
Abstract
Chimeric antigen receptor (CAR) T-cell therapy has revolutionized the treatment paradigms for hematological malignancies. However, more than half of these patients cannot achieve sustainable tumor control, partially due to the inadequate potency of CAR-T cells in eradicating tumor cells. T cells are crucial components of the anti-tumor immune response, and multiple intrinsic T-cell features significantly influence the outcomes of CAR-T cell therapy. Herein, we review progressing research on T-cell characteristics that impact the effectiveness of CAR-T cells, including T-cell exhaustion, memory subsets, senescence, regulatory T-cells, the CD4+ to CD8+ T-cell ratio, metabolism, and the T-cell receptor repertoire. With comprehensive insight into the biological processes underlying successful CAR-T cell therapy, we will further refine the applications of these novel therapeutic modalities, and enhance their efficacy and safety for patients.
Similar content being viewed by others

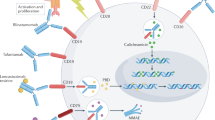
Introduction
Chimeric antigen receptor (CAR) T-cell therapy, a treatment with T cells expressing antibody-based fusion proteins targeting tumor antigens, has brought tremendous breakthroughs in the treatment of hematological malignancies, including B-cell leukemia, lymphoma, and multiple myeloma (MM) [1,2,3,4,5,6,7]. To date, six CAR-T cell products have been approved by the Food and Drug Administration, including four products targeting CD19 and two targeting B-cell maturation antigen (BCMA) (Table 1). From observations in multicenter clinical trials, complete response/remission (CR) rates of relapsed and/or refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL), large B-cell lymphoma (LBCL), follicular lymphoma (FL), mantle cell lymphoma, and MM generated by these CAR-T cells have reached 71–90%, 39–66%, 79–94%, 67–82% and 33–73%, respectively [1, 2, 5, 7,8,9,10,11,12,13,14].
However, the clinical outcomes of these treatments are inconsistent and mixed. Within one year after CAR-T cell therapy, progressive disease can be observed in roughly 50% of B-cell leukemia and LBCL, 20–30% of FL, 40% of mantle cell lymphoma, and 20–40% of MM patients [2,3,4,5,6,7,8,9,10,11, 13, 15]. Meanwhile, in R/R CLL, the most common leukemia in adults, the reported CR rates of anti-CD19 CAR-T cell therapy range from 18 to 29%, which are lower than in other B-cell malignancies [2, 5, 7, 10,11,12,13, 15, 16]. Furthermore, the most common CAR-T cell-specific side effects, cytokine release syndrome and neurotoxicity, are observed in 42–100% and 2–64% of patients in CAR-T cell clinical trials [17]. Severe cytokine release syndrome and neurotoxicity (grade ≥3) can occur in up to 46 and 50% of treated patients, respectively [17]. In addition, rates of unsuccessful CAR-T cell manufacture are approximately 25% for non-Hodgkin’s lymphoma (NHL) patients, and 6.8% for B-ALL and CLL patients, which could be a significant obstacle to the treatment [18].
T cells are the most crucial immune cells in the anti-tumor immune response. T cells orchestrate the adaptive immune system by producing cytokines with chemotactic, proinflammatory, and immunoprotective properties, and also carry out direct cytotoxic reactions towards neoplastic cells [19]. CAR-T cells, which are manufactured from the patients’ T cells and capable of targeting tumor cells by circumventing major histocompatibility complex (MHC) restriction, have become the pillar of successful adoptive cell therapies. Emerging evidence indicates that multiple characteristics of patients’ T cells and CAR-T cells can significantly impact the effectiveness of CAR-T cell therapy. Herein, we summarize various aspects of T-cell biological characteristics related to CAR-T cell therapy outcomes, including exhaustion, memory differentiation, senescence, T-cell subsets, metabolism, and T-cell receptor (TCR) repertoire (Fig. 1), with the aim of improving CAR-T cell efficacy and safety profiles in hematological malignancies.

a TEX cells form during cancer where antigen persists, and are characterized by loss of cytokine production, poor proliferation, and high expression of inhibitory receptors. CAR-TEX have similar features, and the exhaustion molecules are related to poor efficacy of CAR-T therapy. b T cells linearly differentiate to various memory subsets following cancer antigen stimulation, losing proliferation potential, but gaining effector functions. In CAR-T cell therapy, T-cell products with early memory phenotypes exhibit superior proliferative capacity, anti-tumor ability, and persistence. c The metabolic phenotypes and memory status of T cells are interconnected. TN and early memory T cells mainly rely on OXPHOS, but shift to glycolysis as they differentiate into TEFF. TEM utilizes both OXPHOS and glycolysis, and can rapidly increase glycolytic activity to support effector functions. d TREG suppress the function of CAR-T cells, and their numbers are negatively correlated with treatment response and survival. e A defined ratio of CD4+/CD8+ CAR-T cells improves therapeutic response and reduces toxicity in CAR-T cell-treated patients. f Senescent CAR-T cells exhibit terminal differentiation, loss of proliferative capacity, and compromised cytotoxic function. g TCR repertoire can be used as a surrogate for T-cell clonotype and expansion. A highly diverse baseline TCR repertoire is associated with favorable responses and improved survival outcomes in CAR-T cell therapy. CAR, chimeric antigen receptor; TEX, exhausted T cells; TN, naïve T cells; TSCM, T memory stem cells; TCM, central memory T cells; TEM, effector memory T cells; TEFF, effector T cells; TREG, regulatory T cells; TSEN, senescent T cells; OXPHOS, oxidative phosphorylation; TCR, T cell receptor.
T-cell exhaustion
T-cell exhaustion refers to post-thymic T-cell dysfunction in response to persistent antigen stimulation during chronic infections and cancer. Functionally, exhausted T cells (TEX) exhibit a progressive loss of cytokine production, poor proliferative capacity, and compromised killing function. Most importantly, they display high expression of inhibitory receptors, which have become the hallmarks of TEX. They include cytotoxic T lymphocyte-associated antigen 4 (CTLA4), programmed cell death protein 1 (PD-1), lymphocyte activation gene 3 (LAG3), T-cell immunoglobulin mucin receptor 3 (TIM3), and T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) [20, 21]. The gene regulatory programs governing exhaustion are largely conserved across disease settings, and epigenetic findings support TEX representing a distinct T-cell chromatin state rather than merely the isolated expression of inhibitory receptors [22]. While T-cell exhaustion is indispensable to limit damage to healthy cells and prevent autoimmune diseases, it also restricts the effectiveness of cancer immunotherapy [20, 21].
The molecules related to T-cell exhaustion can serve as biomarkers to predict CAR-T cell therapy efficacy and persistence. Several studies have reported that lower frequencies of PD-1, LAG3, and TIM3 expressed by infused and engrafted CAR-T cells are correlated with better disease control [23,24,25,26]. In patients with CLL, Fraietta et al. found that those achieving CR received lower percentages of anti-CD19 CAR-T cells expressing PD-1 combined with either TIM3 or LAG3, compared with patients achieving partial or no response. They also found the presence of a group of PD-1 negative CAR-T cells (PD-1-CD27+CD8+) in the infusion products was associated with good response [23]. In patients with B-ALL, Finney et al. observed that engrafted anti-CD19 CAR-T cells from minimal residual disease (MRD)-negative and durable leukemia-free patients expressed lower levels of LAG3 and TIM3 compared with those from MRD-positive or non-durable leukemia-free patients [26]. In patients with LBCL, researchers confirmed that those who did not achieve an early molecular response had a higher fraction of exhausted CD8+ T cells in CAR-T cell infusion products, featuring increased expression of LAG3 and TIM3 [24]. Consistently, studies have also shown that higher frequencies of inhibitory receptors in leukapheresis T cells, peripheral or bone marrow T cells, or tumor microenvironment T cells correlate with worse responses in CAR-T cell-treated patients [26,27,28]. Furthermore, Huang et al. recently documented CD38 as another hallmark of exhaustion, co-expressed with exhaustion-related transcription factors and LAG3 in anti-CD19 CAR-T cells. Inhibiting CD38 activity could reverse the exhaustion and improve the antitumor response [29].
Interestingly, recent evidence has also suggested that CAR structures affect the exhaustion status of CAR-T cells [30, 31]. For example, incorporating a 4-1BB costimulatory domain reduced the exhaustion induced by CAR signaling, whereas a CD28 costimulatory domain augmented the exhaustion [31]. Moreover, CAR-T cells containing MYD88/CD40 costimulatory domains had lower expression of PD-1 and remained less differentiated than CD28 CAR-T cells [31]. Additionally, higher levels of tonic signaling resulting from self-aggregation of CARs have been reported to cause CAR-T cell exhaustion [30, 32, 33]. For instance, a CAR with an IgG1 CH2–CH3 region as a spacer generates stronger tonic signals than those with the CH3 region only [33]. Another type of tonic signaling activation involves self-aggregation via the framework region of the single-chain variable fragment of CAR on CAR-T cells, which impairs proliferation and promotes exhaustion [30].
Combating CAR-T cell exhaustion represents a promising therapeutic avenue with broad applicability. In addition to the abovementioned CAR structure modification, recent advances have demonstrated that PD-1 signaling interference through PD-1 antibody checkpoint blockade, CRISPR/Cas9 mediated PD-1 knockout, and anti-PD-1 antibody secreting CAR-T cells, restores the effector function of CAR-T cells [34,35,36]. In clinic, the feasibility and safety of anti-CD19 CAR-T cells combined with a PD-1 inhibitors (pembrolizumab or nivolumab) in patients with R/R B-ALL and NHL were confirmed in different studies [37, 38]. Some other engineering strategies have focused on restoring activator protein 1 (AP-1) function to prevent T-cell exhaustion by overexpressing the transcription factor c-Jun and the basic leucine zipper ATF-like transcription factor (BATF) [39, 40]. Additionally, Weber et al. reported ameliorating tonic CAR signaling by using dasatinib, a clinically available tyrosine kinase inhibitor, which subjected CAR-TEX to transient rest to restore functionality [41].
T-cell memory differentiation
Memory differentiation of T cells is largely linear and unidirectional. After activation, circulating naive T cells (TN) massively divide (up to 50,000-fold) into effector T cells (TEFF), which disseminate broadly to eliminate antigens. After antigens are cleared, most TEFF undergo apoptosis, while a small subset survive and persist for years as memory T cells (TMEM), providing recall responses to previously encountered antigens [20, 42]. TMEM are heterogeneous, including central memory cells (TCM) trafficking through lymphoid tissues, effector memory cells (TEM) recirculating through blood and nonlymphoid tissues and surveying the sites of infection, and memory stem cells (TSCM) which are rare but possess the self-renewal and multipotent capacity to reconstitute the entire spectrum of TMEM and TEFF subsets [43]. CAR-T cell products manufactured by ex vivo expansion are predominantly composed of antigen-experienced T cells, with the two major subsets being TEM and TCM [44, 45].
Infusion products with a high content of early memory CAR-T cells display superior expansion, anti-tumor response, and in vivo persistence [23, 24, 44, 46,47,48,49]. For instance, Xu et al. performed an analysis in a group of lymphoma patients and found that a subset of CAR-TSCM (CD8+CD45RA+CCR7+) within infused product positively correlated with CAR-T cell expansion in patients [44]. More specifically, Deng et al. conducted single-cell RNA sequencing of axi-cel infusion products from 24 patients with LBCL. Their data identified threefold higher frequencies of TCM (CCR7+CD27+SELL+) subsets in patients who achieved CR than in those with partial response or progressive disease [24]. Similar results were demonstrated by Bai et al. through cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) analysis, showing that frequencies of TSCM and TCM from CAR-T infusion products could distinguish relapse in patients [50]. Finally, Haradhvala et al. observed that responses to tisa-cel were associated with a striking expansion of CD8+ CAR-TCM-like cell populations from the infusion product in LBCL patients [49].
Further evidence has shown that memory signatures of the premanufacture starting T cells influence the efficacy of post-manufacturing CAR-T cells [23, 51,52,53,54]. It is well-established in a murine model that CAR-T cell manufacturing from purified TN and TSCM populations results in longer and enhanced anti-tumor activity, and reduces the tendency of the cells to cause cytokine release syndrome [55]. In line with the lab study, clinical studies have shown comparable results. One study of tisa-cel-treated patients with LBCL and B-ALL suggested that a higher count of TSCM-like cells in the apheresis, characterized by CD8+CD45RA+CD27+ expression, was related to the efficacy of CAR-T cell therapy [51]. In another group of ide-cel-treated MM patients, a higher percentage of naive (CCR7+ and CD45RA+) and early memory (CD28+CD27+) T cells in the apheresed product was found to be statistically correlated with long-term responses [54]. Similarly, in a cohort of 182 B-cell malignancy patients, apheresis analyses confirmed that T cells significantly enriched in naive or early memory features were associated with responding patients, whereas the TEM subpopulation marked the non-responding patients [52].
Intriguingly, several studies have compared the differentiation phenotypes of CAR-T cells with CD28 versus 4-1BB costimulatory domains. They revealed that more differentiated memory phenotypes make up a greater proportion of CAR-T cells containing the CD28 domain than of those possessing the 4-1BB domain [56, 57]. Moreover, another study recently revealed that the ICOS and OX40 tandem costimulatory domain resulted in less differentiated memory CAR-T cells and superior tumor control compared with CD28 and 4-1BB CAR-T cells in preclinical models [58].
Strategies to promote naive and early memory populations in CAR-T cell manufacturing are being investigated. For instance, interleukin-7 (IL-7) and IL-21 induce TSCM differentiation, and together with IL-15, they promote the maintenance and expansion of TSCM. By contrast, IL-2 drives terminal differentiation of CD8+ T cells through the high-affinity IL-2 receptor [59]. On the basis of these cytokine functions, many CAR-T cell preclinical studies and clinical trials have been conducted to search for favorable CAR-T memory subsets [44, 59,60,61]. Another strategy to achieve early memory phenotypes is the use of pharmacological inhibitors targeting T-cell differentiation signaling pathways during the culture of CAR-T cells, such as phosphatidylinositol 3’-kinase (PI3K) inhibitors (duvelisib) and Bruton’s tyrosine kinase inhibitors (ibrutinib) [62, 63]. Other approaches have aimed at maintaining a less differentiated CAR-T cell product. They include shortening the ex vivo culture period of CAR-T cells and manufacturing CAR-T cells from patients in the early stage of cancer [64,65,66]. The latter approach is appealing, as it avoids the depletion of TN caused by cumulative chemotherapy cycles, promotes better CAR-T expansion in vivo, and has the potential to improve survival outcomes [66,67,68].
T-cell senescence
T-cell senescence is an irreversible process of adaptive immunity degeneration, characterized by terminal differentiation and loss of proliferative capacity, yet the immune cells retain their ability to perform their effector functions. Age-related senescence involves telomere shortening, whereas premature senescence is telomere-independent. Senescent T cells have distinct phenotypes, including the loss of costimulatory molecules CD27 and CD28 and expression of CD57 and killer cell lectin-like receptor subfamily G member 1 (KLRG1) [69]. They likewise exhibit a terminally differentiated phenotype with downregulation of the chemokine receptors CCR7 and CD45RO but upregulation of CD45RA [70]. Nevertheless, there is an overlap between T-cell senescence and exhaustion; for example, the novel exhaustion marker TIGIT was also found to be upregulated in classic senescent CD8+ T cells [71].
CAR-T cell therapy has been most successful in pediatric and young adult B-ALL patients, with a high CR rate of 90%; this is in contrast to MM patients, whose median age is over 70 and whose reported CR rate ranges from 33 to 73% [1, 13, 14]. Although there are differences in tumor biology among these indications, dysfunctional senescent T cells are suggested to be one of the reasons for the difference in CR rates [72]. It has been reported that 75% of MM patients exhibit a subpopulation of prematurely senescent T cells. These senescent T cells were KLRG1+CD57+CD160+CD28-, had low levels of PD-1 and CTLA4 phenotypes, and had normal telomere lengths [73]. Moreover, Guha et al. made an in vitro comparison of CAR-T cells manufactured from young and geriatric healthy donors. They revealed that young CAR-T cells had significantly higher transduction efficiency, improved expansion ability, and higher cytotoxicity than geriatric CAR-T cells [74]. Although most of these data indicate that T-cell senescence adversely impacts CAR-T cell therapy, the direct evidence from clinical studies on this topic is rare, and the relationships between exhaustion and senescence require further elucidation.
Regulatory T cell
Regulatory T cells (TREG) play an essential role in maintaining immune homeostasis and self-tolerance. These cells are characterized by the expression of transcription factor forkhead box P3 (FoxP3), the absence of CD127 (IL-7 receptor α), and the presence of CD4 and CD25 (IL-2 receptor α), constituting a small portion of the CD4+ T population. TREG have a distinct ability to suppress the activation, proliferation, and effector functions of a variety of immune cells, such as CD4+ and CD8+ T cells, natural killer cells, B cells, and antigen-presenting cells [75, 76]. Multiple immunosuppressive mechanisms are employed by TREG, including the expression of IL-2 receptor α (CD25) for sequestration of IL-2, thereby inducing effector T-cell anergy and apoptosis; the release of immunosuppressive cytokines such as IL-10, IL-35, and TGFβ; the abundant expression of inhibitory receptor CTLA4, which inhibits antigen-presenting cell function and T cell activation; and the conversion of ATP into adenosine, an immunosuppressive metabolite that compromises T-cell activation [75, 77]. However, this regulatory ability also dampens anti-tumor immune responses and favors tumor progression [76].
Recently, increasing attention has been paid to TREG in CAR-T cell therapies, particularly their elevated levels in peripheral blood and infusion products, which are linked to inferior therapeutic responses [28, 49, 78,79,80,81,82,83]. For instance, in a study involving 46 R/R B-ALL patients, Pan et al. found that the TREG population (CD4+CD25+CD127low) in peripheral blood measured both pre- and post-CAR-T cell therapy was significantly lower in the remission group than in the non-remission group. The authors also observed that TREG levels were negatively correlated with relapse-free survival, overall survival, and persistence of CAR-T cells [79]. In another study, single-cell transcriptome sequencing of CAR-T cell infusions in R/R LBCL patients revealed an elevation of the CAR-TREG population among non-responders [49]. This study further proved that CAR-TREG suppressed the expansion of other CAR-T cells and drove relapses in an in vivo model [49]. Similar results were indicated by an independent study of LBCL patients, which showed that increased levels of early engrafted CAR-TREG in patients predicted clinical progression but less severe neurotoxicity [78]. Likewise, some other researchers have observed that increased levels of TREG in peripheral blood before and after CAR-T cell infusion were associated with nonresponse and inferior overall survival [28, 80,81,82,83]. Notably, it is well established that IL-2 administration can lead to a profound expansion (nearly 4-fold) of TREG in humans, which hinders the efficacy of immunotherapy [84, 85]. It is also known that CAR-T cells manufactured in the presence of IL-7 and IL-15 have superior expansion and antitumor activity to those given IL-2, partly due to a smaller increase in CAR-TREG [86]. Consequently, new combinations of cytokines such as IL-7, IL-15, and/or low doses of IL-2 are becoming more appealing in CAR-T cell manufacture strategies [59, 86, 87].
CD4+/CD8+ T cell ratio
CD4+ and CD8+ T cells represent two broad classes of T cells crucial for immune responses against infection and cancers and distinguished by divergent recognition and effector mechanisms. Upon antigenic stimulation, CD4+ T cells undergo proliferation and secretion of cytokines that stimulate antibody responses or lead to macrophage activation via interactions with class II MHC molecules, whereas CD8+ T cells kill the antigen-bearing cells through class I MHC molecules [88]. CD4+ T cells help CD8+ T cells in secondary expansion, memory response, and acquisition of effector functions during chronic antigen stimulation [89,90,91]. Without the assistance of CD4+ T cells, CD8+ T cells become exhausted by increasing their expression of inhibitory molecules and transcriptional programs [92].
The frequency of CD4+ and CD8+ T cell subsets in the blood can differ markedly in cancer patients because of age, the effects of chemotherapy, and thymic function [93, 94]. Nevertheless, it remains feasible to manufacture CAR-T cells with a defined CD4+/CD8+ ratio, even in heavily pretreated groups of patients [94,95,96]. Moreover, the CD4+/CD8+ ratio generally decreases during the CAR T cell manufacturing period due to the higher expansion rate of CD8+ T cells compared with that of CD4+ T cells [97]. A recent study identified a CD4+/CD8+ ratio less than 1:3 in peripheral blood at apheresis as a risk factor for CAR-T cell manufacturing failure [98].
The CD4+/CD8+ ratio in CAR-T cell infusion products is also believed to be an important aspect of the overall therapeutic impact of CAR-T therapy. The synergistic activity between CD4+ and CD8+ CAR-T cells was first demonstrated in murine models [99,100,101]. In a B-cell lymphoma murine model, Sommermeyer et al. demonstrated that the infusion of CAR-T cells with a 1:1 ratio of CD4+and CD8+ outperformed unselected CAR-T cells as well as CD8+ or CD4+ CAR-T cells alone in terms of anti-tumor effect. This synergistic efficacy is suggested to be mediated by the cytokines released by CD4+ T cells [99]. Consistent results were also observed in murine models of melanoma lung metastasis and mammary carcinoma [100, 101]. In the clinical setting, a defined CD4+/CD8+ ratio of CAR-T cell infusion has been shown to improve therapeutic efficiency and reduce toxicity [94,95,96, 102]. For instance, Turtle et al. demonstrated in a study of 29 adult B-ALL patients that low doses (2×105/kg) of CAR-T cells with a 1:1 CD4+/CD8+ ratio achieved an impressive 93% CR rate and an 86% MRD-negative rate [95]. Another study involving children and young adults with B-ALL reported that a defined CD4+/CD8+ ratio retained an effective response while reducing the severity of CRS [102]. In B-cell lymphoma, Galli et al. found that patients with complete or partial responses at three and six months after CAR-T cell infusion had a lower CD4+/CD8+ ratio in the infused CAR-T products compared with non-responders. They also observed that patients with a CD4+/CD8+ ratio of less than 1.12 had a three-fold higher risk of developing neurotoxicity compared to those of a higher ratio [103].
Accordingly, liso-cel, one of the Food and Drug Administration-approved commercial products, is manufactured from leukapheresis-selected CD8+ and CD4+ T cells, followed by independent CD8+ and CD4+ T cell activation, transduction, expansion, and formulated into 1:1 ratio [104]. This product can provide high response rates in patients with R/R hematological malignancies [96, 104, 105]. However, Lee et al. to raised concerns about the suboptimal expansion and hypofunction of CD8+ CAR-T cells when grown in the absence of CD4+ T cells. They suggested manufacturing and growing both CD4+ and CD8+ T cells together and then formulating the defined ratio that will be infused [106]. A direct comparative trial will be necessary to verify the clinical superiority of this fixed CD4:CD8 approach.
T-cell metabolism
Metabolism powers T cells by providing the cellular energy and biochemical molecules needed for their proliferation, cytokine production, and cytotoxic activity. During each differentiation stage, T cells adjust their metabolism to meet biosynthetic and energetic demands. TN primarily have slow glycolysis and mainly rely on oxidative phosphorylation (OXPHOS) and fatty acid oxidation, but undergo a metabolic shift from OXPHOS to aerobic glycolysis as they become TEFF [107,108,109,110]. After antigen clearance, the memory T cells that switched from TEFF, including TSCM, TCM and TEM, return to relying on OXPHOS, while TEM can rapidly shift to high glycolytic activity, supporting the effector function [107, 108]. In addition to glucose, T cells rely on extracellular amino acids, such as glutamine, serine, and proline, for proliferation, activation, and effector functions [111,112,113].
The metabolic phenotypes and memory status of T cells are tightly interconnected, and metabolism could shape memory subsets and functions of CAR-T cells [114]. For instance, Fraietta et al. observed that anti-CD19 CAR-TEFF cells from partial-responders and non-responders with CLL exhibited elevated expression of aerobic glycolysis genes and increased uptake of a glucose analog. Furthermore, inhibition of glycolysis using 2-deoxy-D-glucose resulted in increased frequencies of CAR-TCM [23]. Another study documented that T-cell expansion under acidic conditions promoted stem-like T cells [115]. Consistently, recent research has indicated that CAR-T cells maintained at a lower range of normal pH at the beginning of the manufacturing process show enhanced T-cell proliferation and reduced glycolysis [116]. Moreover, IL-7 and IL-15 have been shown to facilitate triglyceride storage, thereby providing TMEM with a stable and long-term energy supply [117]. CAR-TN and CAR-TCM cultured with IL-7/IL-15 demonstrated superior expansion compared with those cultured with IL-2 [86].
Advances in genome editing have also helped overcome metabolic restriction in tumor microenvironments. For instance, Fultang et al. engineered CAR-T cells to resist arginine depletion by knocking in the genes encoding the arginine resynthesis enzymes argininosuccinate synthase and ornithine transcarbamylase [118]. The insertion of both genes increased CAR-T cells’ proliferation and improved their ability to clear leukemia and solid tumors in vivo [118]. Similarly, Ye et al. used a genome-scale gain-of-function CRISPR screen to identify the enzyme proline dehydrogenase 2 (PRODH2], a key enzyme in the proline catabolism pathway [113]. They showed that overexpressing PRODH2 in CAR-T cells promoted memory formation and enhanced cytotoxic activity in leukemia, multiple myeloma, and breast cancer models, both in vitro and in vivo [113].
Additionally, recent evidence suggests that CAR structure may impact the metabolic program in CAR-T cells. A study demonstrated that the CD28 costimulatory domain acts through the PI3K-AKT pathway to increase glucose uptake and glycolysis in response to T-cell activation and differentiation [119]. In contrast, the 4-1BB domain was found to activate both glucose and fatty acid metabolism via the liver kinase B1-AMP-activated protein kinase-acetyl-CoA carboxylase signaling pathway, and it also promotes mitochondrial biogenesis through the PGC1α-mediated pathway [120, 121]. These distinct signaling activities of costimulatory domains, at least in part, drive the T cells carrying CARs with the 4-1BB domain towards TCM, whereas the presence of CD28 domains yields TEFF [57]. Importantly, the mere expression of CARs can increase proliferation and metabolic activity [122], and different single-chain variable fragments can lead to various rates of glucose and amino acid consumption in CAR-T cells [122].
TCR repertoire
TCR is a lineage-defining heterodimeric transmembrane receptor, which is central to initiating ligand-dependent activation of T cells. The diversity of the TCR repertoire is primarily generated by random VDJ recombination of separated gene segments, imprecise joining at gene junction sites, and different pairing of TCRα and TCRβ chains [123]. The estimated lower limit to human TCRαβ repertoire diversity stands at 2.5 × 107 to 1 × 108 [ref 123, 124]. This process results in a varied T-cell population with a highly diverse TCR repertoire poised to respond to foreign antigenic peptides presented by MHC molecules.
TCR repertoire can be used as a surrogate for the T-cell clonotype and expansion [125]. Cancer studies have reported that the diversity and clonality of the TCR repertoire have significant potential in predicting the treatment responses and survivals [126,127,128,129,130,131,132,133,134,135]. In studies of peripheral blood TCR repertoire, higher baseline TCR diversity has been reported to correlate with better immunotherapy response [126, 131, 132], and longer progression-free survival (PFS) in both solid and blood malignancies [126, 129, 132, 133]. Additionally, increased TCR clonality after immunotherapy has been associated with improved treatment responses and may serve as predictor for longer PFS [126, 129, 134]. Meanwhile, studies focusing on the TCR repertoire within the tumor microenvironment have shown that the high and evenly distributed TCR diversity at baseline predicts superior survival [127, 130, 135, 136], whereas pre-treatment high TCR clonality is associated with better response to checkpoint blockade therapies [136]. Recently, further studies have indicated that changes of peripheral blood TCR repertoire could serve as a biomarker for disease progression in advanced lung and bladder cancer, and even for the early detection of ovarian cancers [137,138,139].
In CAR-T therapy, similar power of the TCR repertoire in predicting therapeutic responses and prognosis is observed in CAR-negative T cells [140,141,142]. A study utilizing TCR sequencing and CITE-seq/transcriptome analysis in MM showed that greater baseline TCR diversity was associated with longer PFS, while increased clonal expansion of terminally differentiated T cell clones correlated with shorter PFS [140]. A follow-on study revealed that MM patients achieving CR harbored higher counts of T cell clonotypes both before and after CAR-T cell infusion compared to those of non-CR patients [141]. However, research exploring TCR diversity in CAR-T cells remains limited. Rade et al. found that hyperexpanded CAR-T cells (>100 cells) were predominantly CD8+, and observed the coexistence of CAR-T and non-CAR-T cells with the same clonotype after CAR-T cell infusion [141]. Ledergor et al. revealed a more evenly distributed TCR clonality (lower Gini index) in CAR-T cells compared with non-CAR-T cells [143]. The same trend was found by another study reporting that most CAR-T cells in bone marrow after infusion in MM patients were dominated by a single clone [140]. Despite these findings, the specific clinical implications of TCR repertoire diversity and clonality remain to be fully elucidated.
Conclusion
Since its first clinical application in 2010, CAR-T cell therapy has shown notable success in the treatment of B-cell malignancies. However, achieving sustainable long-term response and remission remains a challenge, for resistance and disease relapse persist as common outcomes for many patients. T cells are the main adaptive immune cells against tumors and serve as the cellular basis for CAR-T cell therapy. Better exploration and characterization of T-cell properties, such as exhaustion, memory status, subsets, senescence, metabolism, and TCR repertoire will enable the dissection of CAR-T cell function in preclinical models and patients.
Importantly, recent developments in multi-omics profiling techniques, including genomics, transcriptomics, proteomics and beyond, have greatly facilitated the exploration of the remarkable diversity of T-cell phenotypes and expanded our collective knowledge in CAR-T cell therapy. It is to be expected that our growing understanding of T-cell characteristics will allow the optimization of CAR-T cell manufacturing, the prediction of treatment responses, and the personalization of therapy.
Change history
22 August 2025
Open Access lisence has been updated to CC BY.
References
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl J Med. 2014;371:1507–17.
Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398:491–502.
Westin JR, Oluwole OO, Kersten MJ, Miklos DB, Perales MA, Ghobadi A, et al. Survival with Axicabtagene Ciloleucel in Large B-Cell Lymphoma. N. Engl J Med. 2023;389:148–57.
Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. 2022;28:325–32.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl J Med. 2020;382:1331–42.
Siddiqi T, Maloney DG, Kenderian SS, Brander DM, Dorritie K, Soumerai J, et al. Lisocabtagene maraleucel in chronic lymphocytic leukaemia and small lymphocytic lymphoma (TRANSCEND CLL 004): a multicentre, open-label, single-arm, phase 1-2 study. Lancet. 2023;402:641–54.
Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N. Engl J Med. 2023;388:1002–14.
Schuster SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22:1403–15.
Kamdar M, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet. 2022;399:2294–308.
Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23:91–103.
Morschhauser F, Dahiya S, Palomba ML, Martin Garcia-Sancho A, Reguera Ortega JL, Kuruvilla J, et al. Lisocabtagene maraleucel in follicular lymphoma: the phase 2 TRANSCEND FL study. Nat Med. 2024;30:2199–207.
Wang Y, Jain P, Locke FL, Maurer MJ, Frank MJ, Munoz JL, et al. Brexucabtagene Autoleucel for Relapsed or Refractory Mantle Cell Lymphoma in Standard-of-Care Practice: Results From the US Lymphoma CAR T Consortium. J Clin Oncol. 2023;41:2594–606.
San-Miguel J, Dhakal B, Yong K, Spencer A, Anguille S, Mateos MV, et al. Cilta-cel or Standard Care in Lenalidomide-Refractory Multiple Myeloma. N. Engl J Med. 2023;389:335–47.
Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl J Med. 2021;384:705–16.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl J Med. 2018;378:439–48.
Amatya C, Weissler KA, Fellowes V, Lam N, Cutmore LC, Natrakul DA, et al. Optimization of anti-CD19 CAR T cell production for treatment of patients with chronic lymphocytic leukemia. Mol Ther Methods Clin Dev. 2024;32:101212.
Xiao X, Huang S, Chen S, Wang Y, Sun Q, Xu X, et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer Res. 2021;40:367.
Wang X, Borquez-Ojeda O, Stefanski J, Du F, Qu J, Chaudhari J, et al. Depletion of high-content CD14(+) cells from apheresis products is critical for successful transduction and expansion of CAR T cells during large-scale cGMP manufacturing. Mol Ther Methods Clin Dev. 2021;22:377–87.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–68.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Baessler A, Vignali DAA. T Cell Exhaustion. Annu Rev Immunol. 2024;42:179–206.
Belk JA, Daniel B, Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol. 2022;23:848–60.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–71.
Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med. 2020;26:1878–87.
Garcia-Calderon CB, Sierro-Martinez B, Garcia-Guerrero E, Sanoja-Flores L, Munoz-Garcia R, Ruiz-Maldonado V, et al. Monitoring of kinetics and exhaustion markers of circulating CAR-T cells as early predictive factors in patients with B-cell malignancies. Front Immunol. 2023;14:1152498.
Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. 2019;129:2123–32.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl J Med. 2019;380:45–56.
Leblay N, Maity R, Barakat E, McCulloch S, Duggan P, Jimenez-Zepeda V, et al. Cite-seq profiling of T cells in multiple myeloma patients undergoing BCMA targeting CAR-T or bites immunotherapy. Blood. 2020;136:11–2.
Huang Y, Shao M, Teng X, Si X, Wu L, Jiang P, et al. Inhibition of CD38 enzymatic activity enhances CAR-T cell immune-therapeutic efficacy by repressing glycolytic metabolism. Cell Rep. Med. 2024;5:101400.
Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–90.
Prinzing B, Schreiner P, Bell M, Fan Y, Krenciute G, Gottschalk S. MyD88/CD40 signaling retains CAR T cells in a less differentiated state. JCI Insight. 2020;5:e136093.
Calderon H, Mamonkin M, Guedan S. Analysis of CAR-Mediated Tonic Signaling. Methods Mol Biol. 2020;2086:223–36.
Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 2016;5:e1253656.
Hu Y, Zu C, Zhang M, Wei G, Li W, Fu S, et al. Safety and efficacy of CRISPR-based non-viral PD1 locus specifically integrated anti-CD19 CAR-T cells in patients with relapsed or refractory Non-Hodgkin’s lymphoma: a first-in-human phase I study. EClinicalMedicine. 2023;60:102010.
Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36:847–56.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44.
Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood. 2018;132:556.
Cao Y, Lu W, Sun R, Jin X, Cheng L, He X, et al. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front Oncol. 2019;9:767.
Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300.
Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol. 2021;22:983–95.
Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372:eaba1786.
Kim C, Williams MA. Nature and nurture: T-cell receptor-dependent and T-cell receptor-independent differentiation cues in the selection of the memory T-cell pool. Immunology. 2010;131:310–7.
Jameson SC, Masopust D. Understanding Subset Diversity in T Cell Memory. Immunity. 2018;48:214–26.
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–9.
Monfrini C, Stella F, Aragona V, Magni M, Ljevar S, Vella C, et al. Phenotypic Composition of Commercial Anti-CD19 CAR T Cells Affects In Vivo Expansion and Disease Response in Patients with Large B-cell Lymphoma. Clin Cancer Res. 2022;28:3378–86.
Wang Y, Tong C, Lu Y, Wu Z, Guo Y, Liu Y, et al. Characteristics of premanufacture CD8(+)T cells determine CAR-T efficacy in patients with diffuse large B-cell lymphoma. Signal Transduct Target Ther. 2023;8:409.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6.
Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020;4:4898–911.
Haradhvala NJ, Leick MB, Maurer K, Gohil SH, Larson RC, Yao N, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat Med. 2022;28:1848–59.
Bai Z, Woodhouse S, Zhao Z, Arya R, Govek K, Kim D, et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci Adv. 2022;8:eabj2820.
Štach M, Pytlík R, Šmilauerová K, Rychlá J, Mucha M, Musil J, et al. Characterization of the input material quality for the production of tisagenlecleucel by multiparameter flow cytometry and its relation to the clinical outcome. Pathol Oncol Res. 2023;29:1610914.
Melenhorst JJ, June CH, Porter DL, Grupp S, Stadtmauer EA, Schuster SJ, et al. Identification and Validation of Predictive Biomarkers to CD19- and BCMA-Specific CAR T-Cell Responses in CAR T-Cell Precursors. Blood. 2019;134:622.
Chen GM, Chen C, Das RK, Gao P, Chen CH, Bandyopadhyay S, et al. Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discov. 2021;11:2186–99.
Lin Y, Raje NS, Berdeja JG, Siegel DS, Jagannath S, Madduri D, et al. Idecabtagene vicleucel for relapsed and refractory multiple myeloma: post hoc 18-month follow-up of a phase 1 trial. Nat Med. 2023;29:2286–94.
Arcangeli S, Bove C, Mezzanotte C, Camisa B, Falcone L, Manfredi F, et al. CAR T cell manufacturing from naive/stem memory T lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J Clin Invest. 2022;132:e150807.
Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin Cancer Res. 2019;25:4014–25.
Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:712.
Moreno-Cortes E, Franco-Fuquen P, Garcia-Robledo JE, Forero J, Booth N, Castro JE. ICOS and OX40 tandem co-stimulation enhances CAR T-cell cytotoxicity and promotes T-cell persistence phenotype. Front Oncol. 2023;13:1200914.
Luo M, Gong W, Zhang Y, Li H, Ma D, Wu K, et al. New insights into the stemness of adoptively transferred T cells by γc family cytokines. Cell Commun Signal. 2023;21:347.
Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res. 2019;7:759–72.
Tian Y, Zajac AJ. IL-21 and T Cell Differentiation: Consider the Context. Trends Immunol. 2016;37:557–68.
Funk CR, Wang S, Chen KZ, Waller A, Sharma A, Edgar CL, et al. PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood. 2022;139:523–37.
Fan F, Yoo HJ, Stock S, Wang L, Liu Y, Schubert ML, et al. Ibrutinib for improved chimeric antigen receptor T-cell production for chronic lymphocytic leukemia patients. Int J Cancer. 2021;148:419–28.
Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat Biomed Eng. 2022;6:118–28.
Dickinson MJ, Barba P, Jäger U, Shah NN, Blaise D, Briones J, et al. A Novel Autologous CAR-T Therapy, YTB323, with Preserved T-cell Stemness Shows Enhanced CAR T-cell Efficacy in Preclinical and Early Clinical Development. Cancer Discov. 2023;13:1982–97.
Garfall AL, Dancy EK, Cohen AD, Hwang WT, Fraietta JA, Davis MM, et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019;3:2812–5.
Das RK, Vernau L, Grupp SA, Barrett DM. Naïve T-cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov. 2019;9:492–9.
Dubnikov Sharon T, Assayag M, Avni B, Kfir-Erenfeld S, Lebel E, Gatt ME, et al. Early lymphocyte collection for anti-CD19 CART production improves T-cell fitness in patients with relapsed/refractory diffuse large B-cell lymphoma. Br J Haematol. 2023;202:74–85.
Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J Hematol Oncol. 2018;11:91.
Zhang J, He T, Xue L, Guo H. Senescent T cells: a potential biomarker and target for cancer therapy. EBioMedicine. 2021;68:103409.
Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8:e000957.
Noll JH, Levine BL, June CH, Fraietta JA. Beyond youth: Understanding CAR T cell fitness in the context of immunological aging. Semin Immunol. 2023;70:101840.
Suen H, Brown R, Yang S, Weatherburn C, Ho PJ, Woodland N, et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016;30:1716–24.
Guha P, Cunetta M, Somasundar P, Espat NJ, Junghans RP, Katz SC. Frontline Science: Functionally impaired geriatric CAR-T cells rescued by increased α5β1 integrin expression. J Leukoc Biol. 2017;102:201–8.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500.
Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol. 2020;38:541–66.
Kumagai S, Itahashi K, Nishikawa H. Regulatory T cell-mediated immunosuppression orchestrated by cancer: towards an immuno-genomic paradigm for precision medicine. Nat Rev Clin Oncol. 2024;21:337–53.
Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Ehlinger ZJ, Kurra S, et al. Post-infusion CAR T(Reg) cells identify patients resistant to CD19-CAR therapy. Nat Med. 2022;28:1860–71.
Pan Y, Wang H, An F, Wu F, Tao Q, Li Y, et al. CD4(+)CD25(+)CD127(low) regulatory T cells associated with the effect of CD19 CAR-T therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Int Immunopharmacol. 2021;96:107742.
An F, Wang H, Liu Z, Wu F, Zhang J, Tao Q, et al. Influence of patient characteristics on chimeric antigen receptor T cell therapy in B-cell acute lymphoblastic leukemia. Nat Commun. 2020;11:5928.
Fischer L, Grieb N, Born P, Weiss R, Seiffert S, Boldt A, et al. Cellular dynamics following CAR T cell therapy are associated with response and toxicity in relapsed/refractory myeloma. Leukemia. 2024;38:372–82.
Beider K, Besser MJ, Schachter J, Grushchenko-Polaq AH, Voevoda V, Wolf I, et al. Upregulation of Senescent/Exhausted Phenotype of CAR T Cells and Induction of Both Treg and Myeloid Suppressive Cells Correlate with Reduced Response to CAR T Cell Therapy in Relapsed/Refractory B Cell Malignancies. Blood. 2019;134:3234.
Pu L, Wang H, Wu F, An F, Xiao H, Wang Y, et al. Predictive model for CAR-T cell therapy success in patients with relapsed/refractory B-cell acute lymphoblastic leukaemia. Scand J Immunol. 2024;99:e13352.
Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–14.
Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2014;124:99–110.
Zhou J, Jin L, Wang F, Zhang Y, Liu B, Zhao T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell. 2019;10:764–9.
Watanabe N, Mo F, McKenna MK. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front Immunol. 2022;13:876339.
Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2:309–22.
Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6.
Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–9.
Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–68.
Provine NM, Larocca RA, Aid M, Penaloza-MacMaster P, Badamchi-Zadeh A, Borducchi EN, et al. Immediate Dysfunction of Vaccine-Elicited CD8+ T Cells Primed in the Absence of CD4+ T Cells. J Immunol. 2016;197:1809–22.
Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N. Engl J Med. 1995;332:143–9.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–38.
Abramson, Palomba JS, Gordon LI ML, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–52.
Aleksandrova K, Leise J, Priesner C, Melk A, Kubaink F, Abken H, et al. Functionality and Cell Senescence of CD4/ CD8-Selected CD20 CAR T Cells Manufactured Using the Automated CliniMACS Prodigy® Platform. Transfus Med Hemother. 2019;46:47–54.
Jo T, Yoshihara S, Okuyama Y, Fujii K, Henzan T, Kahata K, et al. Risk factors for CAR-T cell manufacturing failure among DLBCL patients: A nationwide survey in Japan. Br J Haematol. 2023;202:256–66.
Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500.
Moeller M, Kershaw MH, Cameron R, Westwood JA, Trapani JA, Smyth MJ, et al. Sustained antigen-specific antitumor recall response mediated by gene-modified CD4+ T helper-1 and CD8+ T cells. Cancer Res. 2007;67:11428–37.
Moeller M, Haynes NM, Kershaw MH, Jackson JT, Teng MW, Street SE, et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood. 2005;106:2995–3003.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–31.
Galli E, Bellesi S, Pansini I, Di Cesare G, Iacovelli C, Malafronte R, et al. The CD4/CD8 ratio of infused CD19-CAR-T is a prognostic factor for efficacy and toxicity. Br J Haematol. 2023;203:564–70.
Teoh J, Johnstone TG, Christin B, Yost R, Haig NA, Mallaney M, et al. Lisocabtagene maraleucel (liso-cel) manufacturing process control and robustness across CD19+ hematological malignancies. Blood. 2019;134:593.
Sehgal A, Hoda D, Riedell PA, Ghosh N, Hamadani M, Hildebrandt GC, et al. Lisocabtagene maraleucel as second-line therapy in adults with relapsed or refractory large B-cell lymphoma who were not intended for haematopoietic stem cell transplantation (PILOT): an open-label, phase 2 study. Lancet Oncol. 2022;23:1066–77.
Lee SY, Lee DH, Sun W, Cervantes-Contreras F, Basom RS, Wu F, et al. CD8(+) chimeric antigen receptor T cells manufactured in absence of CD4(+) cells exhibit hypofunctional phenotype. J Immunother Cancer. 2023;11:e007803.
Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–82.
Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14:1064–72.
Rangel Rivera GO, Knochelmann HM, Dwyer CJ, Smith AS, Wyatt MM, Rivera-Reyes AM, et al. Fundamentals of T Cell Metabolism and Strategies to Enhance Cancer Immunotherapy. Front Immunol. 2021;12:645242.
Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 2016;24:104–17.
Chang WK, Yang KD, Chuang H, Jan JT, Shaio MF. Glutamine protects activated human T cells from apoptosis by up-regulating glutathione and Bcl-2 levels. Clin Immunol. 2002;104:151–60.
Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017;25:482.
Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022;34:595–614.
Rial Saborido J, Völkl S, Aigner M, Mackensen A, Mougiakakos D. Role of CAR T Cell Metabolism for Therapeutic Efficacy. Cancers (Basel). 2022;14:5442.
Cheng H, Qiu Y, Xu Y, Chen L, Ma K, Tao M, et al. Extracellular acidosis restricts one-carbon metabolism and preserves T cell stemness. Nat Metab. 2023;5:314–30.
Prochazkova M, Dreyzin A, Shao L, Garces P, Cai Y, Shi R, et al. Deciphering the importance of culture pH on CD22 CAR T-cells characteristics. J Transl Med. 2024;22:384.
Ma S, Ming Y, Wu J, Cui G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol Immunol. 2024;21:419–35.
Fultang L, Booth S, Yogev O, Martins da Costa B, Tubb V, Panetti S, et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood. 2020;136:1155–60.
Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–77.
Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215:1091–100.
Choi BK, Lee DY, Lee DG, Kim YH, Kim SH, Oh HS, et al. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8(+) T cell proliferation. Cell Mol Immunol. 2017;14:748–57.
Lakhani A, Chen X, Chen LC, Hong M, Khericha M, Chen Y, et al. Extracellular domains of CARs reprogramme T cell metabolism without antigen stimulation. Nat Metab. 2024;6:1143–60.
Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4:123–32.
Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA. 2014;111:13139–44.
Pai JA, Satpathy AT. High-throughput and single-cell T cell receptor sequencing technologies. Nat Methods. 2021;18:881–92.
Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, et al. TCR Repertoire Diversity of Peripheral PD-1(+)CD8(+) T Cells Predicts Clinical Outcomes after Immunotherapy in Patients with Non-Small Cell Lung Cancer. Cancer Immunol Res. 2020;8:146–54.
Pothuri VS, Hogg GD, Conant L, Borcherding N, James CA, Mudd J, et al. Intratumoral T-cell receptor repertoire composition predicts overall survival in patients with pancreatic ductal adenocarcinoma. Oncoimmunology. 2024;13:2320411.
Reuben A, Zhang J, Chiou SH, Gittelman RM, Li J, Lee WC, et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat Commun. 2020;11:603.
Abbas HA, Hao D, Tomczak K, Barrodia P, Im JS, Reville PK, et al. Single cell T cell landscape and T cell receptor repertoire profiling of AML in context of PD-1 blockade therapy. Nat Commun. 2021;12:6071.
Keane C, Gould C, Jones K, Hamm D, Talaulikar D, Ellis J, et al. The T-cell Receptor Repertoire Influences the Tumor Microenvironment and Is Associated with Survival in Aggressive B-cell Lymphoma. Clin Cancer Res. 2017;23:1820–8.
Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23.
Abed A, Beasley AB, Reid AL, Law N, Calapre L, Millward M, et al. Circulating pre-treatment T-cell receptor repertoire as a predictive biomarker in advanced or metastatic non-small-cell lung cancer patients treated with pembrolizumab alone or in combination with chemotherapy. ESMO Open. 2023;8:102066.
Charles J, Mouret S, Challende I, Leccia MT, De Fraipont F, Perez S, et al. T-cell receptor diversity as a prognostic biomarker in melanoma patients. Pigment Cell Melanoma Res. 2020;33:612–24.
Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9:eaah3560.
Weber J, Horak C, Hodi F, Chang H, Woods D, Sanders C, et al. Baseline tumor T cell receptor (TcR) sequencing analysis and neo antigen load is associated with benefit in melanoma patients receiving sequential nivolumab and ipilimumab. Ann Oncol. 2016;27:vi359.
Valpione S, Mundra PA, Galvani E, Campana LG, Lorigan P, De Rosa F, et al. The T cell receptor repertoire of tumor infiltrating T cells is predictive and prognostic for cancer survival. Nat Commun. 2021;12:4098.
Liu YY, Yang QF, Yang JS, Cao RB, Liang JY, Liu YT, et al. Characteristics and prognostic significance of profiling the peripheral blood T-cell receptor repertoire in patients with advanced lung cancer. Int J Cancer. 2019;145:1423–31.
Kjær A, Kristjánsdóttir N, Nordentoft I, Juul RI, Birkenkamp-Demtröder K, Ahrenfeldt J, et al. Peripheral T cell receptor repertoire diversity is associated with outcome in bladder cancer. Cancer Res. 2024;84:5216.
Yu X, Pan M, Ye J, Hathaway CA, Tworoger SS, Lea J, et al. Quantifiable TCR repertoire changes in prediagnostic blood specimens among patients with high-grade ovarian cancer. Cell Rep. Med. 2024;5:101612.
Dhodapkar KM, Cohen AD, Kaushal A, Garfall AL, Manalo RJ, Carr AR, et al. Changes in Bone Marrow Tumor and Immune Cells Correlate with Durability of Remissions Following BCMA CAR T Therapy in Myeloma. Blood Cancer Discov. 2022;3:490–501.
Rade M, Grieb N, Weiss R, Sia J, Fischer L, Born P, et al. Single-cell multiomic dissection of response and resistance to chimeric antigen receptor T cells against BCMA in relapsed multiple myeloma. Nat Cancer. 2024;5:1318–33.
Faruqi AJ, Wang P, Bansal R, Corraes ADMS, Zhang H, Shao Z, et al. T-cell receptor repertoire changes associated with clinical response in patients with B-cell non-Hodgkin’s lymphoma receiving CD19 CAR-T therapy. Cancer Res. 2024;84:53.
Ledergor G, Fan Z, Wu K, McCarthy E, Hyrenius-Wittsten A, Starzinski A, et al. CD4+ CAR T-cell exhaustion associated with early relapse of multiple myeloma after BCMA CAR T-cell therapy. Blood Adv. 2024;8:3562–7.
Acknowledgements
This work was supported by the student’s grant system of the University of Ostrava (SGS03/LF/2024); MH CZ-DRO-FNOs/2023 from the Ministry of Health of the Czech Republic; European Union project LERCO (No.CZ.10.03.01/00/22_003/0000003) via the Operational Program Just Transition; SALVAGE project CZ.02.01.01/00/22_008/0004644, supported by OP JAK, with co-financing from the EU and the State Budget. We thank Jana Klepáčová, MSc., and Tereza Ševčíková, Ph.D., for their assistance with proofreading and all the lab members for their support. The figure was created with BioRender.
Author information
Authors and Affiliations
Contributions
RH conceptualized the review. ZT designed and drafted the manuscript and prepared the figure. All authors, ZT, ZC, JK, PC, JM, SC, and RH reviewed, revised, and approved the final version of the review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tao, Z., Chyra, Z., Kotulová, J. et al. Impact of T cell characteristics on CAR-T cell therapy in hematological malignancies. Blood Cancer J. 14, 213 (2024). https://doi.org/10.1038/s41408-024-01193-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-024-01193-6
This article is cited by
-
CAR-engineered cell therapies: current understandings and future perspectives
Molecular Biomedicine (2026)
-
Burixafor, a CXCR4 inhibitor with a differentiated kinetics profile: results of a phase 2 study for rapid cell mobilization in multiple myeloma and lymphoma patients undergoing transplant
Annals of Hematology (2026)
-
Multi-omic profiling and preclinical efficacy of fratricide-driven, unedited CD7 CAR-T cells in T-cell leukemia
Journal of Translational Medicine (2026)
-
AI-guided CAR designs and targeted pathway modulation to enhance multi-antigen CAR T cell durability and overcome antigen escape
Nature Communications (2026)
-
BCMA-directed mRNA CAR-T cell therapy for myasthenia gravis: exploratory biomarker analysis of a placebo-controlled phase 2b trial
Nature Medicine (2026)
