
Abstract
NEXMIF is an X-linked gene implicated in encephalopathy with symptoms of autism spectrum disorder (ASD), intellectual disability, and seizures. Our previous work demonstrated that Nexmif knockout (KO) male mice show autistic-like behaviors and memory deficits, accompanied by significant abnormalities in neuronal development and function. However, to date there are no available therapeutics for NEXMIF-related disorders. Here, as a proof-of-concept study, we examined the effect of postnatal reintroduction of the NEXMIF gene as a strategy for rescuing the impaired cellular and behavioral phenotypes in KO mice. We find that injection of a human NEXMIF lentivirus into KO mouse brains at postnatal day 1 (P1) leads to a restoration in synaptic protein expression and formation of dendritic spines. More importantly, postnatal NEXMIF expression ameliorated behavioral defects in repetitive behavior, sociability, social novelty preference, and cognition at adolescent ages, in addition to restoring dysregulated gene expression. These findings suggest that gene reintroduction at a postnatal stage may serve as a rescue strategy for neurodevelopmental and behavioral deficits caused by NEXMIF deficiencies.
Similar content being viewed by others


Introduction
Autism spectrum disorders (ASD) are characterized by altered language and communication, repetitive behaviors, and impaired social interactions [1]. The prevalence of ASD has been on the rise, currently affecting 1 in 31 children (8-year-olds) in the United States [2]. Comorbidities of ASD include epilepsy, facial dysmorphisms, motor deficits, intellectual disability (ID), attention-deficit/hyperactivity disorder (ADHD), and other psychiatric disorders [3, 4]. Several ASD-associated genes have been identified on the X chromosome, and many of these genes are associated with the development of X-linked intellectual disability (XLID), particularly in males [5,6,7]. Our previous studies and work from others found that loss of function of the X-linked gene NEXMIF leads to a developmental encephalopathy known as XLID98 with symptoms of ASD, ID, and seizures [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24]. The NEXMIF gene, located at Xq13.3, encodes a large ~170 kilodalton nuclear protein, which is strongly expressed in the fetal brain, as well as in the adult hippocampus, cerebellum, cerebral cortex, and olfactory bulb [12, 13]. Early studies on the gene have linked NEXMIF to the regulation of neural circuit formation during early development, as well as the regulation of cytoskeletal dynamics involved in neurite outgrowth [25, 26]. Indeed, we have previously shown that loss of Nexmif leads to defects in neuron migration, dendrite growth, spine density, synaptic protein expression, and synapse formation and function [11, 27,28,29]. Furthermore, NEXMIF has recently been implicated in the maintenance of genomic integrity in the brain via regulating the expression of genes responsible for the suppression of retrotransposon LINE1 activity [30]. These findings suggest that NEXMIF plays a crucial gene regulation role during early brain development.
To further study the function of NEXMIF and its role in ASD, we generated Nexmif knockout (KO) male mice (X-Y) which demonstrate typical ASD features and comorbidities, including impairments in social behavior and vocalization, repetitive behavior, anxiety, deficits in learning and memory function, and frequent seizures at mature ages [28]. Furthermore, in line with clinical reports in females [10], we find that Nexmif heterozygous (HET) female mice (X+X-) demonstrate similar behavioral phenotypes [29]. Given the critical role of NEXMIF during early neurodevelopment and the detrimental effects following its loss, it is important to determine whether these impaired cellular and behavioral phenotypes can be rescued, especially at a stage during early brain development.
Over the past decade, studies have demonstrated that postnatal reintroduction of target genes can sufficiently rescue impaired cellular and behavioral phenotypes in mouse models of neurodevelopmental disorders, such as adeno-associated virus (AAV)-based restoration of Ube3a in Angelman syndrome mice [31], Cre-mediated Mecp2 restoration in Rett syndrome mice [32], conditional knock-in of SHANK3 expression in adult Shank3 KO mice [33], and several others [34,35,36,37,38,39,40,41]. While some of these studies were only able to achieve partial rescue of the impaired phenotypes, the findings nonetheless demonstrate the feasibility of utilizing gene restoration techniques to rescue neuronal deficits and autistic-like behaviors. Here, we generated a lentivirus (LV) containing a human NEXMIF transgene and performed intracerebroventricular (ICV) brain injections in postnatal day 1 (P1) Nexmif KO male mice. We found that reintroduction of NEXMIF in the brain rescued molecular and behavioral abnormalities in adolescent (P30-P70) KO mice, in addition to restoring dysregulated gene expression in the hippocampus.
Materials & methods
Animal care and use
Mouse colonies were maintained in the Laboratory Animal Science Center (LASC) at the Boston University Charles River Campus on a C57BL/6 J genetic background. Female mice heterozygous for Nexmif (X+X-) were crossed with wild-type (WT) male mice to obtain knockout (KO) male mice. All WT (X+Y) mice used were randomized male littermate controls of the KO mice (X-Y) between the ages of P30-P70, or P90 for RNA sequencing. Mice were randomly assigned to control and treatment groups using a random number generator, ensuring equal representation from each litter and cage. All experiments were conducted during the light phase. Animals that did not exhibit normal behavior or health at the onset of the experiment, or showed signs of injury and sickness during experiments, were excluded from the analyses. Following all in vivo injection experiments and behavioral tests, IACUC-approved euthanasia methods were used to sacrifice mice prior to tissue collection, specifically via CO2 inhalation at a flow rate of 4 liters/min in the home-cage for 3 min. Cervical dislocation was performed two minutes after cessation of breathing.
Genotyping
DNA was isolated from tail snips using the Hot Shot Method. A single tail snip was collected from each mouse at either postnatal day 0–1 (P0-1, prior to injections), or at the time of weaning on P21, and placed in an Eppendorf tube. Tail snips were incubated for 30 min at 95 °C in 75 µl of alkaline lysis buffer (25 mM NaOH, 0.2 M EDTA). The tubes were allowed to cool at room temperature for 5 min before 75 µl of neutralization buffer (40 mM Tris-HCl, pH 5) was added. The tubes were mixed by vortexing and 1 µl of DNA extract was used in the following PCR protocol run on a Biorad DNA engine Tetrad 2 Peltier Thermal Cycler: 94 °C for 5 min followed by 30 cycles of 98 °C for 15 s, 55 °C (WT) or 59 °C (KO) for 30 s and 72 °C for 60 s. The resulting PCR fragments were run on a 1.2% agarose gel with ethidium bromide to label and visualize DNA under ultraviolet light. Two sets of primers were used to genotype Nexmif KO mice. One set (WT) was targeted against the exon 4 region using the primers: 5’−aggacttgcttaggttgcttcatggaa−3’ and 5’−cttaaattgctctacctcaagaccacca−3’ with an expected PCR fragment of 949 bp. The other set (KO) was targeted against the KO cassette using the primers: 5’−cacacctccccctgaacctgaaag−3’ and 5’−cccacgaagggatcataccctgta−3’ with an expected PCR fragment of 794 bp.
Primary neuron culture
Cortical brain tissue was dissected from E18 wildtype rat fetus brains of either sex and prepared for primary culture. Brains were digested in a digestion buffer [papain (15 mg/ml in Hanks balanced salt solution, Sigma-Aldrich #4762), L-Cysteine (4 mg/ml in Hanks balanced salt solution, Sigma-Aldrich #C7352), and 0.5 M EDTA pH 7.0] for 20 min at 37 °C, then triturated in a trituration buffer [0.1% DNase (Thermo Fisher #PA5-22017), 1% ovomucoid (Sigma-Aldrich #T2011)/1% bovine serum albumin (Sigma-Aldrich #05470) in Dulbecco’s modified Eagle’s medium (DMEM)] to fully dissociate neurons. Dissociated neurons were counted and plated on 18-mm circular coverslips (Carolina #633013) in 60-mm Petri dishes (five coverslips/dish) and 6-well culture plates that had been coated in poly-l-lysine (Sigma-Aldrich #P2636; 100 μg/ml in borate buffer) overnight at 37 °C then washed three times with sterile deionized water and left in plating medium [minimal essential medium (500 mL) containing 10% fetal bovine serum (Atlanta Biologicals #S11550), 5% horse serum (Atlanta Biologicals #S12150), 31 mg L-cysteine, 1% penicillin/streptomycin (Corning #30-002-Cl), and 1% L-glutamine (Corning #25-005-Cl) before cell plating. The day after plating, plating medium was replaced by feeding medium (Neurobasal medium supplemented with 1% horse serum, 2% SM1, and 1% penicillin/streptomycin and 1% L-glutamine) which was supplemented with 5′-fluoro-2′-deoxyuridine (10 μm; Sigma-Aldrich #F0503) after 7 d in vitro to suppress glial growth.
Plasmids
A full-length human NEXMIF ORF sequence (4548 bp, Origene #RC212264) was enzymatically excised from its expression vector and cloned into the FUW lentiviral expression vector, and the resulting FUW-NEXMIF plasmid was packaged into a lentivirus. As a control, the empty FUW vector was packaged into a separate lentivirus. FUW was a gift from David Baltimore (Addgene plasmid #14882). All unique/stable reagents generated in this study are available from the lead contact without restriction.
Virus preparation
Lentiviruses (LV) were produced by co-transfecting HEK293T cells with the DNA constructs and viral packaging and envelope proteins (pRSV/REV, pMDLg/pRRE, and pCMV-VSV-G) using polyethylenimine reagent (Polysciences #23966). Sodium pyruvate (Thermo Fisher #11360070) was added to the medium 24 h later to supplement the cells. Conditioned medium containing the viral particles was harvested 48 h later and filtered through a 0.45-μm filter. PEG-it Virus Precipitation Solution (System Biosciences #LV810A) was added to the medium, and the mixture was left to incubate at 4 °C for 72 hr. The mixture was centrifuged at 1500 g for 30 min at 4 °C and the viral pellet was resuspended in sterile 1X phosphate buffered saline. The virus was divided into 10 μl aliquots and stored at −80 °C. The LV packaging constructs were gifts from Didier Trono (Addgene plasmids #12251 and #12253) and Bob Weinberg (Addgene plasmid #8454).
Neuron transduction
For viral transductions, wildtype rat cortical neurons plated on coverslips were infected with LV. 24 h later, the medium was replaced with fresh medium, and the cells were incubated at 37 °C in a 5% CO2 incubator for the desired amount of time.
Mouse brain injections
For intracerebroventricular (ICV) injections of LVs, P0-1 WT and Nexmif KO male mouse pups were cryo-anesthetized on wet ice for 3 min prior to bilateral ventricular injection (1.13 × 106 viral particles per ventricle) on a chilled stage using a 10-μl syringe with a sterile 32-gauge needle (Hamilton #7653-01). Fast Green FCF dye (1 μl, Thermo Scientific #A16520-14) was added to the virus aliquots to visualize and confirm successful injection. Following injection, pups were warmed on an isothermal heating pad with home-cage bedding before being returned to the dam.
Immunocytochemistry (ICC) of cultured neurons
Cortical neurons were fixed for 8 min in a 4% paraformaldehyde (PFA) / 1X Phosphate- buffered saline (PBS) solution at room temperature (RT). Cells were rinsed two times in 1X PBS followed by membrane permeabilization for 10 min in 0.3% Triton-X-100 (Sigma Aldrich #T8787) in 1X PBS. Cells were then rinsed two times in 1X PBS followed by incubation with primary antibodies overnight at 4 °C, washed three times with cold 1X PBS, and incubated with Alexa Fluor-conjugated fluorescent secondary antibodies (1:500, Thermo Fisher) for 1 h at RT. Cells were then washed three times with cold 1X PBS, with the first wash containing Hoechst (1:10,000, Thermo Fisher #62249) and mounted to microscopy glass slides with Prolong Gold antifade mounting reagent (Thermo Fisher #P36930) for subsequent visualization. Mounted coverslips were kept overnight in the dark at RT before imaging. For ICC, primary and secondary antibodies were diluted in IHC-Tek™ Antibody Diluent pH 7.4 (IHCWorld, #IW-1000), which does not require the serum blocking step.
Immunohistochemistry (IHC) of brain slices
P40 mice underwent transcardial perfusion with ice cold 1X PBS followed by 4% PFA prior to collection of the brain. Brains were post-fixed in 4% PFA for 4 h and cryoprotected in 30% sucrose/1X PBS for 48 h. Brains were then placed in molds to be rapidly frozen in Tissue-Tek OCT (Sakura #4583) with dry ice and stored at −80 °C until they were cut into 30 μm sections using a LEICA CM1850 cryostat (LEICA Biosystems). Sections were mounted onto SuperFrost microscope slides (Fisher Scientific #12-550-15) and stored at −20 °C until staining. To prepare for immunostaining, sections were hydrated in 1X PBS for 30 min followed by permeabilization in 1% Triton X-100/1X PBS for 1 h. Sections were then incubated in primary antibodies overnight at 4 °C in a humidity chamber, washed three times 10 min each with cold 1X PBS, and incubated in Alexa Fluor-conjugated fluorescent secondary for 1 h at RT. Brain slices were then washed three times with cold 1X PBS, with the first wash containing Hoechst (1:10,000) and mounted under a rectangular coverslip with Prolong Gold anti-fade mounting reagent. Slides were allowed to dry in the dark at room temperature overnight and stored at −20 °C prior to subsequent visualization. For IHC, primary and secondary antibodies were diluted in IHC-Tek™ Antibody Diluent, which does not require the serum blocking step.
Golgi staining
Whole brains from P70 mice were subjected to Golgi neuron staining using the FD Rapid GolgiStain Kit (FD Neurotechnologies #PK401) according to the manufacturer’s instructions. Mice were sacrificed in a 4% CO2 chamber and brains were collected and rinsed in ice-cold PBS. Brains were immersed in a Golgi-Cox solution containing potassium dichromate, mercuric chloride, and potassium chromate. The solution was replaced after 24 h of immersion with fresh solution and stored at RT in the dark for 2 weeks. After immersion, the brains were placed in Tissue Freezing Medium (Electron Microscopy Sciences #72592), rapidly frozen in a dry ice/methanol bath, and stored at –20 °C prior to slicing. Brain slices were sectioned coronally at a 100 µm thickness on a cryostat and were mounted on gelatin-coated slices (FD Neurotechnologies #PO101) and air dried at RT before further processing. Sections were then rinsed in distilled water, incubated in staining solution for 10 min, and dehydrated with 50%, 75%, 95%, and finally 100% ethanol. Sections were defatted in xylene and mounted onto coverslips with Permount mounting medium (Fisher Scientific # SP15-100). Sections were stored at RT in the dark for 3 weeks prior to visualization.
Western blot
Brains were dissected on ice immediately after sacrificing animals at the appropriate time points. For a ∼30 mg piece of cortical/hippocampal tissue, or 1 × 106 primary rat cortical neurons, ∼750 μl of ice-cold lysis buffer [50 mM Tris-HCl pH 8, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate (SDOC), 0.1% sodium dodecyl sulfate (SDS), supplemented with 100X protease inhibitor cocktail (Apex Bio #K1011)] was added and samples were homogenized mechanically with a pestle, followed by sonication (for 10 s). Samples were then centrifuged for 15 min at 13,000 rpm at 4 °C in a microcentrifuge. The tubes were placed on ice and the supernatant was carefully aspirated and placed into a fresh tube kept on ice. Samples were subjected to a BCA assay according to the manufacturer’s protocol (Thermo Fisher #23225) to determine protein concentrations. Protein levels were normalized with the lysis buffer, and an equal volume of 2X sample reducing buffer [5% SDS, 150 mM Tris-HCl pH 6.8, 0.05% Bromophenol Blue (Sigma-Aldrich #B0126), 5% fresh 2-mercaptoethanol (Sigma-Aldrich #M3148)] was added to the samples. The lysates were then boiled for 10 min at 95 °C. SDS-PAGE was performed to separate proteins of interest using standard procedures. Samples were run on 6–12% gels at 110 V for 1 hr. Proteins were transferred at 150 mA overnight to PVDF membranes and blocked for 1 h in 5% bovine serum albumin (BSA, Sigma Aldrich #A2153) prepared in 1X tris-buffered saline supplemented with 0.1% Tween (TBST). After blocking, membranes were probed with the appropriate primary antibody diluted in Signal Enhancer Hikari Buffer (Nacalai USA #NU00101) overnight at 4 °C. Membranes were washed 3 × 5 min each in 1X TBST and then incubated with the appropriate secondary antibody for 1 h. After secondary incubation, membranes were washed 3X with 1X TBST. Blots were visualized using the Azure Radiance Plus chemiluminescence detection system (Azure Biosystems #AC2103) on the Sapphire Biomolecular Imager (Azure Biosystems) and analyzed using NIH Fiji (ImageJ, RRID:SCR_002285) [42].
Antibodies
Primary antibodies to the following proteins were used: rabbit anti-KIAA2022 [1:100 (brain slice for IHC) and 1:300 (cultured neurons for ICC), Sigma #HPA000407], rabbit anti-KIAA2022 (1:750 for WB; Biorbyt Orb312213), rabbit anti-β-tubulin III (1:1000 for WB, Sigma-Aldrich T2200), rabbit anti-GluA1 (1:1000 for WB, Homemade), mouse anti-SynDIG1 (1:1000 for WB, Antibodies Inc #75-251), rabbit anti-TrkB (1:1000 for WB, Cell Signaling #4603), rabbit-anti BAIAP3 (1:1000 for WB, Synaptic Systems 256 003), rabbit-anti Cerebellin-1 (1:1000 for WB; Boster Bio A09176-1), rabbit anti-Neurotensin (1:1000 for WB, Abcam #ab233107), rabbit anti-Filip1 N-terminal (1:1000 for WB, Aviva Systems Biology #ARP79028_P050), rabbit anti-NMDAR1 (1:1000 for WB, Sigma #G8913), rabbit anti-GLRA1 (1:1000 for WB, Proteintech #17951-1-AP), rabbit anti-PSD-95 (1:1000 for WB, Cell Signaling #3450), mouse anti-RGS14 (1:1000 for WB, Antibodies Inc # 75-170-020), rabbit anti-MAGEL2 (1:1000 for WB, Bioss Antibodies bs-6828R), and mouse anti-CACNA1G (CaV3.1, 1:1000 for WB, Novus Biologicals #NBP2-59322). The following secondary antibodies were used: IgG-HRP for WB [1:10000; Thermo Fisher, mouse (#62-6520) and rabbit (#31460)] and Alexa Fluor 555 [1:500, Thermo Fisher, rabbit (#A32732)] for ICC and IHC.
Reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from ~30 mg of mouse hippocampal tissue using the RNeasy Mini kit (Qiagen #74106). RNA concentrations were diluted with nuclease-free water (NEB #B1500S) to 1 μg, followed by reverse transcription to cDNA using the EasyQuick RT MasterMix (CoWin Biosciences #CW2019). 1 μl of the cDNA was added to the HotStart™ Universal 2X SYBR Green qPCR Master Mix (ApexBio Technology #K1170) in a 20 µl volume and real-time fluorescence quantitative PCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems) to detect mRNA levels of mouse Cerebellin-1 and β-actin with the appropriate primers. The primers used: Cerebellin-1 (Forward: gctttctctgccatcaggagca Reverse: ggcgatgaaagtgctgcgttct) and β-actin (Forward: cattgctgacaggatgcagaagg Reverse: tgctggaaggtggacagtgagg). Target gene expression was normalized to β-actin prior to using the delta delta Ct method to determine the fold change in gene expression in KO + CTRL and KO + NEX mouse tissue relative to WT + CTRL.
RNA sequencing
Hippocampal brain tissue collected from P90 WT + CTRL, KO + CTRL, and KO + NEX mice were preserved in RNAlater™ Stabilization Solution (Invitrogen #AM7020) prior to being sent out for commercial RNA-sequencing services provided by GENEWIZ from Azenta Life Sciences. RNA-seq datasets were generated and analyzed by GENEWIZ to identify differentially expressed genes (DEGs) and Gene Ontology (GO) Enrichment Analysis.
Novel object recognition test
The novel object recognition (NOR) test was conducted in an arena constructed from a 0.75 in-thick white plastic box measuring 28 × 28 x 28 cm. Mice were habituated to the environment for 5 min on days 1 and 2. On the test day (day 3), two identical objects (25 cm2 cell culture bottles filled with fresh bedding) were placed diagonally from each other on the floor of the box environment. Each mouse was singly placed into the center of the box and allowed to freely explore the two objects for 10 min. Following the exploration, mice were placed back into their home cage. Four hours later, one of the identical objects was replaced with a salient, “novel” object (10 × 5 x 5 cm LEGO® block) and placed diagonally from the “familiar” object. Each mouse was again singly placed into the center of the box and allowed to freely explore the two objects for 5 min. Movement was analyzed for the time spent actively interacting with each object (licking, sniffing, touching, etc.) during each session (nose ≤ 2 cm). Since mice often climb the objects, the amount of time spent standing on top of an object was excluded from the analysis. Between test mice, the apparatus was wiped with 70% ethanol to eliminate odor cues.
Grooming test
Grooming was assessed in the animal’s home cage with a 3 in. thick layer of fresh pine chip bedding. Mice were briefly moved into a holding cage prior to being singly placed into their home cage and allowed to move freely for 20 min. Video recordings were captured from the side of the cage. The amount of time spent grooming and the number of grooming events during the last 10 min of the session were manually quantified. Grooming was considered as licking of the paws followed by cleaning of the snout, eyes, whiskers, body, and tail with backward and upward sweeps.
Three-chamber social test
A three-chambered box measuring 65 × 28 × 28 cm was constructed from 0.75 in. thick white plastic board with 4 × 4 in. cut-out doors in the walls to the center chamber allowing movement between chambers. A small wire cage was placed in each of the side chambers to later house stranger mice. Test mice were habituated to the apparatus with empty cages in both side chambers and allowed to move freely between all three chambers for 5 min on days 1 and 2. On the testing day (day 3), the side doors were blocked with white plastic boards and test mice were singly placed into the center chamber with an age- and sex-matched stranger mouse (Mouse 1) placed under the wire cage in either of the side chambers. Once the doors were unblocked, the test mouse was allowed to move freely within the apparatus for 5 min. The test mouse was then returned to the center chamber, the doors were blocked again, and a second age- and sex-matched mouse (Novel Mouse) was placed in the wire cage on the other side chamber. The center doors were un-blocked again, and the test mouse was allowed to move freely within the apparatus for 5 min. Between test mice, the entire apparatus was wiped with 70% ethanol to eliminate odor cues. Movement was analyzed for the time spent actively interacting with each chamber (licking, sniffing, touching, etc.) during each session (nose ≤ 2 cm). The interaction times were used to calculate the social Preference Index (PI) for each test: Sociability [(Time at Mouse 1 – Time at Empty Cage) / (Time at Mouse 1 + Time at Empty Cage)*100] and Social Novelty [(Time at Novel Mouse – Time at Mouse 1) / (Time at Novel Mouse + Time at Mouse 1)*100].
Elevated zero maze
Mice were singly placed on a circular track that was elevated 2 ft. above the ground. Two sides of the track had closed arms, while the rest of the track was open. A camera was placed ~5 ft. above the track to record animals’ movements. The test mouse was placed at an open/closed arm boundary facing towards the closed arm. The mouse was allowed to freely explore the open and closed arms of the track for 10 min. The amount of time spent in the open arms relative to the total distance traveled was recorded as a measure of anxiety.
Microscopy
Exposure time for the fluorescence signals was adjusted manually so the signals were within a full dynamic range. Once the parameters were set, they were fixed and used throughout image acquisition for each experiment.
For ICC
Fluorescent images were collected with a 63x oil-objective on a ZEISS Axio Imager Z2 Upright Microscope using the ZEISS ZEN software.
For IHC
Brain sections were imaged with 10x and 20x air-objectives using a DS-Fi2 Color Camera on a Nikon Eclipse NiE using NIS-Elements software.
For Golgi staining
Images were acquired using brightfield transmitted light with a 63x oil-objective on a ZEISS Axio Imager Z2 Upright Microscope using the ZEISS ZEN software. The number and density of basolateral spines on neurons were measured with Fiji software using the Dendritic Spine Counter plugin [43].
Statistical analysis
For IHC, western blot, and golgi spine density
A One-Way ANOVA with Tukey’s or Bonferroni’s Post Hoc Test was used to determine significant differences in fluorescent intensity, protein expression, and spine density between the three test groups: WT + CTRL, KO + CTRL, and KO + NEX. A two-tailed student’s t test was used to determine significant differences in protein expression between LV-Control- and LV-NEXMIF-treated neurons.
For ICC
A two-tailed student’s t test was used to determine significant differences in nuclear intensity between LV-Control- and LV-NEXMIF-treated neurons.
For behavioral tests
Video recordings were captured with a Logitech c920 webcam during each test. Locomotion tracks were generated using the Mouse Activity Analyzer in MATLAB software [44]. These tracks were to quantify the desired aspects of the mouse movement. For the Elevated Zero Maze test, DeepLabCut [45] was used to extract precise coordinates of animal movement, which were subsequently analyzed using custom MATLAB scripts [46]. A One-Way ANOVA with Tukey’s or Bonferroni’s Post Hoc Test or a Two-Way ANOVA with Sidak’s Post Hoc Test was used to determine significant differences in behavior between three test groups: WT + CTRL, KO + CTRL, and KO + NEX.
Experimenters were not blinded to the experimental conditions; however, standardized protocols were used for mouse handling, behavioral testing, and scoring. Testing conditions, such as the time of day, location, noise and lighting, were kept consistent, and the same experimenter was used for all tests to reduce bias. The Novel Object Recognition test, Three-Chamber Social test, and Grooming assay were manually scored. The Elevated Zero Maze test was scored unbiasedly in MATLAB.
All data are expressed as mean ± SEM (standard error of the mean) and were analyzed using GraphPad Prism 10 statistical software (GraphPad Software, Boston, MA). Sample sizes for neuronal and behavioral experiments were based on a power of 0.8 and an α = 0.05, with effect sizes estimated based on our previous studies [27,28,29, 47,48,49,50,51]. All data were treated as parametric for statistics, and equal variances were assumed when comparing means across multiple groups. Outliers were defined as individual data points with a value larger or smaller than 2 standard deviations from the mean. p < 0.05 is considered statistically significant. p values are presented as p > 0.05 (ns, not significant), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Postnatal reintroduction of NEXMIF restores NEXMIF expression in KO mice
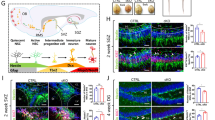
We previously developed a Nexmif knockout (KO) male mouse model which demonstrates cognitive impairments and ASD-like behaviors, accompanied by severe neuronal deficits including reduced synaptic protein expression and impaired spine morphology [27,28,29]. We wanted to determine whether early postnatal reintroduction of NEXMIF in Nexmif KO male mice could reverse the associated neuronal and behavioral deficits. To this end, we generated a human NEXMIF transgene construct driven under the human ubiquitin C (hUbC) promoter, which was packaged into lentiviral particles. To assess the efficacy of the NEXMIF lentivirus (LV-NEXMIF), wildtype rat cortical neurons cultured onto coverslips or 6-well plates and were infected with either a control lentivirus (LV-Control) or LV-NEXMIF at 7 days in vitro (DIV) and immunostained or collected for protein quantification at DIV 14. As expected, NEXMIF immunosignal was detected in the nucleus and was significantly increased in the LV-NEXMIF-infected neurons, relative to control (Fig. 1A, B). Consistently, western blot quantification showed that NEXMIF protein levels were also significantly increased in LV-NEXMIF-treated neurons (Fig. 1C). To assess the expression and rescue effects of LV-NEXMIF in vivo, we performed bilateral intracerebroventricular (ICV) injections of LV-NEXMIF in postnatal (P) day 1 Nexmif KO male mice (KO + NEX), while WT and Nexmif KO mice injected with LV-Control served as controls (WT + CTRL and KO + CTRL, respectively). Behavioral tests were conducted during the adolescent period between P30 to P70, and the brains of injected mice were collected for various biochemical analyses at either P40 or P70 (Fig. 1D). Western blotting revealed that NEXMIF protein expression was restored back to WT levels in the medial prefrontal cortical (mPFC) and hippocampal (HPC) tissue of KO + NEX mice (Fig. 1E, F). In line with protein levels, imaging of brain slices from KO + NEX mice showed significantly increased NEXMIF immunointensity in layer 2/3 medial prefrontal cortical neurons, relative to that of KO + CTRL mice (Fig. 1G, H). Taken together, these findings validate the efficacy of LV-NEXMIF in postnatally restoring NEXMIF expression in the brains of Nexmif KO mice.

A Validation of the human FUW-hUbC-NEXMIF lentivirus (LV-NEXMIF). Cultured wildtype (WT) rat cortical neurons were infected with LV-NEXMIF or FUW control lentivirus (LV-Control) at 7 days in vitro (DIV) and immunostained for NEXMIF at DIV 14. Scale bar = 5 µm. B Quantification of (A) revealed that NEXMIF nuclear intensity was significantly increased in LV-NEXMIF-treated neurons (n = 97), relative to neurons infected with LV-Control (n = 84). C Western blot from DIV 14 neuronal lysates (left) and quantification (right) showing that NEXMIF protein was also significantly increased in LV-NEXMIF-treated neurons (n = 3 wells), relative to control (n = 3 wells). D Schematic illustration of the experimental timeline. LV-Control or LV-NEXMIF virus was bilaterally injected into the ventricles of WT or Nexmif knockout (KO) mouse brains at postnatal (P) days 0–1. The three injected groups were: WT + LV-Control (WT + CTRL), KO + LV-Control (KO + CTRL), and KO + LV-NEXMIF (KO + NEX) mice. From P30-P70, injected mice were subjected to a series of behavioral tests to examine rescue of ASD-like phenotypes. Following behavioral testing, mice were sacrificed and perfused for brain slice immunostaining, or brain tissue was collected for protein quantification and Golgi staining for spine morphology. E Western blot of NEXMIF and β-tubulin protein confirming successful restoration of NEXMIF expression in the medial prefrontal cortex (mPFC, left panel) and hippocampus (HPC, right panel) of KO + NEX mice. F Quantification of the western blots in (D) normalized to β-tubulin loading control revealed significantly increased expression of NEXMIF protein in the mPFC and HPC of KO + NEX mice, relative to KO + CTRL mice (n = 4 WT + CTRL, 3 KO + CTRL, 4 KO + NEX mice). G Representative brain slice images showing NEXMIF immunostaining (top row) merged with DAPI nuclear counterstain (bottom row) from layer 2/3 neurons of the mPFC in WT + CTRL (left panel), KO + CTRL (middle panel), and KO + NEX (right panel) mice. Scale bar = 100 µm. H Quantification of brain slice immunostaining of NEXMIF in WT + CTRL, KO + CTRL, and KO + NEX mice revealed significantly increased NEXMIF immunointensity in Layer 2/3 neurons of the mPFC in KO + NEX mice, relative to that in KO + CTRL mice (WT + CTRL: n = 91 cells, KO + NEX: n = 63 cells). Data are represented as average ± SEM. Two-tailed student’s t test (B-C) or One-way ANOVA (F, H) with Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. ns, Not significant. Figure 1D created with Biorender.com.
NEXMIF restoration rescues novel object discrimination in Nexmif KO mice
At the behavioral level, we previously established that Nexmif KO male and HET female mice display reduced social preferences, overgrooming, anxiety, and impaired learning and memory in the Barnes maze and fear conditioning tests [28, 29]. To determine whether postnatal ICV injection of LV-NEXMIF could rescue the observed memory and ASD-like phenotypes in Nexmif KO mice, we conducted a series of behavioral tests between P30 and P70, the adolescent period in mice. We first examined cognitive function using the Novel Object Recognition (NOR) test to assess deficits in short-term recognition memory. During the NOR test, mice are first habituated to the NOR arena, familiarized to two identical objects, and then tasked with discriminating a novel object from the familiar object 4 h later (Fig. 2A) [47, 48, 52, 53]. The amount of time spent sniffing and interacting with each object (i.e. the exploration time) was recorded as a measure of short-term recognition memory. During the familiarization phase, WT + CTRL, KO + CTRL, and KO + NEX mice spent a similar amount of time on average exploring each object, indicating no bias towards either of the familiar objects (Fig. 2B, C). During the test phase, while the WT + CTRL mice expectedly spent a significantly longer amount of time exploring the novel object over the familiar object, the KO + CTRL mice spent an approximately equal amount of time exploring both novel and familiar objects, indicating a lack of novelty discrimination (Fig. 2D, E). Strikingly, KO + NEX mice spent a significantly longer time exploring the novel object than the familiar object, indicating a rescue of the short-term recognition memory deficit in KO mice (Fig. 2D, E). These results suggest that the short-term memory deficit present in Nexmif KO mice can be rescued by early postnatal NEXMIF restoration.

A Paradigm for the Novel Object Recognition Test. During the two-day habituation phase (left panel), the test mouse was placed in the center of the arena and allowed to move freely within the environment for 5 min. During the familiarization phase on the third day (middle panel), two identical objects were placed diagonally from each other, and the test mouse was allowed to explore the objects for 10 min. Four hours later during the test phase (right panel), one of the identical (familiar) objects was replaced with a novel object, and the test mouse was allowed to interact with both objects for 5 min. B Traces of mouse track paths during the familiarization phase for WT + CTRL, KO + CTRL, and KO + NEX mice. C Quantification of the exploration time during familiarization revealed no preference for either of the identical objects within each group of injected mice (n = 9 WT + CTRL, 6 KO + CTRL, 7 KO + NEX mice). D Traces of mouse track paths during the test phase for WT + CTRL, KO + CTRL, and KO + NEX mice. The star indicates the location of the novel object. E Quantification of the exploration time during the test phase revealed that adolescent KO + CTRL mice showed impaired novel object recognition, which was rescued by ICV injection of LV-NEXMIF in the KO + NEX mice. F, Grooming assay: mice were singly recorded for grooming behavior (paw licking, face cleaning, and ear/back scratching) in their home cage for 10 min. G Quantification of the time spent grooming revealed that relative to WT + CTRL mice (n = 7), KO + CTRL mice (n = 5) spent an increased amount of time grooming. However, this repetitive overgrooming phenotype was rescued in KO + NEX mice (n = 6). Data are represented as average ± SEM. Two-way ANOVA with Sidak’s multiple comparisons test (C,E) or One-way ANOVA with Tukey’s multiple comparisons test (G). *p < 0.05; **p < 0.01; ****p < 0.0001. ns, Not significant. Figure 2A, F created with Biorender.com.
NEXMIF restoration rescues repetitive grooming and anxiety in Nexmif KO mice
Repetitive behaviors are one of the hallmark features of ASD in humans [2]. Indeed, we previously found that loss of NEXMIF in adult mice is associated with repetitive overgrooming behavior, which has been linked to anxiety [28, 29, 54,55,56]. To assess for the presence of ASD-like repetitive behaviors, we performed the grooming assay, in which mice are video recorded in their home cage for 10 min and the total amount of time spent grooming is measured (Fig. 2F). Consistent with our previous findings in adult mice, adolescent KO + CTRL mice spent a significantly increased amount of time grooming, relative to WT + CTRL mice (Fig. 2G). Remarkably, this repetitive overgrooming phenotype was markedly rescued in KO + NEX mice, almost matching that of WT + CTRL mice (Fig. 2G). In addition, we examined mouse performance on the elevated zero maze (EZM) assay given the established sensitivity of the EZM test for measuring innate anxiety [57]. The EZM is a circular track version of the standard elevated plus maze (Suppl. Figure 1A), in which the amount of time spent in the open arms relative to the total distance travelled is measured. We found that while WT + CTRL mice spent a normal amount of time exploring both the open and closed arms of the EZM, adolescent KO + CTRL mice spent significantly less time in the open arms (Suppl. Figure 1B, C). Strikingly, KO + NEX mice trended towards more time spent exploring the open arms, indicating reduced anxiety-like behavior in the EZM (Suppl. Figure 1B, C). These findings demonstrate the efficacy of early postnatal NEXMIF restoration in rescuing repetitive and anxiety-like behaviors in Nexmif KO mice.
NEXMIF restoration rescues social novelty preference in Nexmif KO mice
We then performed the Three-Chamber Social test (3CST) to assess mouse sociability and interest in social novelty [28, 29, 47,48,49,50,51, 58,59,60]. We previously found that NEXMIF loss is associated with impairments in sociability, accompanied by a lack of interest in social novelty in adult Nexmif KO and HET mice [28, 29]. To examine the extent of social behavior deficits in adolescent Nexmif KO mice, as well as to determine whether postnatal NEXMIF restoration could rescue such deficits, WT + CTRL, KO + CTRL, and KO + NEX mice were subjected to the 3CST. The 3CST is divided into three parts: 1) habituation, during which mice are habituated to the three-chamber apparatus to test for bias towards either of the side chambers containing empty cages; 2) sociability, during which preference for exploring an empty chamber versus a stranger mouse (mouse 1) is measured; and 3) social novelty, during which preference for exploring a familiar mouse (mouse 1) versus a novel mouse is measured (Fig. 3A). The amount of time spent sniffing and interacting with each empty cage, mouse 1, and the novel mouse (i.e. the exploration time) was analyzed and used to calculate the social preference index (see methods). During the habituation phase, we found that WT + CTRL, KO + CTRL, and KO + NEX mice spent a similar amount of time on average exploring both empty cages, indicating that the mice showed no bias towards exploring either side chamber (Fig. 3B, E). During the sociability test, while WT + CTRL mice showed intact social preference for mouse 1, KO + CTRL mice displayed significantly reduced social preference for mouse 1, indicating an impairment in sociability behavior (Fig. 3C, F). However, this sociability deficit was partially rescued by NEXMIF restoration, as demonstrated by a trend towards increased social preference for mouse 1 in KO + NEX mice (Fig. 3C, F). Similarly, during the social novelty test, while WT + CTRL mice showed intact social preference for the novel mouse, KO + CTRL mice exhibited significantly reduced social preference for the novel mouse (Fig. 3D, G). Strikingly, we found that this impaired social novelty preference was significantly rescued in KO + NEX mice (Fig. 3D, G). These results demonstrate that adolescent Nexmif KO mice display impairments in social behavior, which can be rescued by early postnatal NEXMIF restoration.

A Paradigm for the Three-Chamber Social Test. During the two-day habituation phase (left panel), the test mouse was released from the center chamber, with empty cages in either of the side chambers, and allowed to move freely within the environment for 5 min. During the sociability test on the third day (middle panel), an unfamiliar mouse (Mouse 1) was placed into either of the side chambers and the test mouse was allowed to move freely within the environment for 5 min. During the social novelty test (right panel), a second mouse (Novel Mouse) was placed into the remaining empty chamber and the test mouse was allowed to interact with both mice for 5 min. B Traces of mouse track paths on the first day of habituation for WT + CTRL, KO + CTRL, and KO + NEX mice. C Traces of mouse track paths in the sociability test for WT + CTRL, KO + CTRL, and KO + NEX mice. The star indicates the location of Mouse 1. D Traces of mouse track paths in the social novelty test for WT + CTRL, KO + CTRL, and KO + NEX mice. The star indicates the location of the Novel Mouse. E Quantification of the exploration time during habituation revealed no preference for either side chamber within each group of injected mice (n = 8 WT + CTRL, 6 KO + CTRL, 6 KO + NEX). F Quantification of the Sociability Preference Index (PI): KO + CTRL mice showed a reduced social PI for Mouse 1, while KO + NEX mice trended towards an increased social PI for Mouse 1. G Quantification of the Social Novelty PI: KO + CTRL mice displayed an attenuated social PI for the novel mouse, which was rescued by ICV injection of LV-NEXMIF in the KO + NEX mice. Data are represented as average ± SEM. Two-way ANOVA with Sidak’s multiple comparisons test (E) or One-way ANOVA with Tukey’s multiple comparisons test (F-G). *p < 0.05; ***p < 0.001; ns, Not significant.
Effects of NEXMIF restoration on synaptic protein expression and spine morphology
In line with a role for NEXMIF in brain development, our previous work showed that loss of NEXMIF results in defects in dendritic spine formation and maturation, accompanied by altered expression of synaptic proteins [28, 29]. To determine the rescue effect of early postnatal NEXMIF restoration on spine density and synaptic protein expression in KO mice, mouse brains were collected following behavioral testing for either Golgi staining or protein expression quantification. First, we quantified spine density on the basolateral dendrites of Golgi-stained neurons in the medial PFC (Layer II/III), HPC (CA1), and dentate gyrus (DG) of WT + CTRL, KO + CTRL, and KO + NEX mice. In all three brain regions, the neurons of KO + CTRL mice showed significantly reduced spine density when compared to that of WT + CTRL neurons (Fig. 4A–D). Consistent with the restoration of NEXMIF expression in the mPFC and HPC (Fig. 1E–H), the neurons of KO + NEX mice exhibited significantly rescued spine density in the mPFC and HPC (Fig. 4B, C). A lack of rescue in the DG (Fig. 4D) was possibly due to low expression of LV-NEXMIF in this region. Next, we examined the expression of synaptic proteins involved in crucial processes such as synaptic plasticity, synaptogenesis, synapse maintenance, and learning and memory function [61,62,63]. We found significant reductions in the protein levels of GluA1, SynDIG1, and TrkB in the mPFC and HPC of KO + CTRL mice, relative to that of WT + CTRL mice (Fig. 4E–H). However, NEXMIF reintroduction was sufficient to restore GluA1, SynDIG1, and TrkB expression back to WT + CTRL levels in the mPFC and HPC of KO + NEX mice (Fig. 4E–H), in addition to several other synaptic and neuronal proteins (Suppl. Figure 2). Overall, these findings indicate that loss of NEXMIF results in aberrant spine morphogenesis and reduced synaptic protein expression in adolescent Nexmif KO mice, which may contribute to the development of memory deficits and ASD-like behaviors. Importantly, early postnatal restoration of NEXMIF in the brain can effectively correct these cellular and molecular impairments in Nexmif KO mice.

A Representative Golgi stain images of dendritic spines in the mPFC (Layer 2/3, left column), Hippocampus (CA1, middle column), and Dentate Gyrus (right column) from P70 WT + CTRL (top row), KO + CTRL (middle row), and KO + NEX mice (bottom row). Scale bar = 2 µm. B–D, Quantification of spine density (# of spines / length of dendrite) revealed that NEXMIF restoration reversed impaired spine density in (B) layer 2/3 cortical neurons and in (C) hippocampal CA1 neurons, but not in (D) dentate gyrus neurons likely due to lack of viral penetrance in this region (B: n = 12, 13, 13; C: n = 18, 18, 17; D: n = 12, 15, 13 dendrites). E Representative western blot images showing the expression of the various synaptic proteins (GluA1, SynDIG1, TrkB) examined in the medial prefrontal cortex (mPFC, left) and hippocampus (HPC, right) of WT + CTRL (n = 3–4), KO + CTRL (n = 3), and KO + NEX (n = 3–4) mice. F–H, Quantification of the mPFC and HPC western blots shown in (E) normalized to β-tubulin loading control revealed that postnatal LV-NEXMIF injection in Nexmif KO mice significantly rescued the expression of the GluA1, SynDIG1, and TrkB synaptic proteins, with all three proteins restored to WT levels. Data are represented as average ± SEM. One-way ANOVA (B-D, F-H) with Bonferroni’s or Tukey’s Multiple Comparisons. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, Not significant.
NEXMIF restoration globally rescues the expression of dysregulated genes associated with Nexmif loss
Because NEXMIF is enriched in the nucleus and thus presumably plays an important role in gene regulation, we previously conducted RNA sequencing on hippocampal tissue from P90 Nexmif KO mice and found significant dysregulation in the expression of several genes relative to WT control tissue. Several of the observed downregulated genes were involved in crucial synapse and morphological development pathways, such as glutamate receptor signaling, synaptogenesis signaling, and synaptic long-term potentiation and depression (data not shown). To examine whether postnatal NEXMIF restoration could rescue the overall landscape of the transcriptome in Nexmif KO mice, we conducted RNA sequencing on hippocampal tissue extracted from P90 WT + CTRL, KO + CTRL, and KO + NEX mice. As expected, the analysis revealed significant dysregulation of gene expression in KO + CTRL hippocampal tissue: out of the top 125 differentially expressed genes (DEGs), 78 (62%) were downregulated and 47 (38%) were upregulated relative to WT + CTRL tissue (Fig. 5A). Remarkably, we found that NEXMIF reintroduction in KO + NEX mice was sufficient to restore the expression of 58 of the top DEGs by 70% or more. We selected 3 candidate genes from the top DEGs and validated their changes in expression by RT-qPCR and western blot. Indeed, we confirmed that expression levels of Cerebellin-1, Magel2, and Glycine receptor alpha-1 (Glra1) in the hippocampus were significantly reduced in KO + CTRL tissue and recovered in KO + NEX tissue (Suppl. Figure 3). Moreover, gene ontology (GO) enrichment analysis revealed that the majority of DEGs were strongly involved in biological processes such as regulation of post-synaptic membrane potential, learning or memory, cognition, neuropeptide signaling, G-protein coupled receptor signaling pathway, axon guidance, central nervous system neuron differentiation, axon development, chemical synaptic transmission, generation of neurons, neuron development, regulation of behavior, brain development, and neuron migration (Fig. 5B). Indeed, the majority of DEGs in each biological process were downregulated, and the expression of 20–80% of the genes within each biological process were restored by over 70% in KO + NEX mice (Fig. 5C). These results further implicate NEXMIF as a crucial regulator of gene transcription in the brain since its restoration in KO mice was sufficient to rescue the expression of a large majority of hippocampal genes, along with significant cellular and behavioral rescue.

A RNA sequencing heat map showing the normalized expression values of the top 125 differentially expressed genes (DEGs) in the hippocampus (HPC) of a WT + CTRL mouse (left column), a KO + CTRL mouse (middle column), and a KO + NEX mouse (right column). 78 DEGs (62%) were downregulated and 47 DEGs (38%) were upregulated in the KO + CTRL mouse brain relative to the WT + CTRL mouse brain. Of the 125 DEGs, 58 were restored in expression by greater than 70% in the KO + NEX mouse brain. B Gene Ontology (GO) analysis bar chart depicting the most enriched biological processes. The total percentage of each bar reflects the total number of DEGs from the dataset within each GO biological process. Each bar is partitioned by the percentage of downregulated (green) versus upregulated (red) DEGs in the KO + CTRL mouse brain relative to that of WT + CTRL. C Gene Ontology (GO) analysis bar chart indicating the percentage of “Rescued” versus “Not Rescued” DEGs. The total percentage of each bar reflects the total number of DEGs from the dataset within each GO biological process. Each bar is partitioned into the percentage of “Rescued” (red) versus “Not rescued” (gray) DEGs in the KO + NEX mouse brain relative to that of WT + CTRL. A “Rescued” DEG is defined as a gene whose expression was restored in the KO + NEX brain to within ± 30% of its WT + CTRL expression level. For example, Gene A has an expression level of 100 arbitrary units (AU) in the WT + CTRL condition. Gene A is downregulated to 15 AU in the KO + CTRL condition. In the KO + NEX condition, Gene A has an expression level of 90 AU. Gene A is considered as “rescued” because its expression level of 90 AU in the KO + NEX condition is within the ± 30% range (70–130) of Gene A’s basal expression level in the WT + CTRL condition.
Discussion
In this proof-of-concept study, we demonstrated that early postnatal restoration of NEXMIF protein using a human NEXMIF lentivirus can successfully rescue cellular, molecular, and behavioral phenotypes in Nexmif KO male mice by adolescent (P30-70) ages. In our previous study, we examined cellular impairments and autistic-like behaviors only in adult (3-6–month-old) KO male mice [28]. Here, we observed similar neuronal and behavioral impairments in the adolescent KO mice, including reduced synaptic protein expression, altered spine morphology, dysregulated gene expression, attenuated novel object discrimination, decreased sociability, lack of preference for social novelty, repetitive overgrooming, and anxiety-like behavior, which can be reversed by NEXMIF restoration. These encouraging findings lay the groundwork for future studies aimed at developing therapeutic strategies for NEXMIF-dependent ASD.
There is a growing literature concerning the use, feasibility, and timing of gene restoration techniques in ASD mouse models [31,32,33,34,35,36,37,38,39,40,41, 64, 65]. These studies support the underlying conclusion that viral-, CRISPR/Cas9-, or Cre-based gene reinstatement should preferably be administered during the early postnatal period in order to achieve the highest therapeutic effect at the cellular and behavioral levels. For example, postnatal activation of Mecp2 between ages P15-P30 in a Rett syndrome mouse model had a much less robust rescue effect on motor activity and life span than when activated at P0 [32]. Additionally, viral delivery of MeCP2 at neonatal ages led to increased survival and motor function in Mecp2 null mice [36], while viral CRISPR/Cas9-induced knockout of the UBE3A antisense transcript during embryonic and neonatal periods in Angelman syndrome mice rescued anatomical and cognitive phenotypes by 3–7 months-of-age [38].
The Nexmif gene is especially crucial during the early neurodevelopmental period, with peak mRNA expression at P3 [25]. We found that ICV injection of LV-NEXMIF as early as P0-1 was sufficient to either reverse or impede the development of neural and behavioral impairments that would otherwise occur during early development of the Nexmif KO mouse brain. The P0-3 timepoint in mice has been shown to be equivalent to the 17–20-week gestational period in humans [66]. From the time of lentiviral ICV injection in neonatal mice, it takes roughly 2–3 weeks to observe transgene expression. While it is difficult to know how the 2-3–week expression period in mice would translate to expression in the human neonatal condition, our findings and that of others nonetheless support that early postnatal intervention via gene restoration can effectively rescue impaired phenotypes in ASD mouse models. Interestingly, however, Cre-based restoration of SHANK3 in 4-month-old Shank3 haploinsufficient female mice led to reversal of social deficits that lasted 5–8 weeks following restoration [37], and engrafting of human stem cells containing viral UBE3A at adolescent ages (P30-40) in Ube3a-immunodeficient mice significantly rescued motor, cognitive, and brain activity deficits by 4–5 months-of-age [34], suggesting that gene therapy intervention at adult ages can also likely reverse ASD-like phenotypes in mice.
To reintroduce NEXMIF, a lentiviral vector was used, which successfully increased NEXMIF expression in the brain. However, because the vector contained a ubiquitous promoter, it is possible that viral NEXMIF also expressed in non-neuronal cell types (e.g. glia). The NEXMIF virus led to robust rescue of the KO phenotypes, and no new phenotypes were detected in the KO + NEX mice. These findings are in line with restoration of NEXMIF in neurons and suggest that any side effects from the possible infection of glia were insignificant. Nevertheless, in our future studies, we will employ an adenoviral NEXMIF construct under the neuron-specific SYNAPSIN1 promoter to avoid potential nonspecific effects. Moreover, the use of an adenoviral vector also serves as a more clinically viable viral alternative for future therapeutic strategies.
In this study, we examined the effects of NEXMIF reinstatement only in KO males; however, the effectiveness of this strategy in females remains to be tested. We have previously shown that our heterozygous (HET) female mouse model demonstrates NEXMIF haploinsufficiency [29]. Due to X-chromosome inactivation, the HET brain exhibits a mosaic pattern of NEXMIF expression: some cells express NEXMIF (WT), while other cells lack NEXMIF (KO) [29]. Therefore, while the introduction of viral NEXMIF may restore its expression in the KO neurons, overdosage of NEXMIF is likely to occur in the WT neurons, which could potentially lead to functional dysregulation. Thus, in our future work, a cell type-specific rescue strategy will be considered for HET females. It is also of interest to determine the latest time point at which NEXMIF restoration could still result in cellular and behavioral rescue in KO mice. Findings from such a study would have major implications for families with teenagers and young adults with NEXMIF-dependent ASD who seek effective intervention at these ages.
Because the primary effect of NEXMIF loss is on transcriptional dysregulation, it is of value to determine whether modulating the expression of significantly dysregulated downstream genes in Nexmif KO mice can rescue specific cellular and/or behavioral phenotypes. In our study, several synapse function genes, such as Cerebellin-1 (excitatory synapse development) [67,68,69], Magel2 (endocytosis and receptor recycling) [70,71,72], and Glra1 (postsynaptic inhibition) [73,74,75] were downregulated by over 16-fold in the KO mouse hippocampus. It is likely that postnatal re-expression of these downstream candidate genes in KO mice may specifically attenuate synaptic dysfunction and ameliorate the associated cognitive impairments, anxiety, and social deficits [76,77,78,79,80]. Such knowledge would further our understanding of the molecular basis of NEXMIF-dependent ASD and provide guidance for alternative rescue strategies.
Data availability
The data that support the findings of this study are available upon request from the corresponding author. The RNA sequencing data is available at https://doi.org/10.6084/m9.figshare.29242037.v2 [81].
References
Landa RJ. Diagnosis of autism spectrum disorders in the first 3 years of life. Nat Clin Pract Neurol. 2008;4:138–47.
Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, et al. Autism spectrum disorder. Nat Rev Dis Primer. 2020;6:5.
Bougeard C, Picarel-Blanchot F, Schmid R, Campbell R, Buitelaar J. Prevalence of autism spectrum disorder and co-morbidities in children and adolescents: a systematic literature review. Front Psychiatry. 2021;12:744709.
Khachadourian V, Mahjani B, Sandin S, Kolevzon A, Buxbaum JD, Reichenberg A, et al. Comorbidities in autism spectrum disorder and their etiologies. Transl Psychiatry. 2023;13:1–7.
Piton A, Redin C, Mandel JL. XLID-Causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet. 2013;93:368–83.
Nguyen TA, Lehr AW, Roche KW. Neuroligins and neurodevelopmental disorders: X-linked genetics. Front Synaptic Neurosci. 2020;12:33.
Wang S, Wang B, Drury V, Drake S, Sun N, Alkhairo H, et al. Rare X-linked variants carry predominantly male risk in autism, Tourette syndrome, and ADHD. Nat Commun. 2023;14:8077.
Kuroda Y, Ohashi I, Naruto T, Ida K, Enomoto Y, Saito T, et al. Delineation of the KIAA2022 mutation phenotype: two patients with X‐linked intellectual disability and distinctive features. Am J Med Genet A. 2015;167:1349–53.
Oliver KL, Scheffer IE, Bennett MF, Grinton BE, Bahlo M, Berkovic SF. Genes4Epilepsy: an epilepsy gene resource. Epilepsia. 2023;64:1368–75.
Stamberger H, Hammer TB, Gardella E, Vlaskamp DRM, Bertelsen B, Mandelstam S, et al. NEXMIF encephalopathy: an X-linked disorder with male and female phenotypic patterns. Genet Med Off J Am Coll Med Genet. 2021;23:363–73.
Van Maldergem L, Hou Q, Kalscheuer VM, Rio M, Doco-Fenzy M, Medeira A, et al. Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Hum Mol Genet. 2013;22:3306–14.
Cantagrel V. Disruption of a new X linked gene highly expressed in brain in a family with two mentally retarded males. J Med Genet. 2004;41:736–42.
Cantagrel V, Haddad MR, Ciofi P, Andrieu D, Lossi AM, Maldergem LV, et al. Spatiotemporal expression in mouse brain of Kiaa2022, a gene disrupted in two patients with severe mental retardation. Gene Expr Patterns. 2009;9:423–9.
Lange, de IM, Helbig KL, Weckhuysen S, Møller RS, Velinov M, Dolzhanskaya N, et al. De novo mutations of KIAA2022 in females cause intellectual disability and intractable epilepsy. J Med Genet. 2016;53:850–8.
Zhong L, Liu C, Lin L. Infantile spasms caused by NEXMIF mutation: A case report and literature review. Appl Neuropsychol Child. 2023;12:380–5.
Chen S, Deng X, Xiong J, Chen B, He F, Yang L, et al. NEXMIF mutations in intellectual disability and epilepsy: a report of 2 cases and literature review. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2022;47:265–70.
Qi H, Pan D, Zhang Y, Zhu Y, Zhang X, Fu T. NEXMIF combined with KIDINS220 gene mutation caused neurodevelopmental disorder and epilepsy: one case report. Actas Esp Psiquiatr. 2024;52:588–94.
He W, Liang Y, Yan H, Wan L, Yang G. [Clinical and genetic analysis of a child with West syndrome due to a de novo variant of NEXMIF gene]. Chin J Med Genet. 2024;41:725–9.
Jogi SV, Kumar VS, Kamble N, Rangaswamy DR. A CHILD WITH X LINKED INHERITED INTELLECTUAL DISABILITY. J Chitwan Med Coll. 2024;14:94–5.
Gamirova RG, Barkov AI, Shaimuchametova VA, Liukshina NG, Volkov IV, Tomenko TR, et al. Epilepsy and other phenotypic features of X-linked intellectual disability due to mutations in the KIAA2022 gene. Neurosci Behav Physiol. 2023;53:767–71.
Caraballo RH, Reyes Valenzuela G, Fortini S, Espeche A, Gamboni B, Silva W, et al. Cannabidiol in children with treatment-resistant epilepsy with myoclonic-atonic seizures. Epilepsy Behav. 2023;143:109245.
Ye ZL, Yan HJ, Guo QH, Zhang SQ, Luo S, Lian YJ, et al. NEXMIF variants are associated with epilepsy with or without intellectual disability. Seizure Eur J Epilepsy. 2024;116:93–9.
Alarcon-Martinez T, Khan A, Myers KA. Torpedo maculopathy associated with NEXMIF mutation. Mol Syndromol. 2019;10:229–33.
Lorenzo M, Stolte-Dijkstra I, van Rheenen P, Smith RG, Scheers T, Walia JS. Clinical spectrum of KIAA2022 pathogenic variants in males: Case report of two boys with KIAA2022 pathogenic variants and review of the literature. Am J Med Genet A. 2018;176:1455–62.
Ishikawa T, Miyata S, Koyama Y, Yoshikawa K, Hattori T, Kumamoto N, et al. Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience. 2012;214:181–91.
Magome T, Hattori T, Taniguchi M, Ishikawa T, Miyata S, Yamada K, et al. XLMR protein related to neurite extension (Xpn/KIAA2022) regulates cell–cell and cell–matrix adhesion and migration. Neurochem Int. 2013;63:561–9.
Gilbert J, Man HY. The X-linked autism protein KIAA2022/KIDLIA regulates neurite outgrowth via N-Cadherin and δ-Catenin signaling. eNeuro. 2016;3:ENEURO.0238-16.2016.
Gilbert J, O’Connor M, Templet S, Moghaddam M, Di Via Ioschpe A, Sinclair A, et al. NEXMIF/KIDLIA knock-out mouse demonstrates autism-like behaviors, memory deficits, and impairments in synapse formation and function. J Neurosci. 2020;40:237–54.
O’Connor M, Qiao H, Odamah K, Cerdeira PC, Man HY. Heterozygous Nexmif female mice demonstrate mosaic NEXMIF expression, autism-like behaviors, and abnormalities in dendritic arborization and synaptogenesis. Heliyon. 2024;10:e24703.
Stekelenburg C, Blouin JL, Santoni F, Zaghloul N, O’Hare EA, Dusaulcy R, et al. Loss of Nexmif results in the expression of phenotypic variability and loss of genomic integrity. Sci Rep. 2022;12:13815.
Judson MC, Shyng C, Simon JM, Davis CR, Punt AM, Salmon MT, et al. Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight. 2021;6:e144712.
Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci USA. 2007;104:1931–6.
Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature. 2016;530:481–4.
Adhikari A, Copping NA, Beegle J, Cameron DL, Deng P, O’Geen H, et al. Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum Mol Genet. 2021;30:1067–83.
Derbis M, Kul E, Niewiadomska D, Sekrecki M, Piasecka A, Taylor K, et al. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat Commun. 2021;12:1265.
Gadalla KKE, Bailey MES, Spike RC, Ross PD, Woodard KT, Kalburgi SN, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther J Am Soc Gene Ther. 2013;21:18–30.
Lee DK, Li SW, Bounni F, Friedman G, Jamali M, Strahs L, et al. Reduced sociability and social agency encoding in adult Shank3-mutant mice are restored through gene re-expression in real time. Nat Neurosci. 2021;24:1243–55.
Wolter JM, Mao H, Fragola G, Simon JM, Krantz JL, Bazick HO, et al. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature. 2020;587:281–4.
Sztainberg Y, Chen Hmei, Swann JW, Hao S, Tang B, Wu Z, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligos. Nature. 2015;528:123–6.
Yu B, Yuan B, Dai JK, Cheng TL, Xia SN, He LJ, et al. Reversal of social recognition deficit in adult mice with MECP2 duplication via normalization of MeCP2 in the medial prefrontal cortex. Neurosci Bull. 2020;36:570–84.
Wang N, Lv L, Huang X, Shi M, Dai Y, Wei Y, et al. Gene editing in monogenic autism spectrum disorder: animal models and gene therapies. Front Mol Neurosci. 2022;15:1043018.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Dendritic Spine Counter. Available from: https://imagej.github.io/plugins/dendritic-spine-counter.
Zhang C, Li H, Han R. An open-source video tracking system for mouse locomotor activity analysis. BMC Res Notes. 2020;13:48.
Mathis A, Mamidanna P, Cury KM, Abe T, Murthy VN, Mathis MW, et al. DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat Neurosci. 2018;21:1281–9.
Fournier LA, Phadke RA, Salgado M, Brack A, Nocon JC, Bolshakova S, et al. Overexpression of the schizophrenia risk gene C4 in PV cells drives sex-dependent behavioral deficits and circuit dysfunction. iScience. 2024;27:110800.
Qiao H, Tian Y, Huo Y, Man HY. Role of the DUB enzyme USP7 in dendritic arborization, neuronal migration, and autistic-like behaviors in mice. iScience. 2022;25:104595.
Tian Y, Qiao H, Odamah K, Zhu LQ, Man HY. Role of androgen receptors in sexually dimorphic phenotypes in UBE3A-dependent autism spectrum disorder. iScience. 2025;28:111868.
Huo Y, Lu W, Tian Y, Hou Q, Man HY. Prkn knockout mice show autistic-like behaviors and aberrant synapse formation. iScience. 2022;25:104573.
Tian Y, Yu F, Yun E, Lin JW, Man HY. mRNA nuclear retention reduces AMPAR expression and promotes autistic behavior in UBE3A-overexpressing mice. EMBO Rep. 2024;25:1282–309.
Gardner Z, Holbrook O, Tian Y, Odamah K, Man HY. The role of glia in the dysregulation of neuronal spinogenesis in Ube3a-dependent ASD. Exp Neurol. 2024;376:114756.
Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, et al. Object recognition test in mice. Nat Protoc. 2013;8:2531–7.
Lueptow LM Novel object recognition test for the investigation of learning and memory in mice. J Vis Exp JoVE. 2017:55718.
CDC. Autism Spectrum Disorder (ASD). 2024. Available from: https://www.cdc.gov/autism/signs-symptoms/index.html.
Maenner MJ. Prevalence and characteristics of autism spectrum disorder among children aged 8 years — Autism and developmental disabilities monitoring network, 11 sites, United States, 2020. MMWR Surveill Summ. 2023. Available from: https://www.cdc.gov/mmwr/volumes/72/ss/ss7202a1.htm.
Kalueff AV, Stewart AM, Song C, Berridge KC, Graybiel AM, Fentress JC. Neurobiology of rodent self-grooming and its value for translational neuroscience. Nat Rev Neurosci. 2016;17:45–59.
Kulkarni SK, Singh K, Bishnoi M. Elevated zero maze: a paradigm to evaluate antianxiety effects of drugs. Methods Find Exp Clin Pharmacol. 2007;29:343–8.
Szabó J, Renczés E, Borbélyová V, Ostatníková D, Celec P. Assessing sociability using the Three-Chamber Social Interaction Test and the Reciprocal Interaction Test in a genetic mouse model of ASD. Behav Brain Funct. 2024;20:24.
Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol Zurich Switz. 2007;17:448–59.
Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL, et al. Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. 2009;8:129–42.
Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–89.
Kalashnikova E, Lorca RA, Kaur I, Barisone GA, Li B, Ishimaru T, et al. SynDIG1: an activity-regulated, AMPA- receptor-interacting transmembrane protein that regulates excitatory synapse development. Neuron. 2010;65:80–93.
Enkavi G, Girych M, Moliner R, Vattulainen I, Castrén E. TrkB transmembrane domain: bridging structural understanding with therapeutic strategy. Trends Biochem Sci. 2024;49:445–56.
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–12.
Markati T, Duis J, Servais L. Therapies in preclinical and clinical development for Angelman syndrome. Expert Opin Investig Drugs. 2021;30:709–20.
Li J, Pan L, Pembroke WG, Rexach JE, Godoy MI, Condro MC, et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun. 2021;12:3958.
Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, et al. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci. 2005;8:1534–41.
Ito-Ishida A, Miura E, Emi K, Matsuda K, Iijima T, Kondo T, et al. Cbln1 regulates rapid formation and maintenance of excitatory synapses in mature cerebellar purkinje cells in vitro and in vivo. J Neurosci. 2008;28:5920–30.
Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science. 2010;328:363–8.
Reichova A, Schaller F, Bukatova S, Bacova Z, Muscatelli F, Bakos J. The impact of oxytocin on neurite outgrowth and synaptic proteins in Magel2-deficient mice. Dev Neurobiol. 2021;81:366–88.
Crutcher E, Pal R, Naini F, Zhang P, Laugsch M, Kim J, et al. mTOR and autophagy pathways are dysregulated in murine and human models of Schaaf-Yang syndrome. Sci Rep. 2019;9:15935.
Ates T, Oncul M, Dilsiz P, Topcu IC, Civas CC, Alp MI, et al. Inactivation of Magel2 suppresses oxytocin neurons through synaptic excitation-inhibition imbalance. Neurobiol Dis. 2019;121:58–64.
Lynch JW. Native glycine receptor subtypes and their physiological roles. Neuropharmacology. 2009;56:303–9.
Samarut E, Chalopin D, Riché R, Allard M, Liao M, Drapeau P. Individual knock out of glycine receptor alpha subunits identifies a specific requirement of glra1 for motor function in zebrafish. PloS One. 2019;14:e0216159.
Ferraroli E, Perulli M, Veredice C, Contaldo I, Quintiliani M, Ricci M, et al. Hereditary hyperekplexia: a new family and a systematic review of GLRA1 Gene-related phenotypes. Pediatr Neurol. 2022;132:45–9.
Fountain MD, Tao H, Chen CA, Yin J, Schaaf CP. Magel2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty. Genes Brain Behav. 2017;16:592–600.
Otsuka S, Konno K, Abe M, Motohashi J, Kohda K, Sakimura K, et al. Roles of Cbln1 in Non-Motor Functions of Mice. J Neurosci. 2016;36:11801–16.
Krishnan V, Stoppel DC, Nong Y, Johnson MA, Nadler MJS, Ozkaynak E, et al. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature. 2017;543:507–12.
Schaefer N, Roemer V, Janzen D, Villmann C. Impaired glycine receptor trafficking in neurological diseases. Front Mol Neurosci. 2018;11:291.
Schaefer N, Signoret-Genest J, von Collenberg CR, Wachter B, Deckert J, Tovote P, et al. Anxiety and startle phenotypes in Glrb spastic and Glra1 spasmodic mouse mutants. Front Mol Neurosci. 2020;13:152.
Odamah K, Man H. Nexmif knockout mice RNA sequencing data. figshare. 2025. https://doi.org/10.6084/m9.figshare.29242037.v2.
Acknowledgements
We would like to thank the Man Lab members for their insightful input and Chris Shortreed for her assistance with mouse husbandry. This work was supported by R01 MH130600, R21 MH133014, and R21 MH134174. The authors declare no competing financial interests.
Author information
Authors and Affiliations
Contributions
KO: Data Curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – Original Draft, Writing – Review & Editing, Conceptualization; HYM: Conceptualization, Data Curation, Funding Acquisition, Investigation, Methodology, Project Administration, Supervision, Validation, Visualization, Writing – Review & Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All of the procedures and methods involving animal use followed the policies of the National Institutes of Health (NIH) and have been approved by the Boston University Institutional Animal Care and Use Committee (IACUC, PROTO201800574).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Odamah, K., Man, HY. Restoration of NEXMIF expression rescues abnormalities in gene transcription, neuron maturation and autistic-like behaviors in Nexmif knockout mice. Transl Psychiatry 15, 361 (2025). https://doi.org/10.1038/s41398-025-03537-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03537-7

{kind=link}
{kind=link}
{kind=link}