
Abstract
Acute lymphoblastic leukemia (ALL) survivors are at risk for developing subsequent neoplasms, but there is limited information on long-term risks and risk factors for both subsequent malignant neoplasms (SMNs) and subsequent non-malignant neoplasms (SNMNs). We analyzed long-term risk and risk factors for SMNs and SNMNs among 3291 5-year ALL survivors from the Dutch Childhood Cancer Survivor Study-LATER cohort (1963–2014). We calculated standardized incidence ratios (SIRs) and cumulative incidences and used multivariable Cox proportional hazard regression analyses for analyzing risk factors. A total of 97 survivors developed SMNs and 266 SNMNs. The 30-year cumulative incidence was 4.1% (95%CI: 3.5–5.3) for SMNs and 10.4%(95%CI: 8.9–12.1) for SNMNs. Risk of SMNs was elevated compared to the general population (SIR: 2.6, 95%CI: 2.1–3.1). Survivors treated with hematopoietic stem cell transplantation (HSCT) with total body irradiation (TBI) (HR:4.2, 95%CI: 2.3–7.9), and without TBI (HR:4.0,95%CI: 1.2–13.7) showed increased SMN risk versus non-transplanted survivors. Cranial radiotherapy (CRT) was also a risk factor for SMNs (HR:2.1, 95%CI: 1.4–4.0). In conclusion, childhood ALL survivors have an increased SMN risk, especially after HSCT and CRT. A key finding is that even HSCT-treated survivors without TBI treatment showed an increased SMN risk, possibly due to accompanied chemotherapy treatment. This emphasizes the need for careful follow-up of HSCT and/or CRT-treated survivors.
Similar content being viewed by others

Introduction
Acute lymphoblastic leukemia (ALL) is the most common type of childhood cancer [1,2,3], with a 5-year survival rate currently exceeding 90% [4]. However, ALL survivors are at risk for long-term adverse health outcomes including the development of subsequent neoplasms [5, 6]. Compared to the general population, childhood ALL survivors have a 2.6 to 13.5 times higher risk of developing subsequent malignant neoplasms (SMNs) [5,6,7,8]. The most frequently observed SMNs in ALL survivors are central nervous system (CNS) tumors [6, 9]. In addition to SMNs, some types of subsequent non-malignant neoplasms (SNMNs) can also cause serious morbidity, such as subsequent meningiomas [5, 10].
Treatment protocols for ALL patients have changed over time. Major adjustments in the Netherlands were 1) the substitution of cranial radiotherapy (CRT) by CNS prophylaxis with intrathecal high-dose methotrexate since to the DCOG-ALL VI protocol in December 1984 [11, 12] and 2) trials with replacing TBI with a chemotherapy conditioning regimen for HSCT between 2011 and 2021 [13, 14] Several studies examined treatment-related risk factors for subsequent neoplasms in ALL survivors [5, 15,16,17]. Although many studies were limited by short follow-up times [15,16,17] or the limited availability of specific treatment data [5, 15,16,17], several risk factors have been suggested. The risk of developing a subsequent neoplasm was found to be higher in patients who were treated with radiotherapy [18], especially CRT [7, 16, 19]. Furthermore, patients who received HSCT also showed an increased risk of subsequent neoplasms as compared to non-transplanted leukemia survivors [20,21,22,23], which is often suggested to be due to TBI [17, 21, 24, 25]. However, the separate impacts of HSCT and TBI are not fully clear.
In the current study, we aimed to analyze the long-term risk and associated risk factors for developing SMNs and SNMNs in 5-year survivors of childhood ALL diagnosed between 1963 and 2014.
Methods
Patients
In this multicenter study, 11 704 5-year survivors diagnosed under the age of 18 in any of the seven former pediatric oncology/stem cell centers in the Netherlands, in the period 1963–2014 were included in the Dutch Childhood Cancer Survivor Study (DCCSS)-LATER cohort [26, 27]. Data collection from both the original cohort (1963–2001) [26] and the expansion cohort (2002–2014) has been previously documented [28]. In the current study, we included 3291 survivors diagnosed with ALL according to the International Classification of Disease for Oncology, Third Edition (ICD-D-O-3) [29] (ICD-O-3 morphology code 9835/3, 9836/3 or 9837/3).
Data collection
Information about demographics, diagnosis, and childhood cancer treatment, including relapses, was collected by trained data managers. For 262 (8%) survivors who objected to adding additional linkage data, we only had basic yes/no treatment data available. For the other 3029 ALL survivors, detailed treatment data were available including type and doses of chemotherapy and radiotherapy and information about hematopoietic allogeneic stem cell transplantation (HSCT). For anthracyclines and alkylating agents, cumulative doses were calculated. For anthracyclines, we used the doxorubicin isotoxic equivalent (DIE) to sum doses of agents [30] (Table S1). For alkylating agents, dose was summed according to the cyclophosphamide equivalent dose (CED) [31] (Table S2).
Data on subsequent neoplasms were ascertained by linkages to two nationwide registries: the Netherlands Cancer Registry (NCR) [32], with nationwide coverage since 1989, (although some regional registries attained full local coverage earlier), and the Dutch Nationwide Pathology Databank (Palga) [33], with nationwide coverage since 1991. The linkage procedure for the DCCSS-LATER cohort has been reported previously [26, 28]. The NCR data were used as main source for malignant neoplasms. For malignant tumors diagnosed before 1989, we used the partially available NCR data in combination with data from Palga and from the DCCSS-LATER registry, based on medical record data. Pathology reports were reviewed to resolve discrepancies between multiple SMN sources. SMN data was complete up to January 31st, 2022. Palga were used as source for histologically confirmed non-malignant tumors and basal cell carcinomas (BCCs) of the skin. SNMNs were defined as subsequent benign, borderline malignant, or in situ tumors. Non-malignant skin tumors were excluded. Excerpts were manually reviewed to identify and classify non-malignant neoplasms according to the ICDD-O-3 [29]. Challenging records were discussed with a pathologist (RdK). SNMN data was complete up to April 7th, 2022 for the original cohort and up to November 30th, 2022 for the expansion cohort. BCC data was complete until November 30th, 2022. We included subsequent neoplasms that occurred five years or more after ALL diagnosis and were histologically different from the ALL.
Statistical analyses
Analyses were done separately for SMNs, SNMNs, and BCCs, because of the differences in entry time. For SMNs, follow-up started five years after ALL diagnosis and for SNMNs and BCCs follow-up started five years after ALL diagnosis or January 1, 1991 (start nationwide coverage Palga), whichever occurred last. Follow-up ended on the date of diagnosis of the first subsequent neoplasm of interest (e.g., for analyses on malignant CNS tumors, at date of first CNS tumor, irrespective of a prior SMN, SNMN, or BCC), date of death, date last known vital status (emigration, loss to follow up), or end of study (January 31st, 2022 for SMNs and April 7, 2022, for SNMNs and BCCs), whichever occurred first.
We calculated standardized incidence ratios (SIRs) and absolute excess risks (AERs) of SNMs. The SIR was calculated by dividing the observed number by the expected number based on age-, sex-, and calendar year-specific general population rates from the NCR. The AER was calculated as the excess number of SMNs per 10,000 person-years. SIRs and AERs were calculated for any SMN and for specific subgroups. For SNMNs and BCCs, there are no reference rates for the general population and we could therefore not calculate SIRs and AERs.
For SMNs, SNMNs, and BCCs we calculated cumulative incidences, accounting for death as a competing risk. We also calculated the cumulative incidence for survivors diagnosed before and after 1984. The cut-off of 1984 was based on the switch from protocol ALL-V to ALL-VI, where cranial radiotherapy (CRT) was omitted as standard of care for non-high-risk ALL survivors. Differences between curves were compared using Gray’s tests [34]. Furthermore, we examined potential risk factors by using multivariable Cox proportional hazard regression models, with attained age as time scale [35]. Our base model included sex, age at diagnosis, cranial radiotherapy, HSCT ± TBI as part of the conditioning regimen for HSCT. In addition, we analyzed the following chemotherapy groups and dose categories: alkylating agents, anthracyclines, etoposide. Etoposide was predominantly administered to HSCT patients as part of initial treatment in this high-risk group or/and as conditioning for HSCT and was sparingly administered to patients without HSCT. In order to stratify these risks, mutually exclusive groups were created combining etoposide exposure with HSCT subgroups. Stratification on etoposide exposure was only feasible in the HSCT with TBI group, but not in the HSCT without TBI group due to low numbers. Furthermore, we were not able to analyze effects of platinum agents (not part of standard ALL treatment, and therefore only very few patients in our cohort were treated with this), glucocorticoids, vinca alkaloids, antimetabolites and asparaginases (part of ALL treatment in almost all protocols and therefore almost everyone in our cohort had this as part of therapy). Although we adjusted for all treatments in our main analysis, we did conduct a sensitivity analysis including only survivors with a relapsed ALL to evaluate the effect of HSCT in a more homogenous group of survivors with intensive treatments. The proportional hazard assumption was tested in all models and was not violated. All analyses were performed using SPSS v 29.0 or R studio v 4.2.
Results
Patient characteristics
Among the 3291 childhood ALL survivors, 55.2% were male (Table 1). Median age at diagnosis was 4.7 years (range: 0.0–17.8 years). In total, 72.8% were treated with chemotherapy only, 18.0% with a combination of chemotherapy and any radiotherapy, 1.7% with chemotherapy and HSCT, and 6.9% with a combination of chemotherapy, radiotherapy and HSCT. Of the 3029 survivors with additional treatment data, 24.5% were treated with any type of radiotherapy, of whom 17.4% with only CRT, 5.3% with only TBI, 0.9% with CRT and TBI, and 0.5% with other types of radiotherapy (Table 1). Of the 420 survivors who experienced a relapse, 39.3% received only CRT, 25.2% only TBI, 6.2% CRT and TBI, and 2.4% other types of radiotherapy (Table S3).
Of all survivors, 430 (13.1%) developed at least one subsequent neoplasms, of whom 97 (2.9%) survivors developed at least one SMN, 266 (8.1%) at least one SNMN and 172 (5.2%) at least one BCC. In total, 21 of the 430 survivors who developed a subsequent neoplasm developed both an SMN and SNMN. Among the 420 survivors with relapsed disease, 24 developed at least one SMN.
Subsequent malignant neoplasms
The median follow-up time for SMN was 21.6 (range: 5.0–54.9) years since ALL diagnosis. In total, 106 SMNs were observed in 97 survivors, with 9 survivors developing multiple SMNs. The median latency between childhood ALL diagnosis and occurrence of an SMN was 26.5 (range: 5.8–46.1) years. 87 SMNs were solid tumors. The most frequently observed SMN sites were CNS (n = 15), thyroid (n = 13), and skin (13 melanomas and 4 squamous cell carcinomas) (Table 2).
Overall SMN risk was significantly increased in ALL survivors compared to the age-, sex-, and calendar-year matched general population with an SIR of 2.6 (95% CI 2.1–3.1) and an AER of 10.0 per 10,000 person-years. The AER increased with follow-up time after diagnosis and was 25.5 per 10,000 person-years for follow-up time beyond 30 years. The highest AERs compared to the general population were observed for CNS tumors (AER: 2.2) and thyroid malignancies (AER: 2.0) (Table 2). High SIRs were observed for survivors who were treated with chemotherapy and HSCT (SIR: 8.4, 95%CI: 0.2–47.0) and chemotherapy, HSCT, and radiotherapy (SIR: 10.5, 95%CI: 6.1–16.8) (Table S4). Types of SMNs after HSCT are shown in Table S5. ALL survivors who had a relapse (n = 24; SIR: 5.6, 95%CI: 3.6–8.4) had a higher SIR than those without a relapse (n = 62; SIR: 2.2, 95%CI: 1.7–2.8) (Table S4).
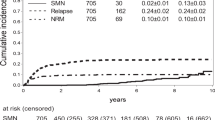
The 30-year cumulative incidence of any SMN was 3.8% (95%CI: 2.9–4.9) (Fig. 1). The cumulative incidence of any SMN was not different between survivors diagnosed ≤ 1984 and survivors diagnosed > 1984 (p = 0.64), the year where CRT was omitted as standard of care (Fig. 2). However, the cumulative incidence of subsequent CNS tumors was significantly lower for survivors diagnosed after 1984 compared to survivors diagnosed in or before 1984 (p = 0.005) (Fig. 2).

A Subsequent malignant neoplasms and non-malignant neoplasms Cumulative incidence of subsequent malignant neoplasms (SMN) and subsequent non-malignant neoplasms (SNMNs). B Basal cell carcinomas Cumulative incidence of Basal cell carcinomas (BCCs).

A Subsequent malignant neoplasms Cumulative incidence of all subsequent malignant neoplasms (SMNs). B Subsequent malignant central nervous system tumors Cumulative incidence of subsequent malignant tumors of the central nervous system. C Subsequent non-malignant neoplasms Cumulative incidence of all subsequent non-malignant neoplasms (SNMNs). D Subsequent non-malignant meningiomas Cumulative incidence of subsequent non-malignant meningiomas.
Subsequent non-malignant neoplasms
In total, 266 survivors developed histologically confirmed SNMNs, with a median latency time between childhood cancer diagnosis and the first SNMN of 25.7 (range: 5.5–48.3) years. The most frequently observed SNMNs were non-malignant meningiomas (n = 81), urogenital system neoplasms (n = 42), and lipomas (n = 36) (Table 2). Types of SNMNs after HSCT are shown in Table S5. The 30-year cumulative incidence of any SNMN was 9.9% (95% CI: 8.5–11.5) and highest for SNMN subtypes meningiomas (2.5%, 95% CI: 1.7–3.5) and urogenital neoplasms (1.9% 95% CI: 1.3–2.8) (Fig. 1, Table 2). For any SNMN, the cumulative incidence was not different for survivors diagnosed ≤ 1984 and 1984 > (p = 0.84), but we did see a significant decrease in the incidence of non-malignant meningiomas for survivors diagnosed after 1984 (p < 0.001) (Fig. 2).
Basal cell carcinoma risk
In total, 172 survivors developed at least one basal cell carcinoma (BCC), with a median latency time of 26.1 (range: 5.6–43.5) years. The 30-year cumulative incidence of BCC was 5.6% (95%CI: 4.5–7.0) (Fig. 1). Among survivors treated with radiotherapy, the 30-year cumulative incidence for BCC was 10.9% (95% CI: 8.6–12.6) compared to 1.2% (95% CI: 0.6–2.4) for survivors treated without radiotherapy (Fig. S1). The HR was 19.3 (95%CI: 12.2–29.8) for survivors treated with TBI and 7.6 (95%CI: 5.5–10.5) for survivors treated with CRT (data not shown).
Risk factors for subsequent neoplasms
We analyzed risk factors for SMNs and SNMNs in multivariable models among 3029 survivors for whom extensive treatment details were available. ALL survivors treated with cranial radiotherapy (CRT) (n = 48 SMNs) had a significantly higher risk of developing any SMN compared to survivors treated without CRT (n = 38 SMNs) (HR: 2.3, 95% CI: 1.4–4.0) (Table 3). Furthermore, HSCT was significantly associated with increased SMN risk, regardless of whether TBI was included in the conditioning regimen (HR for HSCT with TBI: 4.2, 95% CI: 2.3–7.8; HR for HSCT without TBI: 4.0, 95% CI: 1.2–13.7) (Table S6). After adjusting for chemotherapy, we still observed a significant effect of HSCT without TBI (n = 3 SMNs) (HR: 3.8, 95% CI: 1.1–13.8) (Table 3). Survivors treated with HCST, TBI, and etoposide (n = 11 SMNs) appeared to have a higher risk (HR: 5.7; 95% CI: 2.5–12.8) compared to survivors treated with HSCT and TBI without etoposide (n = 2 SMNs) (HR: 1.5; 95% CI: 0.5–6.5); however, this difference was not significant.
ALL survivors treated with CRT also had a higher risk of developing any SNMN compared to survivors treated without CRT (HR: 1.9, 95% CI: 1.3–2.6) (Table 3). Furthermore, compared to survivors treated without HSCT, survivors who received HSCT with TBI showed a significantly increased risk of developing SNMN (HR: 6.4, 95% CI: 3.9–10.4), whereas those treated with HSCT without TBI did not show a significant increase (HR: 1.9, 95%CI: 0.6–7.7) (Table S6). After adjusting for chemotherapy, significant effects were still observed for HSCT with TBI, both with etoposide (HR: 4.9, 95% CI: 2.8–10.3) and without etoposide (HR: 4.9, 95% CI: 2.3–10.3) (Table 3).
ALL survivors treated with radiotherapy were at an increased risk of developing basal cell carcinoma, with HRs of 4.3 (95% CI: 2.8–6.7) for CRT vs. no CRT and 6.4 (95% CI: 3.9–10.4) for HSCT plus TBI vs. no HSCT (Table S6).
In a sensitivity analysis including only survivors who experienced a relapse, HSCT remained a significant risk factor for SMNs (HR: 2.5, 95%CI: 1.0–3.4), and BCCs (HR: 2.7, 95%CI: 1.4–5.4), but not for SNMNs (HR: 4.9, 95%CI: 0.5–2.0) (Table S7).
Discussion
This study demonstrates that 5-year survivors of childhood ALL have an increased risk of developing subsequent neoplasms, especially after HSCT. A significant new finding is that ALL survivors treated with HSCT but without TBI also have an increased risk of SMNs compared to ALL survivors not treated with HSCT, possibly due to accompanying chemotherapy. Furthermore, CRT was a significant risk factor for development of both SMNs and SNMNs. The risk of any SMN and any SNMN did not decrease for survivors treated after 1984, when prophylactic CRT was omitted from standard protocols, compared to those treated in or before 1984. However, the risk of malignant CNS tumors and benign meningiomas decreased significantly among those treated after 1984.
In this study, we showed that ALL survivors who received HSCT, both with and without TBI, had an increased risk of SMNs compared to survivors treated without HSCT. Previous studies that reported on subsequent neoplasms after HSCT vs. no HSCT have also shown significantly increased risk for SMNs and SNMNs after HSCT [20, 21] with most studies attributing this increased risk to TBI conditioning [21, 25]. Recently, similar findings have also been reported for ALL survivors [17]. In our cohort, we observed a similar increased risk of SMNs for ALL survivors who received HSCT with TBI and those who received HSCT without TBI, compared to ALL survivors treated without HSCT. This suggests that aspects of HSCT other than TBI contribute to the elevated risk of SMN development after HSCT. In our multivariable model, we observed a suggestive trend with higher risks among survivors receiving etoposide within the HSCT with TBI group. Due to a limited number of cases, we were unable to stratify by etoposide exposure within the HCST without TBI subgroup and can therefore not analyze whether this increased risk might be due to concurrent etoposide treatment. Other factors beyond chemotherapy could also play a role; for instance, an association between chronic graft versus host disease (GVHD) and oral cavity cancers has been implied [36]. Although we lacked GVHD information, among the nine ALL survivors with malignant oral neoplasms in our cohort, none had received HSCT, and only one out of four with non-malignant oral neoplasms had received HSCT. We could therefore not confirm this previous observation.
Previous studies have indicated that unfractionated and high-dose TBI seemed to be associated with a higher risk of SMN compared to low-dose TBI [21, 25]. Unfortunately, our sample size was too small to further explore the impact of the TBI dose and fractionation. In our cohort, most survivors who received TBI were treated with unfractionated TBI or TBI delivered in 2 fractions.
We observed a significantly lower cumulative incidence of malignant CNS tumors and non-malignant meningiomas among patients diagnosed after 1984. In 1984, CRT was substituted by CNS prophylaxis involving high-dose methotrexate and intrathecal chemotherapy as part of the standard ALL treatment protocols in the Netherlands with the introduction of the DCOG-ALL VI protocol [11, 12]. CRT has been shown to be an important risk factor for CNS neoplasms, particularly meningiomas [27, 37,38,39,40]. We did not observe a decrease in the overall incidence of SMNs and SNMNs for patients treated 1984 > vs. ≤ 1984, which is consistent with the findings of Ishida et al. [16].
Survivors of ALL might also face an increased risk of subsequent neoplasms due to genetic syndromes that could predispose individuals to both ALL and subsequent neoplasms [41, 42]. Well-established associations with childhood leukemia and subsequent neoplasms include conditions such as neurofibromatosis-1 (linked to CNS tumors) [43] and Li-Fraumeni Syndrome (linked to multiple tumors such as sarcomas or breast cancer) [44]. Information on predisposition syndromes within our cohort was incomplete, preventing a detailed examination of their role. Based on the partially available data, anecdotal evidence includes cases of two congenital aberrations potentially related to the development of subsequent neoplasms: one patient with Down syndrome who developed subsequent B-cell leukemia and another with a congenital bone aberration who developed an osteochondroma.
Major strengths of our study include the large cohort size, extensive follow-up duration, and comprehensive treatment data on an individual level. Due to linkage with nationwide registries, we ensure complete follow-up data including objective data on both malignant and histologically-confirmed non-malignant neoplasms. We also need to consider some limitations. Firstly, our non-malignant data only includes pathologically confirmed neoplasms, which might cause a slight underrepresentation of the true SNMN incidence. However, physicians might be more alert in childhood cancer survivors which could lead to increased detection of SNMNs. Secondly, we lacked specific data on protocols and risk groups among survivors, therefore comparison of subsequent neoplasm risks across different protocols could not be conducted. Lastly, we had only limited data on genetic predisposition.
In conclusion, childhood ALL survivors have an increased risk of SMNs. Previous studies have shown that TBI increases SMN risk in HSCT survivors. Our results show that HSCT-treated survivors without TBI conditioning also have increased risk of SMNs. This shows the importance of future studies to further investigate the effects of different conditioning regimes and accompanying therapies in survivors receiving HSCT on the development of SMNs, including more detailed assessments of chemotherapy dose, TBI dose, and fractionation used before and after HSCT. Our results also emphasize the need for careful follow-up of survivors treated with HSCT with or without TBI, or with CRT.
Data availability
The data that support the findings of this study are available on reasonable request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
Ries LAG. Cancer incidence and survival among children and adolescents: United States SEER program, 1975–1995: National Cancer Institute; 1999.
Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28:2625–34.
Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541–52.
Pieters R, Mullighan CG, Hunger SP. Advancing diagnostics and therapy to reach universal cure in childhood ALL. J Clin Oncol. 2023;41:5579–91.
Hijiya N, Hudson MM, Lensing S, Zacher M, Onciu M, Behm FG, et al. Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA. 2007;297:1207–15.
Mody R, Li S, Dover DC, Sallan S, Leisenring W, Oeffinger KC, et al. Twenty-five-year follow-up among survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Blood. 2008;111:5515–23.
Borgmann A, Zinn C, Hartmann R, Herold R, Kaatsch P, Escherich G, et al. Secondary malignant neoplasms after intensive treatment of relapsed acute lymphoblastic leukaemia in childhood. Eur J Cancer. 2008;44:257–68.
Essig S, Li Q, Chen Y, Hitzler J, Leisenring W, Greenberg M, et al. Risk of late effects of treatment in children newly diagnosed with standard-risk acute lymphoblastic leukaemia: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2014;15:841–51.
Kimball Dalton VM, Gelber RD, Li F, Donnelly MJ, Tarbell NJ, Sallan SE. Second malignancies in patients treated for childhood acute lymphoblastic leukemia. J Clin Oncol. 1998;16:2848–53.
Walter AW, Hancock ML, Pui CH, Hudson MM, Ochs JS, Rivera GK, et al. Secondary brain tumors in children treated for acute lymphoblastic leukemia at St Jude Children’s Research Hospital. J Clin Oncol. 1998;16:3761–7.
Veerman AJ, Hahlen K, Kamps WA, Van Leeuwen EF, De Vaan GA, Solbu G, et al. High cure rate with a moderately intensive treatment regimen in non-high-risk childhood acute lymphoblastic leukemia. Results of protocol ALL VI from the Dutch Childhood Leukemia Study Group. J Clin Oncol. 1996;14:911–8.
Kamps WA, Veerman AJ, van Wering ER, van Weerden JF, Slater R, van der Does-van den Berg A. Long-term follow-up of Dutch Childhood Leukemia Study Group (DCLSG) protocols for children with acute lymphoblastic leukemia, 1984-1991. Leukemia. 2000;14:2240–6.
Peters C, Dalle JH, Locatelli F, Poetschger U, Sedlacek P, Buechner J, et al. Total body irradiation or chemotherapy conditioning in childhood all: a multinational, randomized, noninferiority phase III study. J Clin Oncol. 2021;39:295–307.
Versluijs AB, de Koning CCH, Lankester AC, Nierkens S, Kollen WJ, Bresters D, et al. Clofarabine-fludarabine-busulfan in HCT for pediatric leukemia: an effective, low toxicity, TBI-free conditioning regimen. Blood Adv. 2022;6:1719–30.
Bhatia S, Sather HN, Pabustan OB, Trigg ME, Gaynon PS, Robison LL. Low incidence of second neoplasms among children diagnosed with acute lymphoblastic leukemia after 1983. Blood. 2002;99:4257–64.
Ishida Y, Maeda M, Urayama KY, Kiyotani C, Aoki Y, Kato Y, et al. Secondary cancers among children with acute lymphoblastic leukaemia treated by the Tokyo Children’s Cancer Study Group protocols: a retrospective cohort study. Br J Haematol. 2014;164:101–12.
Eichinger A, Poetschger U, Glogova E, Bader P, Basu O, Beier R, et al. Incidence of subsequent malignancies after total body irradiation-based allogeneic HSCT in children with ALL—long-term follow-up from the prospective ALL-SCT 2003 trial. Leukemia. 2022;36:2567–76.
Pui CH, Cheng C, Leung W, Rai SN, Rivera GK, Sandlund JT, et al. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N. Engl J Med. 2003;349:640–9.
Loning L, Zimmermann M, Reiter A, Kaatsch P, Henze G, Riehm H, et al. Secondary neoplasms subsequent to Berlin-Frankfurt-Munster therapy of acute lymphoblastic leukemia in childhood: significantly lower risk without cranial radiotherapy. Blood. 2000;95:2770–5.
Pole JD, Darmawikarta D, Gassas A, Ali M, Egler M, Greenberg ML, et al. Subsequent malignant neoplasms in pediatric cancer patients treated with and without hematopoietic SCT. Bone Marrow Transplant. 2015;50:721–6.
Curtis RE, Rowlings PA, Deeg HJ, Shriner DA, Socie G, Travis LB, et al. Solid cancers after bone marrow transplantation. N Engl J Med. 1997;336:897–904.
Deeg HJ, Socie G. Malignancies after hematopoietic stem cell transplantation: many questions, some answers. Blood. 1998;91:1833–44.
Ades L, Guardiola P, Socie G. Second malignancies after allogeneic hematopoietic stem cell transplantation: new insight and current problems. Blood Rev. 2002;16:135–46.
Bhatia S, Louie AD, Bhatia R, O’Donnell MR, Fung H, Kashyap A, et al. Solid cancers after bone marrow transplantation. J Clin Oncol. 2001;19:464–71.
Baker KS, Leisenring WM, Goodman PJ, Ermoian RP, Flowers ME, Schoch G, et al. Total body irradiation dose and risk of subsequent neoplasms following allogeneic hematopoietic cell transplantation. Blood. 2019;133:2790–9.
Teepen JC, van Leeuwen FE, Tissing WJ, van Dulmen-den Broeder E, van den Heuvel-Eibrink MM, van der Pal HJ, et al. Long-term risk of subsequent malignant neoplasms after treatment of childhood cancer in the DCOG LATER study cohort: role of chemotherapy. J Clin Oncol. 2017;35:2288–98.
Kok JL, Teepen JC, van der Pal HJ, van Leeuwen FE, Tissing WJE, Neggers S, et al. Incidence of and risk factors for histologically confirmed solid benign tumors among long-term survivors of childhood cancer. JAMA Oncol. 2019;5:671–80.
Westerveld ASR, Tytgat GAM, van Santen HM, van Noesel MM, Loonen J, de Vries ACH, et al. Long-term risk of subsequent neoplasms in 5-year survivors of childhood neuroblastoma: A DCCSS-LATER 3 study. J Clin Oncol.
Fritz A, Percy C, Jack A, Shanmugaratnam K, Sobin L, Parkin DM, et al. International classification of diseases for oncology. 3rd ed. World Health Organization; 2020..
Feijen EAM, Leisenring WM, Stratton KL, Ness KK, van der Pal HJH, van Dalen EC, et al. Derivation of anthracycline and anthraquinone equivalence ratios to doxorubicin for late-onset cardiotoxicity. JAMA Oncol. 2019;5:864–71.
Green DM, Nolan VG, Goodman PJ, Whitton JA, Srivastava D, Leisenring WM, et al. The cyclophosphamide equivalent dose as an approach for quantifying alkylating agent exposure: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. 2014;61:53–67.
Bray F, Ferlay J, Laversanne M, Brewster DH, Gombe Mbalawa C, Kohler B, et al. Cancer incidence in five continents: inclusion criteria, highlights from volume X and the global status of cancer registration. Int J Cancer. 2015;137:2060–71.
Casparie M, Tiebosch AT, Burger G, Blauwgeers H, van de Pol A, van Krieken JH, et al. Pathology databanking and biobanking in The Netherlands, a central role for PALGA, the nationwide histopathology and cytopathology data network and archive. Cell Oncol. 2007;29:19–24.
Gray RJ. A class of k-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–54.
Yasui Y, Liu Y, Neglia JP, Friedman DL, Bhatia S, Meadows AT, et al. A methodological issue in the analysis of second-primary cancer incidence in long-term survivors of childhood cancers. Am J Epidemiol. 2003;158:1108–13.
Rizzo JD, Curtis RE, Socie G, Sobocinski KA, Gilbert E, Landgren O, et al. Solid cancers after allogeneic hematopoietic cell transplantation. Blood. 2009;113:1175–83.
Godlewski B, Drummond KJ, Kaye AH. Radiation-induced meningiomas after high-dose cranial irradiation. J Clin Neurosci. 2012;19:1627–35.
Rimm IJ, Li FC, Tarbell NJ, Winston KR, Sallan SE. Brain tumors after cranial irradiation for childhood acute lymphoblastic leukemia. A 13-year experience from the Dana-Farber Cancer Institute and the Children’s Hospital. Cancer. 1987;59:1506–8.
Neglia JP, Robison LL, Stovall M, Liu Y, Packer RJ, Hammond S, et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2006;98:1528–37.
Withrow DR, Anderson H, Armstrong GT, Hawkins M, Journy N, Neglia JP, et al. Pooled analysis of meningioma risk following treatment for childhood cancer. JAMA Oncol. 2022;8:1756–64.
Schmiegelow K. Treatment-related toxicities in children with acute lymphoblastic leukaemia predisposition syndromes. Eur J Med Genet. 2016;59:654–60.
Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 2019;16:227–40.
Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247–53.
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–6.
Acknowledgements
The authors thank all physicians, research nurses, data managers, research assistants, the Dutch National Childhood Cancer Association, and participating patients for their contribution. We would also like to thank the registration team of the Netherlands Comprehensive Cancer Organization (IKNL) for the collection of data for the Netherlands Cancer Registry. Moreover, we thank the staff from Palga Foundation for providing linkage data on subsequent non-malignant neoplasms from their registry.
Author information
Authors and Affiliations
Contributions
AW: Conceptualization, data collection, data analysis, interpretation of data, and writing—original draft, PR: Data collection, data analysis, interpretation of data, and writing—original draft, HvdP: Conceptualization, interpretation of data and writing—Review & Editing, funding acquisition, and supervision, DB, MB, PH, RdK, and RP: Interpretation of data, writing—review & editing, JL, AdV, ML, MK, MvdHvdL, MvdHE and GJ: Data acquisition, writing—review & editing, CR: Conceptualization, interpretation of data, funding acquisition, and writing—review & editing, LK: Conceptualization, data collection, data analysis, interpretation of data, writing—review & editing, funding acquisition, and supervision, JT: Conceptualization, data collection, data analysis, interpretation of data, writing—review & editing, funding acquisition, and supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study is in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Princess Máxima Center for Pediatric Oncology (PMCLAB2020.162). This study is a retrospective study with use of care data that were already collected and there was no active consent needed for survivors according to Dutch legislation. Therefore, this retrospective study is exempted from review of Medical Ethical Committee review in compliance with Dutch law and regulations for health research involving human beings, because the subjects in this retrospective study were not subjected to procedures or to follow rules of behavior. However, informed consent was obtained from survivors who had been invited for active participation in DCCSS-LATER research projects. For survivors who had been invited for active participation in DCCSS-LATER research projects, but did not respond after repeated requests via a standardized protocol, and for survivors who had not yet been invited for active participation in any DCCSS-LATER research projects, specific consent was not needed in accordance with Dutch legislation. For 262 survivors who objected to adding linkage data directly to the DCCSS-LATER registry, but not object against anonymous linkage, we anonymized a minimal dataset via a trusted third party. Survivors who declined use of their healthcare data for research purposes were excluded from the eligible study cohort.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Westerveld, A.S.R., Roesthuis, P., van der Pal, H.J.H. et al. Increased risk of subsequent neoplasm after hematopoietic stem cell transplantation in 5-year survivors of childhood acute lymphoblastic leukemia. Blood Cancer J. 14, 150 (2024). https://doi.org/10.1038/s41408-024-01122-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-024-01122-7
This article is cited by
-
Next-generation immunotherapeutic approaches for blood cancers: Exploring the efficacy of CAR-T and cancer vaccines
Experimental Hematology & Oncology (2025)
