive disorder, GAD, any anxiety disorder), positive and negative affect, and five cognitive measures [RFFT (executive functioning), detection task (psychomotor speed), identification task (attention), one-back task (working memory), and one card learning task (episodic memory)] are illustrated in Table 3, both unadjusted and adjusted for covariates. Notably, CRP was associated with a greater likelihood of meeting criteria for a range of clinical outcomes, with a numerically greater likelihood consistently reported for depression as compared to anxiety at baseline and first follow-up assessment. However, the magnitude of these associations were attenuated after controlling for confounding by age, sex, education, health status, and BMI. Higher CRP was also associated with higher negative affect, lower positive affect, and worse cognitive task performance, although the magnitude of associations was generally very small and negligible after controlling for covariates.
Associations of GRSs for inflammatory markers with affect, depressive and anxiety disorders
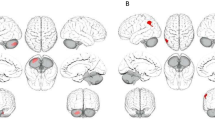
In the primary analysis, CRPGRS (cis) was associated with a higher negative affect score (beta: 0.006; 95% CI: 0.0005 to 0.012, p = 0.035, N = 57,946) and increased risk of any anxiety disorder (beta: 0.002, 95% CI: 0.0001 to 0.004, p = 0.037, N = 57,047). GlycAGRS was associated with higher negative affect score (beta: 0.006, 95% CI: 0.00002 to 0.012, p = 0.049; N = 57,946) and increased risk of MDD (beta: 0.001, 95% CI: 0.0001 to 0.002; p = 0.036; N = 57,047). Other inflammatory marker GRSs were not associated with depressive/anxiety disorders or affect scores (ps ≥ 0.15). In the secondary analysis, there was evidence that CRPGRS (genome-wide) was associated with increased risk of any anxiety disorders (beta: 0.002, 95% CI: 0.0003 to 0.004, p = 0.023, N = 57,047). Other inflammatory marker GRSs were non-significantly associated with depressive/anxiety disorders or affect (ps ≥ 0.16). For all results, see Fig. 1 and Supplementary Table 5. All sensitivity analyses removing differing degrees of related individuals (up to 1st-degree, up to 2nd-degree, up to 3rd-degree) within chips (non-GRAMMAR method) did not substantially alter results, see Supplementary Tables 8–13. However, there was slightly stronger evidence for associations between GlycAGRS and negative affect score (ps ≤ 0.015) and between CRPGRS (cis) and negative affect score (ps ≤ 0.033).

Associations of genetic risk scores for inflammatory markers with mood, anxiety disorders and positive and negative affect scores.
Association of GRS for inflammatory markers and cognition
In primary analyses, inflammatory marker GRSs were not associated with performance on cognitive tasks (ps ≥ 0.14), except for sIL-6RGRS which was negatively associated with episodic memory performance (one card learning task accuracy; beta: −0.009, 95% CI: −0.017 to −0.002, p = 0.018, N = 36,783). In secondary analyses, inflammatory markers GRSs (genome-wide) were not associated with performance on cognitive tasks (ps ≥ 0.22). For all results, see Fig. 2 and Supplementary Table 5. Sensitivity analyses after removing related individuals within chips (non-GRAMMAR method) did not alter the results, see Supplementary Tables 11–13.

Associations of genetic risk scores for inflammatory markers with cognitive task performance.
Testing potential causality between CRP, negative affect, and anxiety disorders using Mendelian randomization with individual level data
CRP genetic instruments had F-statistics >10 (158 for cis GRS, 1045 for genome-wide GRS), indicating adequate instrument strength [74]. For tests on the MR assumptions, see Supplementary Results 2.2. Genetically-proxied CRP (cis) was non-significantly associated with a causally increased risk of any anxiety disorders (beta: 0.12, p = 0.054, N = 22,154) and negative affect (beta: 0.27, p = 0.16; N = 23,268). Sensitivity analysis removing related individuals did not alter overall conclusions.
The overall pattern of results for the non-genetic and genetic analyses are visualized in Fig. 3.

Arrows indicate direction of effect. Solid arrows (p < 0.05); dashed arrows (p > 0.05). Positive arrows = increased liability to mood and anxiety disorders/affect and better cognitive performance, except for reaction time (RT) measures where positive score = higher RT (i.e., slower response). All psychiatric disorders are from the baseline assessment where sample size is largest. Non-genetic results = adjusted analyses.
Discussion
We conducted complementary non-genetic and genetic analyses to interrogate the relationship between inflammatory markers and affect, depressive and anxiety disorders, and cognitive task performance using data from the Lifelines cohort. In non-genetic analyses, higher CRP was associated with diagnosis of any depressive disorder, positive and negative affect scores, figural fluency, attention, and psychomotor speed after adjusting for potential confounders, although the magnitude of these associations was generally small. In genetic analyses, genetic risk scores for CRP (CRPGRS) and GlycAGRS were both associated with higher negative affect score. CRPGRS was associated with any anxiety disorder whereas GlycAGRS was associated with major depressive disorder. Inflammatory marker GRSs were not associated with cognitive task performance, except for soluble IL-6RGRS which was associated with poorer memory performance. Individual level MR was numerically associated with a causal effect of CRP on any anxiety disorder. Genetic and non-genetic analyses provided consistent evidence for an association, albeit small, of CRP on negative affect. Genetic analyses suggest that IL-6 signaling could be relevant for memory, and that the association between CRP and anxiety disorders could be potentially causal.
Affect
Inflammation has generally been associated with higher levels of negative affect and lower levels of positive affect, although findings are primarily based on medical populations [75,76,77] and small community samples [13, 78]. To our knowledge, this is the first large, population-based study to find small but consistent associations of higher CRP with higher negative affect and lower positive affect, both unadjusted and adjusted for potential confounds. Interestingly, both CRP and GlycA genetic risk scores were associated with higher levels of negative affect, but not positive affect. This consistent association across non-genetic and genetic analyses may reflect the effect of inflammation on a range of emotional states beyond the cardinal features of depression (i.e., sadness/anhedonia), which aligns with prior research linking inflammation with fear and irritability [52, 79]. Prior work has shown that inflammation is differentially associated with a specific clinical presentation characterized by anhedonia and somatic/neurovegetative symptoms (e.g., fatigue, altered sleep and appetite changes) and further work is needed that more accurately characterize an inflammatory phenotype in depression [80, 81].
Depression
These data add to a growing body of evidence evaluating the role of inflammation in the etiology of depression. The results of non-genetic analyses broadly aligns with results from the UK Biobank cohort in terms of (i) prevalence estimates of depression and anxiety, (ii) robust univariate associations between CRP and depression and anxiety, which were generally no longer statistically significant when controlling for covariates, and (iii) stronger univariate associations for CRP and depression when compared to anxiety [37]. Observational and experimental research studies investigating the association of inflammation and depression have produced mixed results [15, 82,83,84,85] and the cause of discrepant results remains unclear. On one hand, it may be that inflammation is associated with depression only in specific populations (e.g., treatment-resistant depression) [86] or with specific clinical presentations (e.g., somatic symptoms) [87]. As such, discrepant results may be due to individuals recruited to a given study or the measure of depression being used (e.g., the Hamilton Depression Rating Scale is more likely to detect somatic symptoms of depression than, for example, the PROMIS self-rating questionnaire). On the other, an association of inflammation and depression may merely be a by-product of true causal processes in depression, such as adiposity [88]. Indeed, the association of CRP and depression was substantially attenuated in this study, as well as many others, following adjustment for potential confounders [89]. What remains unclear is interpretation of attenuated results following statistical adjustment of potential confounding factors (e.g., BMI) that may be key mechanisms underlying the pathophysiology of inflammatory depression [26]. As such, attenuation of associations following adjustment for covariates would not, by itself, indicate a non-causal relationship and, in fact, recent research using directed acyclic graphs and a combination of empirical and simulated data has found that controlling for adiposity reduced precision of the association of inflammation and depression, cautioning researchers against controlling for adiposity when modeling the relationship between inflammation and depression [90]. Indeed, inflammation may increase risk for depression via increasing the risk of inflammation-related physical multimorbidity (e.g., cardiovascular disease) [34] – a hypothesis that requires further investigation.
In genetic analyses, there was also little evidence of an association between CRPGRS and depression outcomes, although there was evidence suggesting GlycAGRS increases liability to MDD. The null CRP findings are consistent with previous MR studies showing no evidence of an effect in MDD [42, 43, 91]. However, the MR literature of CRP on depression is mixed with some studies reporting CRP to decrease [37] or increase [34] risk for depression. It is unclear what accounts for these mixed findings, but potential factors may include CRP SNP selection, definition and/or measurement of depression, statistical power, and selection bias (see Supplementary Discussion). In contrast, MR studies have shown more consistent findings for the potential causal role of IL-6 on depression [34,35,36,37]. This is similar to MR findings for coronary heart disease, where IL-6 but not CRP have been shown to play a potential causal role [92, 93].Studies on a broader range of immune markers (e.g. cytokines, immune cells) and specific immune pathways may be more useful to understand the role of inflammation in depression, rather than CRP which is a non-specific marker of systemic immune activation [94]. When considered alongside the results of observational studies, it may be that CRP itself does not play a causal role in the etiology of depression and that it may be a by-product of other processes playing a true causal role in depression, such as the activation and trafficking of monocytes to the central nervous system, metabolic dysregulation, stress reactivity, substance use or adiposity [81, 95].
Cognition
We observed relatively small effects of CRP on cognitive task performance, and in genetic analysis only sIL-6RGRS was associated with poor memory performance. Our findings contribute to inconsistent findings across population-based cohorts assessing circulating inflammatory biomarkers and cognitive performance where associations observed in population-based studies [25] often are not large in magnitude or consistently observed [44]. Few MR studies have been conducted on the role of inflammation on cognition. Consistent with results presented here, our previous MR study examining the role of the same inflammatory markers (i.e., CRP, IL-6, IL-6R, sIL-6R, GlycA) on specific executive functions within the ALSPAC cohort (e.g., emotion recognition, working memory, response inhibition [44]) found little evidence of a potential causal effect. However, Pagoni et al. recently reported that other cytokines and chemokines (i.e., Eotaxin, IL-8, MCP1) may be causally related to lower fluid intelligence (and IL-4 with higher fluid intelligence) [45]. The finding regarding sIL-6R and memory performance is novel and would align with convergent evidence that trans-signaling – in which sIL-6R plays a critical role – may be responsible for the deleterious effect of IL-6 on cognitive functioning [38, 96]. Central IL-6 signaling plays an important and complex role in functions critical to human memory, such as: long-term potentiation, neurogenesis and neuroplasticity [97]. It is worth noting that this relationship is complex and is likely beneficial when IL-6 signaling occurs within normative ranges, but deleterious when substantially elevated (see Yirmiya & Goshen, 2011 for excellent review). Indeed, circulating IL-6 has previously been associated with worse episodic memory in humans [26, 98]. Given the distal and far-reaching impact of trans- IL-6 signaling when compared with the proximal and circumscribed impact of classic IL-6 signaling [96], these results (if replicated) may help explain variability observed in the relationship between IL-6 signaling and memory.
Interpreting the relationship between inflammation and cognitive task performance in population-based studies is difficult for several reasons. First, there is considerable heterogeneity in the cognitive abilities assessed across studies and there is a need for future studies to more uniformly include well-validated measures assessing individual differences, rather than detecting pathological states (e.g., dementia, epilepsy)] [99]. There is a similar need to evaluate the impact that inflammation has on other psychological functions that impact cognition (e.g., reward process, aversive process) – there is strong theoretical work and empirical data to support an indirect effect of inflammation on cognition via, for instance, dysregulated reward circuitry that impact performance on cognitive tasks via decreased motivation or increased fatigue [100]. Moreover, there are a range of sociodemographic factors that may moderate the association between inflammation and cognition – prior work has found that inflammation and cognition may differ based on age and sex [101, 102]. It is reasonable to assume, for instance, that modest increases in inflammation may exert a cumulative effect across the lifespan, and thus may only be detected later in life and/or in specific domains of cognition.
Anxiety
In the non-genetic analyses, circulating CRP levels was associated with a modestly increased likelihood of meeting criteria for anxiety disorders, although this association was substantially attenuated following adjustments for covariates. Prior research in population-based cohorts have found CRP to be associated with an increased risk for anxiety disorders [103, 104], although results are inconsistent and other studies indicate that anxiety prospectively predicts an increase in circulating CRP levels [105]. The MR analysis may be indicative of a potentially causal role of CRP on the likelihood of meeting criteria for an anxiety disorder (which covers a broad range of anxiety-related conditions including panic disorder, social phobia, agoraphobia, GAD); however, it should be noted the p value (p = 0.054) did not meet a threshold for statistical significance and therefore is in particular need of replication in a adequately powered research study. Prior theory has primarily focused on anxiety as a cause of inflammation [106]; however, alternative theories suggest that inflammatory physiology is implicated in both sickness behaviors (e.g., anhedonia, social withdrawal) and anxiety arousal and alarm [107], which would align with the results presented here. That the association of anxiety with CRP is weaker when compared to depression and significantly attenuated following adjustments for covariates may be indicative of, for example, the relatively high levels of comorbid depression and anxiety (i.e., higher CRP in anxiety is confined primarily to anxious individuals with depression) or that the proportion of individuals with inflammatory anxiety is even smaller than the ~25% of depressed individuals with an inflammatory phenotype [108].
Limitations
This study used a large and population-based sample that employed triangulation of genetic and non-genetic analyses, which increases confidence in the inferences drawn. Limitations, however, exist. First, although broadly representative, like other cohort studies (e.g., UK Biobank), the Lifelines cohort predominantly includes individuals of European descent and is less representative of individuals from low socioeconomic status [109], which consequentially limits the generalizability of findings. Second, analyses were not corrected for multiple comparisons and therefore our findings need to be replicated and validated using external data. Moreover, as effect sizes reported are small and reflect associations in the general population, there is a need for studies to investigate whether there are sub-groups for whom these associations may be larger (e.g., older age, clinical populations). Third, in the genetic analysis the CRP GRS explained 1–4% of the variance in CRP [a level of variance consistent with similar analyses in the ALSPAC cohort [44]] and few cases of depression were observed in Lifelines [although the point prevalence of approximately 4% is consistent with reported population point prevalence estimates [1]]. It is possible that this limited our capacity to detect potential causal effects, were they small in magnitude or non-linear. Fourth, the CogState tasks used in the current study may not be optimal for detecting individual differences in healthy individuals, or even deficits in some conditions such as depression; multiple studies have shown that the CogState tasks used in this study do not improve in successful antidepressant trials, even when improvement in other cognitive measures are observed [110,111,112]. This is compounded by more general difficulties that exist in the very substantial variability in the types of cognitive tests used across studies. Aside from the very real and ongoing challenges that exist in developing reliable, valid, and specific measures of discrete cognitive functions, many tests have been developed to identify focal deficits in discrete patient populations as opposed to tests designed to capture individual variability within the general population – considerations that likely contribute greatly to variability in research findings. These considerations gather renewed importance as remote and passive cognitive measures are increasingly being developed. Fifth, although we include multiple instruments related to IL-6 (i.e., genetic variants related to IL-6 and sIL-6R levels), most instruments contain few genetic variants (≤3 SNPs) and genetic variants for IL-6 and sIL-6R overlap. While sIL-6R is involved in IL-6 trans-signaling, the overlap of SNPs makes it challenging to interpret the effect of these genetic variants on different immune phenotypes specifically (i.e., IL-6 levels vs IL-6 signaling). Future studies are needed to better understand the biological role of these genetic variants and develop instruments to proxy specific IL-6 signaling pathways including IL-6 trans-signaling. A further limitation of this study is that we did not apply Steiger Filtering to check whether genetic variants included in our immune marker GRS are more strongly associated with the immune marker than the outcome. Whilst this is less of a concern in primary analyses (using cis variants which are less likely to be pleiotropic than trans variants), it is a limitation in our secondary analyses (and analyses using GlycA for which there is no protein coding gene to enable cis analyses). Finally, it is worth considering that some instruments in the genetic analyses were associated with potential confounds.
Conclusions
Genetic and non-genetic analyses provide consistent evidence for a modest effect of CRP on negative affect. Genetic analyses suggest that IL-6 signaling could be relevant for memory, and that the association between CRP and anxiety disorders could be causal. Overall, these results suggest that inflammation may affect a range of emotional states beyond the cardinal features of depression. However, given the small effect sizes and multiple tests conducted, future studies should investigate whether effects are moderated by sub-groups and whether these findings replicate in other cohorts.
Data availability
Supplementary information is available at Translational Psychiatry’s website.
Code availability
All code for analyses will be made available at: https://github.com/chloeslaney.
References
Vos T, Lim SS, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204–22.
James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–858.
Taylor CA, Bouldin ED, McGuire LC. Subjective cognitive decline among adults aged ≥45 Years - United States, 2015–2016. MMWR Morb Mortal Wkly Rep. 2018;67:753–7.
Janelsins MC, Kesler SR, Ahles TA, Morrow GR. Prevalence, mechanisms, and management of cancer-related cognitive impairment. Int Rev Psychiatry. 2014;26:102–13.
Ceban F, Ling S, Lui LMW, Lee Y, Gill H, Teopiz KM, et al. Fatigue and cognitive impairment in Post-COVID-19 syndrome: a systematic review and meta-analysis. Brain Behav Immun. 2022;101:93–135.
Wang Y, Liu M, Lu Q, Farrell M, Lappin JM, Shi J, et al. Global prevalence and burden of HIV-associated neurocognitive disorder. A meta-analysis. Neurology. 2020;95:e2610–e2621.
Barreira DP, Marinho RT, Bicho M, Fialho R, Ouakinin SRS. Psychosocial and neurocognitive factors associated with hepatitis C - implications for future health and wellbeing. Front Psychol. 2018;9:2666.
Fava M, Mahableshwarkar AR, Jacobson W, Zhong W, Keefe RS, Olsen CK, et al. What is the overlap between subjective and objective cognitive impairments in MDD? Ann Clin Psychiatry. 2018;30:176–84.
Keefe RS & Harvey PD in Essentials of Schizophrenia 1st Ed (eds Lieberman JA, Stroup TS, & Perkins, DO) Ch. 4 (American Psychiatric Publishing, Inc., Washington, 2012).
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR* D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40.
Fesharaki-Zadeh A, Lowe N, Arnsten AFT. Clinical experience with the α2A-adrenoceptor agonist, guanfacine, and N-acetylcysteine for the treatment of cognitive deficits in “Long-COVID19”. Neuroimmunol Rep. 2023;3:100154.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Ironson G, Banerjee N, Fitch C, Krause N. Positive emotional well-being, health Behaviors, and inflammation measured by C-Reactive protein. Soc Sci Med. 2018;197:235–43.
Felger JC, Treadway MT. Inflammation effects on motivation and motor activity: role of dopamine. Neuropsychopharmacology. 2017;42:216–41.
Mac Giollabhui N, Ng TH, Ellman LM, Alloy LB. The longitudinal associations of inflammatory biomarkers and depression revisited: systematic review, meta-analysis, and meta-regression. Mol Psychiatry. 2021;26:3302–14.
Noble JM, Manly JJ, Schupf N, Tang MX, Mayeux R, Luchsinger JA. Association of C-reactive protein with cognitive impairment. Arch Neurol. 2010;67:87–92.
Baune B, Ponath G, Golledge J, Varga G. Association between IL-8 cytokine and cognitive performance in an elderly general population—the MEMO-Study. Neurobiol Aging. 2008;29:937–44.
Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A, et al. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry. 2001;58:445–52.
Singh-Manoux A, Dugravot A, Brunner E, Kumari M, Shipley M, Elbaz A, et al. Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology. 2014;83:486–93.
Crisan AF, Oancea C, Timar B, Fira-Mladinescu O, Crisan A, Tudorache V. Cognitive impairment in chronic obstructive pulmonary disease. PLoS ONE. 2014;9:e102468.
Huang YS, Guilleminault C, Hwang FM, Cheng C, Lin CH, Li HY, et al. Inflammatory cytokines in pediatric obstructive sleep apnea. Medicine. 2016;95:e4944.
Mac Giollabhui N. Inflammation and depression: research designs to better understand the mechanistic relationships between depression, inflammation, cognitive dysfunction, and their shared risk factors. Brain Behav Immun Health. 2021;15:100278.
Krogh J, Benros ME, Jorgensen MB, Vesterager L, Elfving B, Nordentoft M. The association between depressive symptoms, cognitive function, and inflammation in major depression. Brain Behav Immun. 2014;35:70–6.
Goldsmith DR, Haroon E, Woolwine BJ, Jung MY, Wommack EC, Harvey PD, et al. Inflammatory markers are associated with decreased psychomotor speed in patients with major depressive disorder. Brain Behav Immun. 2016;56:281–8.
Mac Giollabhui N, Alloy LB, Hartman CA. Investigating whether depressed youth exhibiting elevated C reactive protein perform worse on measures of executive functioning, verbal fluency and episodic memory in a large, population based sample of Dutch adolescents. Brain Behav Immun. 2021;94:369–80.
Mac Giollabhui N, Swistun D, Murray S, Moriarity DP, Kautz MM, Ellman LM, et al. Executive dysfunction in depression in adolescence: the role of inflammation and higher body mass. Psychol Med. 2020;50:683–91.
McAfoose J, Baune BT. Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev. 2009;33:355–66.
Carvalho AF, Miskowiak KK, Hyphantis TN, Kohler CA, Alves GS, Bortolato B, et al. Cognitive dysfunction in depression - pathophysiology and novel targets. CNS Neurol Disord Drug Targets. 2014;13:1819–35.
Chesnokova V, Pechnick RN, Wawrowsky K. Chronic peripheral inflammation, hippocampal neurogenesis, and behavior. Brain Behav Immun. 2016;58:1–8.
Goldsmith D, Bekhbat M, Mehta ND, Felger JC. Inflammation-related functional and structural dysconnectivity as a pathway to psychopathology. Biol Psychiatry. 2023;93:405–18.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68:748–54.
Harrison NA, Cercignani M, Voon V, Critchley HD. Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology. 2015;40:831–8.
Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22.
Khandaker GM, Zuber V, Rees JMB, Carvalho L, Mason AM, Foley CN, et al. Shared mechanisms between coronary heart disease and depression: findings from a large UK general population-based cohort. Mol Psychiatry. 2020;25:1477–86.
Perry BI, Upthegrove R, Kappelmann N, Jones PB, Burgess S, Khandaker GM. Associations of immunological proteins/traits with schizophrenia, major depression and bipolar disorder: a bi-directional two-sample Mendelian randomization study. Brain Behav Immun. 2021;97:176–85.
Kelly KM, Smith JA, Mezuk B. Depression and interleukin-6 signaling: a Mendelian Randomization study. Brain Behav Immun. 2021;95:106–14.
Ye Z, Kappelmann N, Moser S, Davey Smith G, Burgess S, Jones PB, et al. Role of inflammation in depression and anxiety: Tests for disorder specificity, linearity and potential causality of association in the UK Biobank. EClinicalMedicine. 2021;8:100992.
Rose-John S, Jenkins BJ, Garbers C, Moll JM, Scheller J. Targeting IL-6 trans-signalling: past, present and future prospects. Nat Rev Immunol. 2023;23:666–81.
Crick DC, Khandaker GM, Halligan SL, Burgner D, Mansell T, Fraser A Comparison of the stability of glycoprotein acetyls and high sensitivity C‐reactive protein as markers of chronic inflammation. Immunology 2024;4:497–512.
Kappelmann N, Arloth J, Georgakis MK, Czamara D, Rost N, Ligthart S et al. Dissecting the association between inflammation, metabolic dysregulation, and specific depressive symptoms: a genetic correlation and 2-sample Mendelian randomization study. JAMA Psychiatry 2021;78:161–70.
Crick DCP, Sanderson E, Jones H, Goulding N, Borges MC, Clayton G, et al. Glycoprotein acetyls and depression: testing for directionality and potential causality using longitudinal data and Mendelian randomization analyses. J Affect Disord. 2023;335:431–9.
Bekkevold O-J, Damås JK, Brumpton BM, Åsvold BO The causal role of C-reactive protein and interleukin-6 on anxiety and depression symptoms and life satisfaction: Mendelian randomisation analyses in the HUNT study. Psychol Med. 2023;53:7561–8.
Galan D, Perry BI, Warrier V, Davidson CC, Stupart O, Easton D, et al. Applying Mendelian randomization to appraise causality in relationships between smoking, depression and inflammation. Sci Rep. 2022;12:15041.
Slaney C, Sallis HM, Jones HJ, Dardani C, Tilling K, Munafo MR, et al. Association between inflammation and cognition: triangulation of evidence using a population-based cohort and Mendelian randomization analyses. Brain Behav Immun. 2023;110:30–42.
Pagoni P, Korologou-Linden RS, Howe LD, Davey Smith G, Ben-Shlomo Y, Stergiakouli E, et al. Causal effects of circulating cytokine concentrations on risk of Alzheimer’s disease and cognitive function. Brain Behav Immun. 2022;104:54–64.
Munafò MR, Higgins JPT, Smith GD. Triangulating evidence through the inclusion of genetically informed designs. Cold Spring Harb Perspect Med. 2021;11:a040659.
Fleshner M, Frank M, Maier SF. Danger signals and inflammasomes: stress-evoked sterile inflammation in mood disorders. Neuropsychopharmacology. 2017;42:36–45.
Slavich GM. Social safety theory: a biologically based evolutionary perspective on life stress, health, and behavior. Annu Rev Clin Psychol. 2020;16:265–95.
Murphy K, Weaver C. Janeway’s Immunobiology (9th Edition). New York, New York: Garland Science, Taylor & Francis Group; 2017.
Kiecolt-Glaser JK, Derry HM, Fagundes CP. Inflammation: depression fans the flames and feasts on the heat. Am J Psychiatry. 2015;172:1075–91.
Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34.
Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140:774–815.
Scholtens S, Smidt N, Swertz MA, Bakker SJ, Dotinga A, Vonk JM, et al. Cohort Profile: LifeLines, a three-generation cohort study and biobank. Int J Epidemiol. 2015;44:1172–80.
Ross TP. The reliability and convergent and divergent validity of the Ruff Figural Fluency Test in healthy young adults. Arch Clin Neuropsychol. 2014;29:806–17.
Mac Giollabhui N, Alloy LB, Schweren LJS, Hartman CA. Investigating whether a combination of higher CRP and depression is differentially associated with worse executive functioning in a cohort of 43,896 adults. Brain Behav Immun. 2021;96:127–34.
Overbeek T, Schruers K, Griez E. MINI: Mini international neuropsychiatric interview, Dutch version 5.0. 0 (DSM-IV). Maastricht: University of Maastricht 1999.
Hergueta T, Weiller E MINI-S for DSM-5, Dutch version, 2017.
Lecrubier Y, Sheehan D, Weiller E, Amorim P, Bonora I, Sheehan K, et al. The Mini International Neuropsychiatric Interview (MINI). A short diagnostic structured interview: Reliability and validity according to the CIDI. Eur Psychiatry. 1997;12:224–31.
Wanders RB, van Loo HM, Vermunt JK, Meijer RR, Hartman CA, Schoevers RA, et al. Casting wider nets for anxiety and depression: disability-driven cross-diagnostic subtypes in a large cohort. Psychol Med. 2016;46:3371–82.
Watson D, Clark LA, Tellegen A. Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol. 1988;54:1063.
Crawford JR, Henry JD. The Positive and Negative Affect Schedule (PANAS): Construct validity, measurement properties and normative data in a large non‐clinical sample. Br J Clin Psychol. 2004;43:245–65.
Tang Y, Horikoshi M, Li W. ggfortify: unified interface to visualize statistical results of popular R packages. R J. 2016;8:474.
ieugwasr: Interface to the ‘OpenGWAS’ Database API. R package version 0.2.2-9000. 2024, Accessed Date Accessed 2024 Accessed.
Said S, Pazoki R, Karhunen V, Võsa U, Ligthart S, Bodinier B, et al. Genetic analysis of over half a million people characterises C-reactive protein loci. Nat Commun. 2022;13:2198.
Collaboration IRGCERF, Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379:1205–13.
Project IOG. Glycoprotein acetyls Dataset: met-d-GlycA. 2023.
Swerdlow DI, Kuchenbaecker KB, Shah S, Sofat R, Holmes MV, White J, et al. Selecting instruments for Mendelian randomization in the wake of genome-wide association studies. Int J Epidemiol. 2016;45:1600–16.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–7.
Myers TA, Chanock SJ, Machiela MJ. LDlinkR: an R package for rapidly calculating linkage disequilibrium statistics in diverse populations. Front Genet. 2020;11:157.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Aulchenko YS, De Koning D-J, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–85.
Kleiber C, Zeileis A Applied econometrics with R. Springer Science & Business Media 2008.
Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, et al. Mendelian randomization. Nat Rev Methods Primers. 2022;2:6.
Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755–64.
Slavish DC, Jones DR, Smyth JM, Engeland CG, Song S, McCormick NM, et al. Positive and negative affect and salivary markers of inflammation among young adults. Int J Behav Med. 2020;27:282–93.
Brouwers C, Mommersteeg PM, Nyklíček I, Pelle AJ, Westerhuis BL, Szabó BM, et al. Positive affect dimensions and their association with inflammatory biomarkers in patients with chronic heart failure. Biol Psychol. 2013;92:220–6.
Sepah SC, Bower JE. Positive affect and inflammation during radiation treatment for breast and prostate cancer. Brain Behav Immun. 2009;23:1068–72.
Stellar JE, John-Henderson N, Anderson CL, Gordon AM, McNeil GD, Keltner D. Positive affect and markers of inflammation: discrete positive emotions predict lower levels of inflammatory cytokines. Emotion. 2015;15:129.
Boylan JM, Lewis TT, Coe CL, Ryff CD. Educational status, anger, and inflammation in the MIDUS national sample: does race matter? Ann Behav Med. 2015;49:570–8.
Milaneschi Y, Kappelmann N, Ye Z, Lamers F, Moser S, Jones PB, et al. Association of inflammation with depression and anxiety: evidence for symptom-specificity and potential causality from UK Biobank and NESDA cohorts. Mol Psychiatry. 2021;26:7393–402.
Milaneschi Y, Lamers F, Berk M, Penninx B. Depression heterogeneity and its biological underpinnings: toward immunometabolic depression. Biol Psychiatry. 2020;88:369–80.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31–41.
Baune BT, Sampson E, Louise J, Hori H, Schubert KO, Clark SR, et al. No evidence for clinical efficacy of adjunctive celecoxib with vortioxetine in the treatment of depression: A 6-week double-blind placebo controlled randomized trial. Eur Neuropsychopharmacol. 2021;53:34–46.
Nettis MA, Lombardo G, Hastings C, Zajkowska Z, Mariani N, Nikkheslat N, et al. Augmentation therapy with minocycline in treatment-resistant depression patients with low-grade peripheral inflammation: results from a double-blind randomised clinical trial. Neuropsychopharmacology. 2021;46:939–48.
Hellmann-Regen J, Clemens V, Grözinger M, Kornhuber J, Reif A, Prvulovic D, et al. Effect of minocycline on depressive symptoms in patients with treatment-resistant depression: a randomized clinical trial. JAMA Netw Open. 2022;5:e2230367–e2230367.
Chamberlain SR, Cavanagh J, de Boer P, Mondelli V, Jones DNC, Drevets WC, et al. Treatment-resistant depression and peripheral C-reactive protein. Br J Psychiatry. 2019;214:11–19.
Miller AH, Raison CL. Burning down the house: reinventing drug discovery in psychiatry for the development of targeted therapies. Mol Psychiatry. 2023;28:68–75.
Felger JC, Haroon E, Miller AH. What’s CRP got to do with it? Tackling the complexities of the relationship between CRP and depression. Brain Behav Immun. 2018;73:163–4.
Pitharouli MC, Hagenaars SP, Glanville KP, Coleman JRI, Hotopf M, Lewis CM, et al. Elevated C-Reactive protein in patients with depression, independent of genetic, health, and psychosocial factors: results from the UK biobank. Am J Psychiatry. 2021;178:522–9.
Moriarity DP, Mengelkoch S, Slavich GM. Incorporating causal inference perspectives into psychoneuroimmunology: a simulation study highlighting concerns about controlling for adiposity in immunopsychiatry. Brain Behav Immun. 2023;113:259–66.
Wium-Andersen MK, Ørsted DD, Nielsen SF, Nordestgaard BG. Elevated C-Reactive protein levels, psychological distress, and depression in 73 131 individuals. JAMA Psychiatry. 2013;70:176.
Consortium I-RMRA. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379:1214–24.
Collaboration CRPCHDG. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548.
Foley ÉM, Slaney C, Donnelly NA, Kaser M, Ziegler L, Khandaker GM. A novel biomarker of interleukin 6 activity and clinical and cognitive outcomes in depression. Psychoneuroendocrinology. 2024;164:107008.
Cathomas F, Lin HY, Chan KL, Li L, Parise LF, Alvarez J, et al. Circulating myeloid-derived MMP8 in stress susceptibility and depression. Nature. 2024;626:1108–15.
Mac Giollabhui N, Foster S, Lowry CA, Mischoulon D, Raison CL, Nyer M. Interleukin-6 receptor antagonists in immunopsychiatry: can they lead to increased interleukin-6 in the central nervous system (CNS) and worsening psychiatric symptoms? Brain Behav Immun. 2022;103:202–4.
Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213.
Fard MT, Savage KM, Stough CK. Peripheral inflammation marker relationships to cognition in healthy older adults – A systematic review. Psychoneuroendocrinology. 2022;144:105870.
Boogert NJ, Madden JR, Morand-Ferron J, Thornton A. Measuring and understanding individual differences in cognition. Philos Trans R Soc Lond B Biol Sci. 2018;373:20170280.
Mac Giollabhui N, Mischoulon D, Dunlop BW, Kinkead B, Schettler PJ, Liu RT, et al. Individuals with depression exhibiting a pro-inflammatory phenotype receiving omega-3 polyunsaturated fatty acids experience improved motivation-related cognitive function: Preliminary results from a randomized controlled trial. Brain Behav Immun Health. 2023;32:100666.
Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16:626–38.
Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–22.
Vogelzangs N, Beekman ATF, de Jonge P, Penninx BWJH. Anxiety disorders and inflammation in a large adult cohort. Transl Psychiatry. 2013;3:e249–e249.
Costello H, Gould RL, Abrol E, Howard R. Systematic review and meta-analysis of the association between peripheral inflammatory cytokines and generalised anxiety disorder. BMJ Open. 2019;9:e027925.
Glaus J, von Känel R, Lasserre AM, Strippoli MPF, Vandeleur CL, Castelao E, et al. The bidirectional relationship between anxiety disorders and circulating levels of inflammatory markers: results from a large longitudinal population‐based study. Depress Anxiety. 2018;35:360–71.
O’Donovan A, Slavich GM, Epel ES, Neylan TC. Exaggerated neurobiological sensitivity to threat as a mechanism linking anxiety with increased risk for diseases of aging. Neurosci Biobehav Rev. 2013;37:96–108.
Raison CL, Miller AH. Pathogen–host defense in the evolution of depression: insights into epidemiology, genetics, bioregional differences and female preponderance. Neuropsychopharmacology. 2017;42:5–27.
Osimo EF, Baxter LJ, Lewis G, Jones PB, Khandaker GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med. 2019;49:1958–70.
Klijs B, Scholtens S, Mandemakers JJ, Snieder H, Stolk RP, Smidt N. Representativeness of the LifeLines Cohort Study. PLoS ONE. 2015;10:e0137203.
Freeman MP, Cheng LJ, Moustafa D, Davies A, Sosinsky AZ, Wang B, et al. Vortioxetine for major depressive disorder, vasomotor, and cognitive symptoms associated with the menopausal transition. Ann Clin Psychiatry. 2017;29:249–57.
Mahableshwarkar AR, Zajecka J, Jacobson W, Chen Y, Keefe RS. A randomized, placebo-controlled, active-reference, double-blind, flexible-dose study of the efficacy of vortioxetine on cognitive function in major depressive disorder. Neuropsychopharmacology. 2015;40:2025–37.
Olver JS, Ignatiadis S, Maruff P, Burrows GD, Norman TR. Quetiapine augmentation in depressed patients with partial response to antidepressants. Hum Psychopharmacol. 2008;23:653–60.
Acknowledgements
The Lifelines initiative has been made possible by subsidy from the Dutch Ministry of Health, Welfare and Sport, the Dutch Ministry of Economic Affairs, the University Medical Center Groningen (UMCG), Groningen University and the Provinces in the North of the Netherlands (Drenthe, Friesland, Groningen). The generation and management of GWAS genotype data for the Lifelines Cohort Study is supported by the UMCG Genetics Lifelines Initiative (UGLI). UGLI is partly supported by a Spinoza Grant from NWO, awarded to Cisca Wijmenga. The authors wish to acknowledge the services of the Lifelines Cohort Study, the contributing research centers delivering data to Lifelines, and all the study participants.
Funding
This work was supported by Harvard University’s Mind Brain Behavior Interfaculty Initiative and National Institute of Mental Health grant K23MH132893 (Dr. Mac Giollabhui), and a UK Medical Research Council (MRC) grant to GMK (MC_UU_00032/6) which forms part of the MRC Integrative Epidemiology Unit (IEU) at the University of Bristol. The MRC grant also supports CS and EMF. GMK acknowledges additional funding support from the Wellcome Trust (201486/Z/16/Z and 201486/B/16/Z), MRC (MR/W014416/1; MR/S037675/1; MR/Z50354X/1, and MR/Z503745/1), and the UK National Institute of Health and Care Research (NIHR) Bristol Biomedical Research Centre (NIHR 203315). GH and GDS are supported by the MRC (MC_UU_00032/1), and the NIHR Bristol Biomedical Research Centre (NIHR 203315). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care, UK.
Author information
Authors and Affiliations
Contributions
NMG, CS, GMK, CH conceptualized and designed the study. NMG conducted the non-genetic analysis. CS conducted the genetic analyses. NMG and CS wrote the first draft of the paper. GH, EMF, PJM, IMN, HS, and GDS advised on the project/analysis and critically reviewed the final version of the manuscript. CH and GMK provided overall supervision for this project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mac Giollabhui, N., Slaney, C., Hemani, G. et al. Role of inflammation in depressive and anxiety disorders, affect, and cognition: genetic and non-genetic findings in the lifelines cohort study. Transl Psychiatry 15, 164 (2025). https://doi.org/10.1038/s41398-025-03372-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-025-03372-w
