
Abstract
Systemic inflammation has been linked with major depressive episode (MDE) severity and treatment-resistant depression (TRD), but not for all patients. Brain mechanisms underlying these processes are still under investigation. Objectives: based on an integrative approach, we aimed at identifying clinical, inflammatory and perfusion markers predictive of depression outcome at 6 months. We conducted a longitudinal study including 60 patients diagnosed with MDE, focusing on anxiety and anhedonia as main clinical candidates, inflammation (C-Reactive Protein - CRP) and cerebral blood flow (CBF) using pseudo-continuous arterial spin labeling (pcASL) MRI. A bootstrapped elastic net regression analysis was conducted including clinical, CBF and inflammation as predictors with depressive severity at 6 months as the dependent variable. Our findings exhibited positive association of depression outcome with baseline depression intensity, duration of current episode, CRP, right accumbens, as well as left and right orbito-frontal CBF. Negative predictors were age, disease duration, right and left caudate nuclei, left amygdala, left mid frontal gyrus, and right ventromedial prefrontal cortex CBF. Neither anxiety nor anhedonia were significant predictors. Combining clinical, inflammation and brain imaging outperformed other models in diagnosing depression severity change over time, highlighting the interest of integrative approaches. These results suggested that systemic inflammation and cerebral perfusion abnormalities in key regions involved in emotion, reward processing and decision making, may serve as biomarkers for identifying patients at risk for persistence of depression.
Similar content being viewed by others


Introduction
A putative biological mechanism underlying treatment resistance in depression is the activation of the immuno-inflammatory system [1,2,3,4]. There is now converging evidence on dysregulation in both the innate and adaptive immune systems that may impact the course of mood depressive disorder. Elevated inflammation markers are found in approximately a quarter of patients with major depressive disorder (MDD) [5], which is also associated with poor responses to antidepressants [6]. Studying inflammation processes associated with brain physiology and their clinical correlates could improve our understanding of this subtype of inflamed MDD.
Recent studies in the field of immunopsychiatry have consistently reported that pro-inflammatory mechanisms and systemic inflammation were involved in the onset of psychiatric disorders, including MDD [3, 7]. Furthermore, patients with autoimmune pathologies or receiving pro-inflammatory treatments, such as interferon alpha, have an increased risk of MDD major depressive episode [8], suggesting a bidirectional link between these two conditions [3]. Inconsistent efficacy of anti-inflammatory treatments in depression might arise from variations in the inflammatory alterations amongst depressed patients, pointing out the benefits of targeting specific profiles [9]. The low-grade inflammation in depression relates to increased levels of inflammatory mediator, including peripheral biomarkers such as C-reactive protein (CRP) [10]. Proinflammatory cytokines, responsible for stimulating CRP production, can decrease the availability and metabolism of serotonin and tryptophan [11]. The observation of a common increase in proinflammatory cytokine and the peripheral expression of genes associated with cytokines in treatment-resistant depression has led to the consideration of the specific action of CRP on the antidepressant response [12].
The question of specific clinical profiles in these patients whose inflammation would interfere with persistence of depressive symptoms over time remain unclear [1]. Some works have reported that inflammation markers, such as CRP, may be associated with specific symptoms, foremost among them being anhedonia and anxiety [6, 13]. Individuals exposed to different inflammatory triggers consistently display alterations in reward processing pathways and threat sensitivity, clinically reflected by anxiety and anhedonia [14,15,16]. Inversely, improvement in anhedonia and anxiety symptoms were observed by inhibiting specific inflammatory cytokines [17, 18].
Focusing on the brain regions or neurocircuitry associated with anhedonia and anxiety could help clarify the pathophysiology in these subsets of patients potentially exposed to immune-inflammatory activation. Neuroimaging studies have highlighted the relationship between peripheral and microglial inflammation not only with whole brain activity [19], but also within specific regions relevant to emotional and stress processing [15,20, 21]. Recent research has established significant connections between inflammation markers and various neuroimaging patterns, depressive symptoms and depression subtypes. For instance, Chung et al. found a negative correlation between high-sensitivity CRP (hs-CRP) levels and the volume of the orbitofrontal cortex in euthymic bipolar disorder (BD) patients [22]. Similarly, Tseng et al. reported that increased hs-CRP was associated with increased dorsal corticostriatal connectivity and poorer executive functions in euthymic BD [23]. In MDD, increased hs-CRP and interleukin-6 (IL-6) levels were linked to reduced hippocampal volumes [24]. Furthermore, Byrne et al. demonstrated that experimentally induced inflammation affects the ventral striatum and medial prefrontal cortex (PFC), altered reward processing and emotional stimuli responses in both healthy individuals and MDD patients [25]. Burrows et al. found that MDD patients with high CRP levels showed attenuated reward responses in the dorsal caudate, thalamus, left insula and left precuneus [26]. Additionally, Mehta et al. showed that increased CRP levels predicted decreased amygdala-ventromedial PFC connectivity, correlating with anxiety in MDD patients [27]. Collectively, these studies emphasize the intricated relationship between peripheral inflammation and specific brain structural and functional changes, underscoring the role of inflammation in the pathophysiology of depression and its outcome.
Moreover, inflammation has been reported to have deleterious effects on brain perfusion, which is bidirectionally linked to depressive phenotypes [28]. Microvascular alterations have been causally associated with the onset of late-life depression [29]. Specifically, inflammation triggered by stress has been hypothesized to contribute to microvascular changes through endothelial dysfunction [29, 30]. A common pathological mechanism underlying treatment resistance is thought to involve blood-brain barrier dysfunction, which facilitates an increased influx of inflammatory cells into the brain [30]. This evidence underscores the importance of exploring neurovascular coupling as a potential marker of inflammation’s effects on the brain in relation to depression. In this context, arterial spin labeling perfusion MRI emerges as a particularly relevant imaging modality for investigating these processes.
Indeed, arterial spin labeling (ASL) MRI serves as a dependable substitute for nuclear perfusion imaging methods [31, 32]. Perfusion abnormalities were repeatedly highlighted in MDD [33,34,35,36,37,38] within various cortical and limbic brain regions known to be linked with depressive symptoms. ASL could serve as a valuable indicator for detecting these perfusion variations and link them to various subtypes of depression [33, 39]. However, to our knowledge there are currently no ASL studies examining the cerebral perfusion correlates of specific clinical dimensions such as anhedonia or anxiety and inflammation in depression, which could deepen the understanding of their underlying neurobiological mechanism. Thus, no study has investigated the perfusion correlates of cerebral areas associated with anhedonia, anxiety, inflammation and persistence of depressive symptoms over a 6-month period.
Adopting an integrative approach, we combined clinical, biological and perfusion imaging sets of variables to determine the relationship between clinical dimensions associated with treatment resistant depression - anxiety and anhedonia - inflammation and their perfusion correlates on a priori selected brain regions known to be involved in those symptoms. Specifically, we tested how these associations might predict depressive symptoms over 6 months. We hypothesized that an anhedonic and/or anxious symptom, perfusion characteristics of brain region involved in emotion regulation and reward processing, and CRP peripheral blood level could predict depression severity at 6 months follow-up.
Methods
Participants
Participants, aged of 18 years old and above, were recruited from a prospective monocentric study named LONGIDEP conducted in routine care in the academic psychiatric department of Rennes, from November 2014 to August 2018.
Main inclusion criteria were a current major depressive episode (MDE) as per the criteria outlined in DSM-IV TR, as part of a unipolar or bipolar disorder with a Montgomery-Åsberg Depression Rating Scale (MADRS) score of 15 or higher at the time of enrolment.
Non-inclusion criteria were any other psychiatric or neurodegenerative disorder, except anxiety comorbidities (generalized anxiety, post-traumatic stress disorder, social phobia, disorder, panic disorder), a current concomitant severe medical conditions, a history of severe head trauma, a detectable MRI lesion or an abnormal MRI, or involuntary commitment at the time of inclusion. Additional non-inclusion criteria included possible safety contraindications for MRI scans. Figure 1 represents the study flow chart.

Flow chart: details of enrollment flow.
Patients were routinely followed up by their physicians as described in a previous published study [40], with visit frequency determined by their clinical state. If patients did not achieve a 20% improvement on the MADRS scale, their medication dose was either increased or changed. Medication adjustments were based on international guidelines [41,42,43,44].
All participants provided a written informed consent after a complete description of the study, and the study received approval of its protocol from a national ethics committee (CHU Rennes) before being registered on www.clinicaltrial.gov (NCT02286024).
Clinical assessment
Patients were clinically assessed by a trained psychiatrist. A Mini-International Neuropsychiatric Interview (M.I.N.I.) [45] was performed to rule out any excluding psychiatric comorbidities. Depressive symptoms were assessed using the MADRS [46], a widely used scale in the field based on its psychometric performances, and a good sensitivity to change [47]. A reassessment of the MADRS at 6 months was used as our primary outcome, focusing directly on depressive severity at follow-up rather than the change from baseline.
Anxiety was assessed using the two subscales of the Spielberger State–Trait Anxiety Inventory (STAI), STAI-YA and STAI-YB scales [48]. STAI-YA assessed state anxiety which refers to current apprehension and nervousness. It served as an indicator of transient changes in anxiety caused by threatening or aversive situations. STAI-YB assessed trait anxiety, which refers to usual apprehension and nervousness. Its aim was to identify anxiety as a personality trait, denoting enduring variances in an individual’s inclination towards anxiety across different situations [48].
Anhedonia, a clinical dimension characterized by a loss of the ability to experience pleasure in normally pleasant situations, was measured using the Snaith-Hamilton Pleasure Scale (SHAPS) [49]. The dichotomous scoring (strongly disagree/disagree = 1; strongly agree/agree = 0) for the SHAPS was used. It aimed to evaluate processes linked to the neurological circuits of reward and pleasure, involving mechanisms of evaluation and anticipation of the reward that motivates decision-making [50].
Biological assessment
Fasting blood draw were done within a few days after patient consent. Patients with current infectious condition, with a history of stable or unstable chronic inflammatory disease or undergoing immunomodulatory treatment (including anti-inflammatory drugs or related medications, and antibiotics) were excluded. Based on the existing literature [5, 51], CRP level greater than or equal to 10 mg/L led to patient exclusion, as there was a substantial likelihood that the elevated level could be attributed to a factor unrelated to MDD.
MRI data acquisition
Each participant underwent an MRI scan in the radiology department of Rennes University Hospital on the NeurInfo platform, an imaging and neuro-informatics infrastructure. The acquisition was on a 3-Tesla whole body Siemens MR scanner (Magnetom Verio, Siemens Healthcare, Erlangen, Germany) using a 32- channel head coil, within three days of their clinical assessment. This evaluation included anatomical sequences (3D T1-weighted) and Pseudo-Continuous Arterial Spin Labeling (pCASL) perfusion sequences. The image data were pseudonymized and securely stored in Shanoir®, a platform designed for the management of brain imaging research databases [52]. The MRI acquisition parameters are as follows:
-
3D T1-weighted MPRAGE sequence: TR/TE/TI = 1900/2.26/900 ms, field of view 256 × 256 mm2, 176 sagittal slices, 1 × 1 × 1 mm3 resolution, parallel imaging with GRAPPA factor 2.
-
Pseudo-continuous ASL sequence (pCASL): 4 min scan time, 30 repetitions, TR/TE = 4000/12 ms, flip angle 90°, matrix size 64 × 64, labeling duration/post-labeling delay = 1500/1500 ms, parallel imaging SENSE factor 2, 20 axial slices with 3.5 × 3.5 mm2 in-plane resolution, 5 mm slice thickness, 1 mm gap.
MRI data processing – ROI selection
MRI data processing followed a methodology previously published [33]. Image data were preprocessed using an in-house autoMRIFootnote 1 pipeline with MATLAB (v. R2021b, The MathWorks Inc.) and the SPM12 toolbox (Wellcome Department of Imaging Neuroscience at University College London, UK). The SPM12 routine based on the unified segmentation model [53, 54] was used to correct the anatomical series for intensity inhomogeneity, segment them into tissues probability maps (grey and white matter, cerebrospinal fluid, bone), and warp the 3D T1w volume to the Montreal Neurological Institute (MNI) ICBM152 template. A rigid body transformation was used to correct the ASL series for motion before a rigid co-registration to the anatomical sequence. A pairwise subtraction between labelled and control images of the co-registered ASL series followed by averaging yielded perfusion weighted (PW) maps for each patient. As the arithmetic sample mean is very sensitive to outliers, we instead use Huber’s M-estimator to robustly estimate the PW map [55]. This PW map was quantified to a cerebral blood flow (CBF) map by applying the standard kinetic model [56]. We used the mean of the control images as an estimation of the equilibrium magnetization of arterial blood, the so-called ‘M0’ image in the Buxton’s model.
Based on a non-systematic literature review, two sets of brain regions were defined, corresponding to those most involved respectively in the anxious and anhedonic components of depression (for review, see Supplementary Table 1). To this end, we ran the following search on PubMed: [2003; 2023]; keywords: C-reactive protein OR leukocytosis OR neutrophilia OR inflammation AND major depressive disorder OR treatment depressive disorder OR bipolar disorder OR depression AND functional MRI OR positron emission tomography OR magnetic resonance spectroscopy OR Arterial spin labeling OR cerebral blood flow. A total of 20 ROIs was a priori selected for the left and right hemispheres: accumbens nucleus, amygdala, caudate, anterior cingulate cortex (caudal and rostral parts), orbitofrontal cortex (medial and lateral parts), ventromedial prefrontal cortex, medial frontal cortex (caudal and rostral parts), hippocampus, insula and putamen. The selected ROIs were segmented from the structural images using the Desikan-Killiany Atlas [57] with the Freesurfer software (version 7.4.1). The labels of the ROIs are detailed in Supplementary Table 2.
Mean CBF values for each ROI were calculated from the CBF map in each patient’s MNI space by applying the Freesurfer parcellation. To account for CBF outliers related to the low resolution of ASL acquisition, we used the modified z-score developed by Iglewicz and Hoaglin [58]. CBF values for each ROI were normalized by the mean CBF value over grey matter (mean CBF value over the ROI divided by mean CBF over patient grey matter) to account for inter-individual whole brain perfusion variability.
Statistical analysis
Statistical analyses were performed using R software (version 4.3.3). The significance level for all statistical tests was set at 5% (p < 0.05). Quantitative variables are summarized as mean ± standard deviation, while qualitative variables are presented as counts and percentages for each modality.
Descriptive analysis
For descriptive statistics, continuous variables were analyzed using means and standard deviations, and categorical variables were expressed as proportions. A heatmap of correlations was created for continuous variables, highlighting significant correlations (p < 0.05) based on Pearson’s correlation coefficients.
Predictive analysis – elastic net regression
We chose to identify predictors of depressive severity at 6 months (MADRS(M6)), a machine learning elastic net regression (using glmnet R package – version 4.1-8) was employed to effectively handle multicollinearity while performing variable selection leading to improved prediction accuracy and interpretability [59]. Before regression, continuous variables were standardized. The response variable was MADRS at 6 months (MADRS_M6), rather than change in MADRS score, to reduce sensitivity to baseline severity, and better reflect the clinical state at follow-up. The baseline predictors included clinical characteristics (including anxiety and anhedonia as main clinical interest), inflammatory markers (CRP) and regional brain perfusion metrics (see section 2.5).
A 5-fold cross-validation was used to identify the optimal model with the lowest mean squared error for the mixing parameter, α, and the regularization parameter, λ. The subset of predictors of the resulting model with non-zero coefficients were considered relevant explanatory variables.
The stability of the selected coefficients was assessed using bootstrap resampling with 5000 iterations to compute confidence intervals for each coefficient. Predictors whose confidence intervals did not cross zero were considered significant.
Exploratory analysis – integrative approach assessment
To assess the diagnostic accuracy (depression improvement vs non improvement) of different predictor combinations, an integrative approach combining clinical, inflammatory and neuroimaging data was explored. Delta MADRS was dichotomized into “Improved” (delta MADRS < 0) and “Not Improved” (delta MADRS ≥ 0) to create a binary outcome variable. Logistic regression models were constructed for the following predictor combinations: clinical predictors only, clinical + inflammation, clinical + imaging, clinical + imaging + inflammation. The variables included in each combination were selected based on those identified as significant predictors of depression outcome (MADRS at 6 months) in the prior elastic net regression analysis.
For each model, receiver operating characteristic (ROC) curves were generated and areas under the curve (AUC) were computed to evaluate the predictive performance. Pairwise comparisons between ROC curves were performed with the DeLong’s test (using the pROC R package, version 1.18.5), to assess whether the inclusion of additional data types (e.g., imaging or inflammation) significantly improved diagnostic accuracy (“Improved” vs “Not Improved”). Results are presented as a comparison table reports Z-values and p-values for DeLong’s tests.
This exploratory analysis aimed to assess the potential added value of integrating multimodal data for diagnosing improvement vs no improvement.
Results
A total of 60 patients have been analyzed in the study (Fig. 1).
General sample characteristics
The clinical characteristics of the 60 patients included in the study are summarized in Table 1. Our sample was composed of a higher proportion of female as well as patients suffering from bipolar disorders. Overall, patients underwent several depressive episodes prior to enrollment with a mean disease duration of 17.5 years. At baseline, the depression intensity was moderate to severe with a significant level of anxiety. At 6-month follow-up, the cohort exhibited a mean MADRS score improvement of 12 points~. The biological (inflammation and CBF) are summarized in the Table 2.
Univariate analysis
Correlation heatmap is provided as supplementary material (Supplementary Fig. 1 & Supplementary Table 3). MADRS change – between baseline and 6-month follow-up – correlated negatively with age (r = −0.28, p = 0.03) and positively with left orbito-frontal perfusion (r = 0.28, p = 0.026). CRP correlated positively with left accumbens baseline perfusion (r = 0.26, p = 0.047) and left caudate baseline perfusion (r = 0.26, p = 0.049).
Elastic net regression analysis
Based on the cross validation, the optimal parameters for the elastic-net were α = 0.049 and λ = 1. The final model explained 59.93% of the variance of MADRS at M6.
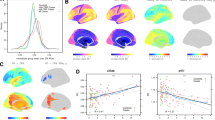
The bootstrap analysis exhibited a substantial variability in coefficients computations across iterations (Supplementary Table 3). Non-zero-coefficients predictors that exhibited a 95% CI not crossing zero were considered as robust significant predictors and are represented in bold color (negative = blue, positive = red) (Fig. 2). Positive robust coefficients—indicating that higher values are associated with increased MADRS scores at 6 months (i.e., worse depressive symptoms)—included CRP, left and right orbitofrontal perfusion, duration of the current depressive episode, right nucleus accumbens perfusion, and baseline MADRS severity. Negative robust coefficients—indicating that lower values are associated with worse depressive symptoms—were observed for age, disease duration, left and right caudate nucleus, left mid-frontal gyrus, right ventromedial prefrontal cortex, and left amygdala perfusion. For visual representation, significant ROI brain predictors were mapped on a brain glass (Fig. 3).

Dots represent mean coefficient over 5000 bootstrap replications for a given predictor and horizontal bar, the associated confidence interval. Colored and bold dots/bars represent relevant non-zero coefficients selected. OFC orbito-frontal cortex, CRP C-reactive protein, MDE_dur duration of mood depressive episode, ACC anterior cingulate cortex, accum accumbens nucleus, MFG mid frontal gyrus, nbr_MDE number of depressive episodes (lifetime), hippo hippocampus, put putamen, vmpfc ventromedial prefrontal cortex, cau caudate, dis_dur disease duration, amy amygdala, unip_bip unipolar/bipolar disorder (categorical variable).

Brain glass visualization of brain ROIs whose baseline perfusion predicts MADRS score at 6 months, independently of baseline depression severity (based on bootstrapped elastic net regression). Dots are plotted according to MNI coordinates, and color reflects the standardized regression coefficients of selected predictors.
Exploratory analysis – integrative approach assessment
Overall, the diagnosis accuracy (as measured by AUC) of the different integrative models ranged from 0.796 (moderate) to 0.977 (excellent) performance. The area under the curve (AUC) improved from 0.796 (clinical only) to 0.860 with inflammation markers, 0.923 with imaging, and 0.977 with all modalities combined. The improvements were statistically tested using DeLong’s method. Significant differences were observed between the clinical model and the full model (p = 0.0096), and between the clinical+inflammation model and the full model (p = 0.0243). These results suggest a synergistic value in combining clinical, imaging, and inflammatory data. (Table 3).
Discussion
Main findings
This longitudinal study following 60 patients over 6 months emphasized that persistence of depressive symptoms was associated with a higher baseline level of inflammation, a higher baseline perfusion of the left and right orbito-frontal cortices as well as right accumbens nucleus. In addition, a younger baseline age, longer episode duration, as well as lower baseline perfusion of left and right caudate nuclei, left mid frontal gyrus, right ventromedial prefrontal cortex, left amygdala were predictive of worst outcome. The strongest effect was seen for the let OFC. Our results confirmed our hypothesis that persistence of depression was associated with higher systemic inflammation level along with perfusion abnormalities in regions involved in emotion regulation and decision making. Contrary to our initial hypothesis, our model did not exhibit any significant contribution of anhedonia or anxiety. Clinical anxiety and anhedonia – as assessed by self-reports– might not be strong predictors of depression outcome.
Clinical predictors of depression outcomes at 6 months
Our study identified baseline age as a significant predictor of depression persistence at 6-month follow-up. This result echoes with works that repeatedly demonstrated that younger age is associated with worst trajectory of depressive disorder but also a higher risk of developing any other mental disorder. A recent study specifically focused on age of onset of MDE and its association with lifetime psychiatric disorders, health-related quality of life and the impact of gender [60]. Aligning with our study, another work explored the moderating effect of age (≤64 years old) on peripheral CRP levels and central markers of inflammation, noting significant differences in inflammatory profiles based on age [61]. With respects to the role of age on inflammation, one hypothesized mechanism has been reported by Lee and Giuliani who emphasized the role of critical developmental windows in adolescence and early adulthood, where neuroplasticity and environmental interactions are most influential [62]. Together, these findings align with the neuropathological characteristics commonly associated with depressive disorders [10] and emphasize the potential moderating effect of age on CRP effects on brain in depressed patients.
Thus, longer duration of the current depressive episode predicted greater depression severity at 6 months. This finding is consistent with the literature linking episode chronicity to poorer prognosis, higher relapse risk, and reduced treatment responsiveness [63, 64]. Prolonged episodes may reflect underlying neuroprogression, marked by cumulative alterations in stress-response systems, inflammatory sensitization, and impaired neuroplasticity. These biological changes can persist beyond symptom resolution, contributing to residual symptoms and functional decline [65, 66]. Thus, episode duration may serve as a clinical marker of illness burden and biological embedding, reinforcing the importance of early intervention to prevent long-term persistence [67].
We did not show that anxiety and anhedonia were significant predictors of depression outcome. One explanation might lie on the use of self-reports to measure these clinical dimensions [68]. Thus, a study found that anhedonia, as measured by SHAPS, predicts poorer recovery in treatment-resistant depression. The authors noted the limitations of self-reported measures in capturing the full scope of the disorder and suggested to integrate biological markers for more accurate predictions which is supported by our findings [69]. Lastly, the same limitations have been pointed out in a preprint that discussed the predictive value of anhedonia and anxiety scores in treatment-resistant depression. While self-reports like SHAPS and STAI are useful, they may not fully account for underlying neural changes associated with treatment resistance, highlighting a limitation in relying solely on these tools [70].
Involvement of prefrontal cortex
We found that lower baseline right VMPFC, higher baseline bilateral OFC and lower MFG perfusion were associated with worse depression outcome.
Higher baseline left OFC perfusion was the strongest predictor of depression outcome at 6 months. OFC plays a significant role in depression by modulating emotional, reward processing [71] and decision-making [72], which are often disrupted in depressive states. OFC structural alterations (grey matter volume) predicted antidepressant response emphasizing the role of this structure in depression outcome [73]. Also, some studies have reported that enhanced connectivity between OFC and subcortical regions [74], as well as change in brain entropy in the OFC [75], were related to the antidepressant treatment response suggesting that this structure is a promising biomarker to predict TRD (for review see [76]). Alterations in OFC activity and connectivity, particularly its interactions with other brain regions involved in mood regulation, have been linked to the severity of depressive symptoms and treatment outcomes [77]. Yin et al. in an above-cited study identified OFC as one of the regions in the widely distributed network that exhibited decreased FC associated with CRP levels [78]. To our knowledge, our study is the first to demonstrate that OFC CBF is robustly associated with depression course over 6 months accounting for inflammation and bring OFC – as a hub of emotional / reward processes as well as decision making – in depression.
VMPFC has been identified to be involved in depression and linked with anhedonia in both animal model and human [79, 80]. Increased activity of VMPFC and decreased activity in accumbens and caudate nuclei have been reported associated to anhedonia [81]. We failed to find any relationship between anhedonia – as measured by SHAPS – and perfusion patterns in our univariate analysis (see Supplementary Materials – Fig. 1). This could be due to our measure of anhedonia as mentioned above or the use of a different modality to measure brain activity. Several foundational works from Jennifer Felger and team have highlighted the central role of VMPFC – as part of a large reward network - in anhedonia and the potential effect of CRP on these functional abnormalities as well as treatment response. First, they demonstrated that higher CRP level was linked to reduced connectivity between the ventral striatum and VMPFC which effect was associated with anhedonia [71]. Moreover, they reported that the effect of CRP on the connectivity patterns mediated the relationship between CRP and anhedonia [71]. Second, they emphasized that not only CRP was associated with largely distributed decrease in functional connectivity, VMPFC was a key hub of this effect [78]. Third, they suggested that FC in reward circuitry was a potential biomarker for target engagement of dopaminergic treatments in MDD patients with elevated inflammation, identifying sub-population that might benefit from this treatment [82]. Although our study converged with these results, we must note that ours was not specifically design to test the effect of CRP on VMPFC CBF and anhedonia. Finally, a study prospectively testing the relationship of perfusion response of VMPFC – as measured by ASL – with treatment response to transcranial magnetic stimulation failed to find any effect following a course of high frequency stimulation whereas DPLFC CBF was negatively correlated with improvement in depression [83]. However, this result is mitigated by another study – using resting state functional connectivity – that demonstrated that core structures involved in default mode network as well as salience network (ACC/VMPFC, dorsal ACC and left insula) are highly predictive of treatment response to a left DLPFC TMS course [84].
Role of subcortical structures, caudate and amygdala
The amygdala plays pivotal roles in emotion regulation and processing, with significant implications for depression. Several works – in animal models and mammals - have pointed out the link between amygdala hyperactivity and peripheral inflammation (as measured by various inflammatory cytokines such as TNF-α, and IL-6) [85]. In line with these data, we should have expected that baseline increased amygdala CBF would be predictive of higher depression scores at follow-up while we found an opposite effect. First, one aspect is that the CBF depends on the structural component of grey matter. But we know that duration of depression can be associated with a decreased of grey matter volume of amygdala which could in turn contribute to a CBF decrease of this region as it has been described in depression and other conditions [86, 87]. Second, one work has reported that blunted amygdala reactivity to an emotional task was associated with greater depression severity in TRD patients. The authors suggested that baseline amygdala reactivity may serve as a biomarker for treatment resistance in depression [88]. Together with our results, our findings could be interpreted as a default of amygdala activity with a perspective of maladaptive limbic response rather than amygdala hyperactivity as a state marker of TRD. Further longitudinal are required to assess the amygdala activity over the course of depressive disorder.
In our elastic net regression model, decreased baseline caudate perfusion—a key component of the striatal reward system—was predictive of worse depressive outcomes. This finding aligns with prior evidence linking inflamed depression to impairments in brain networks associated with reward processing and anhedonia [89]. Inflammation has been increasingly implicated in the disruption of striatal reward circuitry, including the caudate nucleus, in depression. Notably, inflammation-associated disruptions in the dopaminergic corticostriatal circuitry have been observed, including reductions in ventral striatal neural responses to reward anticipation, decreased dopamine (DA) and its metabolites in cerebrospinal fluid, and reduced striatal DA release and availability. These disruptions correlate strongly with symptoms of anhedonia, reduced motivation, and psychomotor retardation [71]. Importantly, inflammation-associated symptoms, such as anhedonia, are often resistant to standard pharmacological treatments. This resistance may stem from inflammation’s impact on dopamine synthesis and availability, further underscoring the need for tailored therapeutic strategies targeting these pathways. Supporting this, Felger and colleagues demonstrated associations between increased CRP levels and decreased resting-state functional connectivity of the dorsal caudate and putamen with the ventromedial prefrontal cortex and presupplementary motor area, linked to psychomotor slowing in depression [71]. Interestingly and consistent with other studies, we found that clinical variables were less predictive of depression outcomes than brain imaging [90]. Together with our study, these findings highlight the critical role of inflammation in striatal dysfunction and its impact on reward-related brain circuitry, particularly in relation to anhedonia and psychomotor slowing in depression. This underscores the potential utility of integrating neuroimaging and inflammatory biomarkers in understanding and addressing treatment-resistant symptoms in depression.
Consistent with the nucleus accumbens’ role in reward processing, higher baseline right accumbens CBF was associated with greater depression severity at 6 months. Prior studies support this view: inflammatory challenges acutely reduce ventral striatal dopamine release and neural activity, leading to anhedonia [71]. Patients with MDD and elevated inflammation (CRP > 3 mg/L) show blunted striatal reward anticipation and weakened corticostriatal connectivity, correlating with higher anhedonia and worse mood [91]. Thus, heightened resting accumbens CBF may reflect compensatory hypermetabolism or neuroinflammatory activation within reward pathways – an attempt to overcome deficient reward signaling. Such a chronic hyperactive state in the ventral striatum has been linked to depression persistence and treatment resistance, indicating a poorer prognosis. It also underscores the need to stratify depressive subtypes by neurocircuit profiles (e.g., an inflammation-related anhedonic subtype), consistent with proposals that targeting reward circuit dysfunction with pro-dopaminergic or anti-inflammatory strategies may improve outcomes [71].
CRP as an inflammation marker in depression
CRP was our main inflammatory marker due to its widespread use across various medical disciplines for gauging systemic inflammation, established CRP thresholds in the literature, and its frequent association with adverse depressive outcomes in prior research [10]. Our results firstly emphasized the relationship of inflammation on the outcome of depressive disorders, as frequently highlighted in the literature [6, 10, 12], and secondly supported the relevance of CRP in representing and monitoring the immunobiological mechanisms of this inflammation. This result is of importance due to the accessibility in routine practice of CRP measurement.
Although CRP has been shown as a replicable marker across studies, its lack of specificity has been frequently discussed [92,93,94]. Even if we demonstrated that CRP was a reliable and stable marker of depression outcome in our sample, some points need to be discussed. First, the close link between the various clinical variables included and CRP, which, when assessed alone in univariate analysis, showed no significant association with changes in depressive severity, but did appear to be associated with the perfusion of only left accumbens nucleus. This suggested that the relationship between inflammation, clinical characteristics and brain perfusion are complex and might lie in a multidimensional interaction. Longitudinal design would be helpful to assess causal relationships between those variables. Second, the influence of inflammation on depression might predominantly occur in a distinct subpopulation with chronic low-grade inflammation as illustrated by the absence of such effect in late life depression [95]. Finally, studies have addressed more specific markers of inflammation in depression, focusing on cytokines such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-alpha), among other pro-inflammatory cytokines and monocytes/lymphocyte T activation markers whose production has been linked with depression episode state [96] and propensity to cross the blood-brain barrier appear to influence neuronal metabolism and synaptic plasticity [97,98,99]. To sharpen the understanding of immune-inflammation’s potential specificity in the pathophysiology of adverse, potentially treatment-resistant depression, a holistic approach encompassing an array of inflammatory biomarkers might be relevant.
Limitations and strengths
Our study has several limitations that warrant consideration. Firstly, the design of our longitudinal study inherently limits its ability to address mechanistic aspects, restricting our findings to an exploratory level. While we gained valuable insights into brain perfusion patterns associated with poor depression outcomes within the context of systemic inflammation, we were unable to investigate causal relationships. Moreover, the study’s limited sample size and absence of a control group limit the robustness of our statistical analyses. A larger sample size would enhance statistical power, reduce the influence of individual variations and allow broader generalization of the findings. In addition, a limitation of our study is that systemic inflammation was assessed solely using C-reactive protein (CRP), which, although widely used and readily available in clinical practice, represents only one component of the broader inflammatory profile. Other markers—such as interleukins (e.g., IL-6), tumor necrosis factor-alpha (TNF-α), and soluble cytokine receptors—may provide complementary insights into immune dysregulation in depression and could be integrated in future studies for a more comprehensive characterization of inflammatory states [16]. Thus, the lack of detailed information regarding medication changes during the 6-month follow-up period. Although all patients were treated according to international guidelines, we could not account for potential adjustments in dosage or treatment switches. Another limitation pertains to symptom-level analyses over time. While baseline assessments of anxiety and anhedonia were included, follow-up data from the corresponding STAI and SHAPS questionnaires were not available for analysis, limiting our ability to examine how these dimensions evolved in relation to inflammation and brain perfusion findings. Despite these limitations, the stability and strength of our model, derived from a routine care design, offer promising avenues for generalization. Thus, our exploratory analysis highlighted the potential added value for integrative models that significantly enhances to diagnostic accuracy of depression persistence over 6-month follow-up. Combining clinical, biological and brain imaging variables seems more promising in the prediction of depression episode course.
Conclusion
Using an integrative approach, the aim of this trial was to investigate the relationship between systemic inflammation, brain perfusion abnormalities and clinical characteristics in predicting depressive outcome. Our findings revealed that higher baseline levels of systemic inflammation, as measured by CRP, and altered perfusion in brain regions implicated in emotion regulation and decision-making were associated with worse depression outcomes. Additionally, clinical variables, including younger age, longer depression episode, further contributed to predicting treatment resistance. These results underscore the potential of integrating biomarkers to characterize a clinical and biological profile of patients with inflammatory depression. This study opens new perspectives on the role of inflammation in depression, particularly its influence on brain circuits associated with reward processing and anhedonia. By combining biological and neuroimaging biomarkers, our findings pave the way for developing personalized therapeutic strategies targeting inflammation-driven mechanisms in depression. Future research should validate these findings in larger cohorts and explore the therapeutic implications of addressing neuroinflammation in clinical practice.
Data availability
The dataset generated and/or analyzed during the current study is not publicly available due to restrictions related to participant privacy and institutional permissions.
References
Almutabagani LF, Almanqour RA, Alsabhan JF, Alhossan AM, Alamin MA, Alrajeh HM, et al. Inflammation and treatment-resistant depression from clinical to Animal study: a possible link? Neurol Int. 2023;15:100–20.
Beckett CW, Niklison-Chirou MV. The role of immunomodulators in treatment-resistant depression: case studies. Cell Death Discov. 2022;8:367.
Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107:234–56.
Branchi I, Viglione A, Vai B, Cirulli F, Benedetti F, Poggini S. Breaking free from the inflammatory trap of depression: regulating the interplay between immune activation and plasticity to foster mental health. Neuroscience Applied. 2024;3:103923.
Osimo EF, Baxter LJ, Lewis G, Jones PB, Khandaker GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med. 2019;49:1958–70.
Chamberlain SR, Cavanagh J, De Boer P, Mondelli V, Jones DNC, Drevets WC, et al. Treatment-resistant depression and peripheral C-reactive protein. Br J Psychiatry. 2019;214:11–19.
Zeng Y, Chourpiliadis C, Hammar N, Seitz C, Valdimarsdóttir UA, Fang F, et al. Inflammatory biomarkers and risk of psychiatric disorders. JAMA Psychiatry. 2024;81:1118.
Su K-P, Lai H-C, Peng C-Y, Su W-P, Chang JP-C, Pariante CM. Interferon-alpha-induced depression: comparisons between early- and late-onset subgroups and with patients with major depressive disorder. Brain Behav Immun. 2019;80:512–8.
Simon MS, Arteaga-Henríquez G, Fouad Algendy A, Siepmann T, Illigens BMW. Anti-inflammatory treatment efficacy in major depressive disorder: a systematic review of meta-analyses. Neuropsychiatr Dis Treat. 2023;19:1–25.
Osimo EF, Pillinger T, Rodriguez IM, Khandaker GM, Pariante CM, Howes OD. Inflammatory markers in depression: a meta-analysis of mean differences and variability in 5166 patients and 5083 controls. Brain Behav Immun. 2020;87:901–9.
Janssen DGA, Caniato RN, Verster JC, Baune BT. A psychoneuroimmunological review on cytokines involved in antidepressant treatment response. Hum Psychopharmacol. 2010;25:201–15.
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22:370–9.
Franklyn SI, Stewart J, Beaurepaire C, Thaw E, McQuaid RJ. Developing symptom clusters: linking inflammatory biomarkers to depressive symptom profiles. Transl Psychiatry. 2022;12:133.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68:748–54.
Harrison NA, Voon V, Cercignani M, Cooper EA, Pessiglione M, Critchley HD. A neurocomputational account of how inflammation enhances sensitivity to punishments versus rewards. Biol Psychiatry. 2016;80:73–81.
Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34.
Davies KA, Cooper E, Voon V, Tibble J, Cercignani M, Harrison NA. Interferon and anti-TNF therapies differentially modulate amygdala reactivity which predicts associated bidirectional changes in depressive symptoms. Mol Psychiatry. 2021;26:5150–60.
Lee Y, Mansur RB, Brietzke E, Carmona NE, Subramaniapillai M, Pan Z, et al. Efficacy of adjunctive infliximab vs. placebo in the treatment of anhedonia in bipolar I/II depression. Brain Behav Immun. 2020;88:631–9.
Sandiego CM, Gallezot J-D, Pittman B, Nabulsi N, Lim K, Lin S-F, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci USA. 2015;112:12468–73.
Goldsmith DR, Bekhbat M, Mehta ND, Felger JC. Inflammation-related functional and structural dysconnectivity as a pathway to psychopathology. Biol Psychiatry. 2023;93:405–18.
Haroon E, Chen X, Li Z, Patel T, Woolwine BJ, Hu XP, et al. Increased inflammation and brain glutamate define a subtype of depression with decreased regional homogeneity, impaired network integrity, and anhedonia. Transl Psychiatry. 2018;8:1–11.
Chung K-H, Huang S-H, Wu J-Y, Chen P-H, Hsu J-L, Tsai S-Y. The link between high-sensitivity C-reactive protein and orbitofrontal cortex in euthymic bipolar disorder. Neuropsychobiology. 2013;68:168–73.
Tseng H-H, Chang HH, Wei S-Y, Lu T-H, Hsieh Y-T, Yang YK, et al. Peripheral inflammation is associated with dysfunctional corticostriatal circuitry and executive dysfunction in bipolar disorder patients. Brain Behav Immun. 2021;91:695–702.
Frodl T, Carballedo A, Hughes MM, Saleh K, Fagan A, Skokauskas N, et al. Reduced expression of glucocorticoid-inducible genes GILZ and SGK-1: high IL-6 levels are associated with reduced hippocampal volumes in major depressive disorder. Transl Psychiatry. 2012;2:e88.
Byrne ML, Whittle S, Allen NB. The role of brain structure and function in the association between inflammation and depressive symptoms: a systematic review. Psychosom Med. 2016;78:389–400.
Burrows K, Stewart JL, Kuplicki R, Figueroa-Hall L, Spechler PA, Zheng H, et al. Elevated peripheral inflammation is associated with attenuated striatal reward anticipation in major depressive disorder. Brain Behav Immun. 2021;93:214–25.
Mehta ND, Haroon E, Xu X, Woolwine BJ, Li Z, Felger JC. Inflammation negatively correlates with amygdala-ventromedial prefrontal functional connectivity in association with anxiety in patients with depression: preliminary results. Brain Behav Immun. 2018;73:725–30.
Corrigan M, O’Rourke AM, Moran B, Fletcher JM, Harkin A. Inflammation in the pathogenesis of depression: a disorder of neuroimmune origin. Neuronal Signal. 2023;7:NS20220054.
van Agtmaal MJM, Houben AJHM, Pouwer F, Stehouwer CDA, Schram MT. Association of microvascular dysfunction with late-life depression: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74:729–39.
Empana J-P, Boutouyrie P, Lemogne C, Jouven X, Van Sloten TT. Microvascular contribution to late-onset depression: mechanisms, current evidence, association with other brain diseases, and therapeutic perspectives. Biol Psychiatry. 2021;90:214–25.
Ferré J-C, Bannier E, Raoult H, Mineur G, Carsin-Nicol B, Gauvrit J-Y. Arterial spin labeling (ASL) perfusion: techniques and clinical use. Diagn Interv Imaging. 2013;94:1211–23.
Hernandez-Garcia L, Lahiri A, Schollenberger J. Recent progress in ASL. Neuroimage. 2019;187:3–16.
Batail JM, Corouge I, Combès B, Conan C, Guillery-Sollier M, Vérin M, et al. Apathy in depression: an arterial spin labeling perfusion MRI study. J Psychiatr Res. 2023;157:7–16.
Chen Z-Q, Du M-Y, Zhao Y-J, Huang X-Q, Li J, Lui S, et al. Voxel-wise meta-analyses of brain blood flow and local synchrony abnormalities in medication-free patients with major depressive disorder. J Psychiatry Neurosci. 2015;40:401–11.
Cooper CM, Chin Fatt CR, Liu P, Grannemann BD, Carmody T, Almeida JRC, et al. Discovery and replication of cerebral blood flow differences in major depressive disorder. Mol Psychiatry. 2020;25:1500–10.
Duhameau B, Ferré J-C, Jannin P, Gauvrit J-Y, Vérin M, Millet B, et al. Chronic and treatment-resistant depression: a study using arterial spin labeling perfusion MRI at 3Tesla. Psychiatry Res Neuroimaging. 2010;182:111–6.
Sun X, Huang W, Wang J, Xu R, Zhang X, Zhou J, et al. Cerebral blood flow changes and their genetic mechanisms in major depressive disorder: a combined neuroimaging and transcriptome study. Psychol Med. 2023;53:6468–6480.
Wang Y, Yang Z. Aberrant pattern of cerebral blood flow in patients with major depressive disorder: a meta-analysis of arterial spin labelling studies. Psychiatry Res Neuroimaging. 2022;321:111458.
Haller S, Zaharchuk G, Thomas DL, Lovblad K-O, Barkhof F, Golay X. Arterial spin labeling perfusion of the brain: emerging clinical applications. Radiology. 2016;281:337–56.
Batail J, Coloigner J, Soulas M, Robert G, Barillot C, Drapier D. Structural abnormalities associated with poor outcome of a major depressive episode: the role of thalamus. Psychiatry Res Neuroimaging. 2020;305:111158.
Bauer M, Severus E, Möller H-J, Young AH. WFSBP task force on unipolar depressive disorders. Pharmacological treatment of unipolar depressive disorders: summary of WFSBP guidelines. Int J Psychiatry Clin Pract. 2017;21:166–76.
Bennabi D, Yrondi A, Charpeaud T, Genty J-B, Destouches S, Lancrenon S, et al. Clinical guidelines for the management of depression with specific comorbid psychiatric conditions French recommendations from experts (the French Association for Biological Psychiatry and Neuropsychopharmacology and the fondation FondaMental). BMC Psychiatry. 2019;19:50.
Kennedy SH, Lam RW, McIntyre RS, Tourjman SV, Bhat V, Blier P, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder: section 3. Pharmacological treatments. Can J Psychiatry. 2016;61:540–60.
Yrondi A, Javelot H, Nobile B, Boudieu L, Aouizerate B, Llorca P-M, et al. French Society for Biological Psychiatry and Neuropsychopharmacology (AFPBN) guidelines for the management of patients with partially responsive depression and treatment-resistant depression: update 2024. L’Encéphale. 2024. https://doi.org/10.1016/j.encep.2023.11.029.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59:22–33. quiz 34-57.
Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9.
Iannuzzo RW, Jaeger J, Goldberg JF, Kafantaris V, Sublette ME. Development and reliability of the HAM-D/MADRS interview: an integrated depression symptom rating scale. Psychiatry Res. 2006;145:21–37.
Spielberger, CD, Gorsuch, RL, Lushene, R, Vagg, PR, Jacobs, GA Manual for the state-trait anxiety inventory. Palo Alto, CA: (Consulting Psychologists Press. 1983).
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167:99–103.
Gaillard R, Gourion D, Llorca PM. [Anhedonia in depression]. Encephale. 2013;39:296–305.
Copeland WE, Shanahan L, Worthman C, Angold A, Costello EJ. Cumulative depression episodes predict later C-reactive protein levels: a prospective analysis. Biol Psychiatry. 2012;71:15–21.
Barillot C, Bannier E, Commowick O, Corouge I, Baire A, Fakhfakh I et al. Shanoir: applying the software as a service distribution model to manage brain imaging research repositories. Frontiers in ICT 2016; 3. https://doi.org/10.3389/fict.2016.00025.
Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–51.
Malone IB, Leung KK, Clegg S, Barnes J, Whitwell JL, Ashburner J, et al. Accurate automatic estimation of total intracranial volume: a nuisance variable with less nuisance. Neuroimage. 2015;104:366–72.
Maumet C, Maurel P, Ferré J-C, Barillot C. Robust estimation of the cerebral blood flow in arterial spin labelling. Magn Reson Imaging. 2014;32:497–504.
Buxton RB, Frank LR, Wong EC, Siewert B, Warach S, Edelman RR. A general kinetic model for quantitative perfusion imaging with arterial spin labeling. Magn Reson Med. 2005;40:383–96.
Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–80.
Iglewicz B, Hoaglin DC How to detect and handle outliers. Milwaukee, WI: Asq Press; (ASQC Quality Press. 1993).
Cleophas TJ, Zwinderman AH Optimal scaling: regularization including ridge, lasso, and elastic net regression. In: Cleophas TJ, Zwinderman AH (eds). Machine learning in medicine. Dordrecht: Springer Netherlands; 2013, pp 39–53.
Jamet C, Dubertret C, Le Strat Y, Tebeka S. Age of onset of major depressive episode and association with lifetime psychiatric disorders, health-related quality of life and impact of gender: a cross sectional and retrospective Cohort study. J Affect Disord. 2024;363:300–9.
Cho SH, Lim J-E, Lee J, Lee JS, Jeong H-G, Lee M-S, et al. Association between high-sensitivity C-reactive protein levels and depression: moderation by age, sex, obesity, and aerobic physical activity. J Affect Disord. 2021;291:375–83.
Lee C-H, Giuliani F The role of inflammation in depression and fatigue. Front Immunol 2019; 10. https://doi.org/10.3389/fimmu.2019.01696.
Kupfer DJ, Frank E, Phillips ML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. The Lancet. 2012;379:1045–55.
Berwian IM, Walter H, Seifritz E, Huys QJM. Predicting relapse after antidepressant withdrawal - a systematic review. Psychol Med. 2017;47:426–37.
Nierenberg AA. Residual symptoms in depression: prevalence and impact. J Clin Psychiatry. 2015;76:e1480–e1480.
Israel JA. The impact of residual symptoms in major depression. Pharmaceuticals (Basel). 2010;3:2426–40.
Liu J, Fan Y, Ling-Li Z, Liu B, Ju Y, Wang M, et al. The neuroprogressive nature of major depressive disorder: evidence from an intrinsic connectome analysis. Transl Psychiatry. 2021;11:102.
Kennedy BL, Schwab JJ, Morris RL, Beldia G. Assessment of state and trait anxiety in subjects with anxiety and depressive disorders. Psychiatr Q. 2001;72:263–76.
Liao A, Walker R, Carmody TJ, Cooper C, Shaw MA, Grannemann BD, et al. Anxiety and anhedonia in depression: associations with neuroticism and cognitive control. J Affect Disord. 2019;245:1070–8.
Lynch CJ, Elbau I, Ng T, Ayaz A, Zhu S, Manfredi N et al. Expansion of a frontostriatal salience network in individuals with depression. bioRxiv 2023; 2023.08.09.551651.
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2016;21:1358.
Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118.
Sämann PG, Höhn D, Chechko N, Kloiber S, Lucae S, Ising M, et al. Prediction of antidepressant treatment response from gray matter volume across diagnostic categories. Eur Neuropsychopharmacol. 2013;23:1503–15.
Tozzi L, Goldstein-Piekarski AN, Korgaonkar MS, Williams LM. Connectivity of the cognitive control network during response inhibition as a predictive and response biomarker in major depression: evidence from a randomized clinical trial. Biol Psychiatry. 2020;87:462–72.
Liu X, Song D, Yin Y, Xie C, Zhang H, Zhang H, et al. Altered Brain Entropy as a predictor of antidepressant response in major depressive disorder. J Affect Disord. 2020;260:716–21.
Lai C-H. Fronto-limbic neuroimaging biomarkers for diagnosis and prediction of treatment responses in major depressive disorder. Prog Neuro-Psychopharmacol Biol Psychiatry. 2021;107:110234.
Goldsmith DR, Bekhbat M, Le N-A, Chen X, Woolwine BJ, Li Z, et al. Protein and Gene Markers of Metabolic Dysfunction and Inflammation Together Associate with Functional Connectivity in Reward and Motor Circuits in Depression. Brain Behav Immun. 2020;88:193–202.
Yin L, Xu X, Chen G, Mehta ND, Haroon E, Miller AH, et al. Inflammation and decreased functional connectivity in a widely-distributed network in depression: centralized effects in the ventral medial prefrontal cortex. Brain Behav Immun. 2019;80:657–66.
Gorwood P. Neurobiological mechanisms of anhedonia. Dialogues Clin Neurosci. 2008;10:291–9.
Roberts AC, Clarke HF. Why we need nonhuman primates to study the role of ventromedial prefrontal cortex in the regulation of threat- and reward-elicited responses. Proc Nat Acad Sci USA. 2019;116:26297–304.
Jung M, Han K-M. Behavioral activation and brain network changes in depression. J Clin Neurol. 2024;20:362.
Bekhbat M, Li Z, Mehta ND, Treadway MT, Lucido MJ, Woolwine BJ, et al. Functional connectivity in reward circuitry and symptoms of anhedonia as therapeutic targets in depression with high inflammation: evidence from a Dopamine Challenge study. Mol Psychiatry. 2022;27:4113–21.
Ikawa H, Tochigi M, Noda Y, Oba H, Kaminaga T, Sakurai K, et al. A preliminary study on predictors of treatment response to repetitive transcranial magnetic stimulation in patients with treatment-resistant depression in Japan. Neuropsychopharmacol Rep. 2022;42:478–84.
Ge R, Blumberger DM, Downar J, Daskalakis ZJ, Dipinto AA, Tham JCW, et al. Abnormal functional connectivity within resting-state networks is related to rTMS-based therapy effects of treatment resistant depression: a Pilot study. J Affect Disord. 2017;218:75–81.
Hu P, Lu Y, Pan B-X, Zhang W-H. New insights into the pivotal role of the amygdala in inflammation-related depression and anxiety disorder. Int J Mol Sci. 2022;23:11076.
Dong H, Guo L, Yang H, Zhu W, Liu F, Xie Y et al. Association between gray matter atrophy, cerebral hypoperfusion, and cognitive impairment in Alzheimer’s disease. Front Aging Neurosci 2023; 15. https://doi.org/10.3389/fnagi.2023.1129051.
Martinot M-LP, Martinot J-L, Ringuenet D, Galinowski A, Gallarda T, Bellivier F, et al. Baseline brain metabolism in resistant depression and response to transcranial magnetic stimulation. Neuropsychopharmacology. 2011;36:2710–9.
Ferri J, Eisendrath SJ, Fryer SL, Gillung E, Roach BJ, Mathalon DH. Blunted amygdala activity is associated with depression severity in treatment-resistant depression. Cogn Affect Behav Neurosci. 2017;17:1221–31.
Wijaya MT, Jin RR, Liu X, Zhang R, Lee TMC. Towards a multidimensional model of inflamed depression. Brain, Behav, Immun - Health. 2022;26:100564.
Liu Q, Ely BA, Simkovic SJ, Tao A, Wolchok R, Alonso CM, et al. Correlates of C-reactive protein with neural reward circuitry in adolescents with psychiatric symptoms. Brain, Behav, Immun - Health. 2020;9:100153.
Han K-M, Ham B-J. How inflammation affects the brain in depression: a review of functional and structural MRI studies. J Clin Neurol. 2021;17:503–15.
Felger JC, Haroon E, Patel TA, Goldsmith DR, Wommack EC, Woolwine BJ, et al. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol Psychiatry. 2020;25:1301–11.
Mac Giollabhui N, Ng TH, Ellman LM, Alloy LB. The longitudinal associations of inflammatory biomarkers and depression revisited: systematic review, meta-analysis, and meta-regression. Mol Psychiatry. 2021;26:3302–14.
Orsolini L, Pompili S, Tempia Valenta S, Salvi V, Volpe U. C-reactive protein as a biomarker for major depressive disorder? IJMS. 2022;23:1616.
Luning Prak ET, Brooks T, Makhoul W, Beer JC, Zhao L, Girelli T, et al. No increase in inflammation in late-life major depression screened to exclude physical illness. Transl Psychiatry. 2022;12:1–9.
Daray FM, Grendas LN, Arena ÁR, Tifner V, Álvarez Casiani RI, Olaviaga A, et al. Decoding the inflammatory signature of the major depressive episode: insights from peripheral immunophenotyping in active and remitted condition, a Case–Control study. Transl Psychiatry. 2024;14:1–17.
Cheng Y, Desse S, Martinez A, Worthen RJ, Jope RS, Beurel E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav Immun. 2018;69:556–67.
Medina-Rodriguez EM, Beurel E. Blood brain barrier and inflammation in depression. Neurobiol Dis. 2022;175:105926.
Ting EY-C, Yang AC, Tsai S-J. Role of interleukin-6 in depressive disorder. IJMS. 2020;21:2194.
Acknowledgements
MRI data acquisition was supported by the Neurinfo MRI research facility from the University of Rennes I. Neurinfo is granted by the European Union (FEDER), the French State, the Brittany Council, Rennes Metropole, Inria, Inserm and the University Hospital of Rennes. The authors thank Mr Stéphane Brousse, Mr Jacques Soulabaille, and Mrs Lydie Fourets for their involvement in the conduct of the study.
Author information
Authors and Affiliations
Contributions
JMB conceived the study, contributed to methodology, performed formal analyses, participated in the investigation and data curation, drafted the original manuscript, and revised it. IC contributed to formal analyses, data curation, and manuscript revision. TB contributed to formal analyses, data curation, and manuscript revision. JCR contributed to formal analyses, participated in the investigation, and revised the manuscript. GR contributed to methodology, participated in the investigation, and revised the manuscript. DD contributed to conceptualization and methodology, supervised the project, participated in the investigation, and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All participants provided a written informed consent after a complete description of the study, and the study received approval of its protocol from a national ethics committee before being registered on www.clinicaltrial.gov (NCT02286024). All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
All authors have reviewed and approved the manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Batail, JM., Corouge, I., Blanchard, T. et al. Inflammatory and MRI perfusion biomarkers in predicting persistence of depression: a 6-month Longitudinal Study. Transl Psychiatry 15, 370 (2025). https://doi.org/10.1038/s41398-025-03587-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03587-x
