
Abstract
Teclistamab, a BCMAxCD3-directed bispecific antibody, has shown high response rates and durable remissions in triple-class-exposed patients with relapsed/refractory multiple myeloma. We performed a retrospective study evaluating the efficacy and safety of teclistamab in 210 patients treated at 9 academic centers from five countries within the IMWG Immunotherapy Working Group Committee. Patients were heavily pretreated, with 83% having triple-class refractory disease and 44% with prior BCMA-targeted therapy. With a median follow-up of 5.3 months, the overall response rate (ORR) was 67% in 188 response-evaluable patients, including 55% with a very good partial response or better. The 6-month progression-free survival (PFS) and overall survival rates were 53% (95% CI, 46–61%) and 73% (67–80%), respectively. Patients who received prior BCMA-directed therapy compared to BCMA-treatment-naïve patients had a lower ORR (58.3 vs 74.0%; P = 0.03) and PFS (6-month PFS 43% [95% CI, 33–55%] vs 63% [54–73%]; logrank P = 0.004). Step-up dosing occurred in an outpatient setting for 23% of patients. CRS occurred in 54% of patients, and infections were reported in 56.2% of patients, with 22% having grade ≥3 infections. In this multicenter real-world study, we found that teclistamab can lead to rapid responses in heavily pretreated myeloma patients with comparable efficacy and safety profiles, as demonstrated in MajesTEC-1.
Similar content being viewed by others


Introduction
Teclistamab is the first approved B-cell maturation antigen (BCMA)xCD3 bispecific antibody for the treatment of triple-class-exposed patients with relapsed/refractory multiple myeloma (RRMM) in select jurisdictions, including the FDA and EMA based on the pivotal MajesTEC-1 study [1, 2]. In the phase 1/2 MajesTEC-1 study, teclistamab demonstrated an overall response rate (ORR) of 63%, including a very good partial response (VGPR) or better in 58.8% at a median follow-up of 14.1 months. After a longer follow-up of 30.4 months, the CR rate increased to 46.1%, and median duration of response (DOR) was 24 months (95% CI, 17.0 – not reached [NR]), and median progression-free survival (PFS) was 11.4 months (95% CI, 8.8–16.4) [2].
Teclistamab is associated with unique side effects, including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), and has been associated with high rates of infection. In MajesTEC-1, 119 (72.1%) patients experienced CRS, with most events being grade ≤2 and one grade 3 event in the setting of grade 3 pneumonia. All CRS cases were resolved, and none led to treatment discontinuation. Five patients had a total of nine ICANS events, all grade ≤2, and resolved without treatment discontinuation or dose reduction [1]. In the primary safety analysis of MajesTEC-1, infections occurred in 76.4% of patients with grade 3/4 infections in 44.8% [1]. With a longer median follow-up (22.8 months), Nooka and colleagues reported infections in 132 patients (80%), with grade 3/4 infections occurring in 55.2% of patients. Opportunistic infections occurred in 15 (9.1%) patients, and 21 (12.7%) patients died from infections [3]. Sixty-one (37%) patients had IgG levels less than 400 mg/dL during teclistamab therapy.
Since teclistamab was the first bispecific antibody approved for use in patients with RRMM, and phase 1/2 studies generally include more fit patients [4, 5], real-world data is needed to understand the outcomes in routine clinical practice. Although there have been other real-world studies early after teclistamab approval [6,7,8], our study is the largest to date evaluating the real-world safety and efficacy of teclistamab in a diverse patient population including specific subgroups treated at International Myeloma Working Group (IMWG) designated centers.
Methods
This is an international multicenter retrospective study of patients with RRMM treated with teclistamab administered outside of a clinical trial at 9 academic centers from five countries (United States, United Kingdom, Greece, Spain, and Canada) within the IMWG Immunotherapy Working Group Committee. Patients started teclistamab between May 24, 2022 to October 1, 2023. The last follow-up date was January 23, 2024. Data underwent peer-based quality checks for completeness and internal consistencies.
Teclistamab was administered according to the FDA/EMA approved label and institutional standards, and patients were included if they received at least 1 dose of teclistamab. Modifications to the dosing schedule, antimicrobial prophylaxis, management of adverse events, and supportive therapies were per institutional guidelines and at the discretion of the treating physician. Cytogenetic information was obtained from samples taken at any timepoint since diagnosis. High-risk cytogenetic abnormalities (HRCA) was defined as one or more of the following abnormalities: 1q+, t(4;14), t(14;16), t(14;20), and del(17p).
Response was assessed using the IMWG consensus criteria for response [9]. Individual patients’ charts were reviewed to assess baseline characteristics, treatment characteristics, and to evaluate for specific adverse events (AEs), including infection, cytopenias, CRS, and ICANS. CRS and ICANS were graded according to the American Society for Transplantation and Cellular Therapy criteria [10]. All other AEs were graded using Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 retrospectively.
Statistical analysis
Descriptive statistics were used to summarize patient and disease characteristics and toxicities for patients with RRMM treated with teclistamab. ORR was compared between subgroups using Fisher’s exact tests and associated with risk factors using logistic regressions. PFS was defined as the time from the start of teclistamab until the progression of disease or death from any cause. Overall survival (OS) was defined as the time from the start of teclistamab to death from any cause. DOR was defined as the time from the first observation of at least partial response (PR) to disease progression or death. Censoring time was defined as the last study contact prior to the data analysis cut-off date. PFS, OS, and DOR were summarized using the Kaplan–Meier method and compared between subgroups using the logrank test. Cox regressions were used to evaluate covariate effects on PFS and OS. All statistical analyses were performed using R software [11]. Variables with P values <0.05 were considered statistically significant.
Results
Patient characteristics
Between May 24, 2022 and October 1, 2023, 210 patients with RRMM received teclistamab at 9 international centers. Baseline characteristics are summarized in Table 1. The median age was 67 (range 33–91) years, with 23% of patients being ≥75 years. At diagnosis, 82/164 (50%) patients had HRCAs, and 37/126 (29%) had extramedullary disease (EMD). At the start of teclistamab, 26/209 (12.4%) patients had creatinine clearance (CrCl) ≤30 ml/min. The median time from diagnosis to the first teclistamab dose was 6.1 (0.60–29.2) years. The patients were heavily pretreated with a median of 6 (range, 1–20) prior lines of therapy. The majority (83%) had triple-class refractory disease, and 44% of patients had penta-drug refractory disease.
When utilizing key eligibility criteria from MajesTEC-1, 71% of patients in this real-world cohort would not have met the defined eligibility criteria. The ineligible cohort included 92 (44%) patients treated with prior BCMA-targeted therapy, 26 (12.4%) with CrCl ≤30 ml/min, 52 (25%) with significant cytopenias, and 35 (27%) with Eastern Cooperative Oncology Group (ECOG) performance status ≥2. Among the 92 patients treated with prior BCMA-directed therapy, 42 (46%) had received prior CAR T-cell therapy alone, 27 (29%) had prior antibody-drug conjugate (ADC) alone, 6 (7%) had prior BCMA-bispecific antibody alone, and 17 (18%) had received multiple BCMA-directed agents. The median time from the last dose of prior BCMA-directed therapy to the start of teclistamab was 249.5 (range, 30–1290) days for 90 patients with available data. Additionally, six patients had G protein-coupled receptor, class C, group 5, member D (GPRC5D)-targeted therapy prior to teclistamab.
Efficacy
Response to therapy
Overall, 188 patients were evaluable for response in this study. The ORR was 67% with 63 (34%) achieving VGPR and 40 (21%) a ≥CR. Responses appear to deepen over time and as patients undergo bone marrow sampling (not done routinely in the real world) (Fig. 1A). Among responders, median time to first response was 1.1 (interquartile range [IQR] 0.8–2.1) months.

A Stacked bar graph illustrating the rate of PR, VGPR, CR, and sCR in 188 response-evaluable patients treated with teclistamab. Responses according to IMWG criteria. B Forest Plot of ORR in Subgroups.
In patients who had prior BCMA-directed therapy, ORR was lower at 58.3% compared to BCMA-treatment naïve patients with an ORR of 74.0% (OR 0.49, 95% CI 0.26–0.91; P = 0.024) (Fig. 1B). Of the 84-response-evaluable patients with prior BCMA-directed therapy, 45.2% had ≥VGPR, including 18% with sCR/CR. ORR based on the type of prior BCMA-targeted agent includes 57.5% for CAR T-cell therapy alone, 73.9% for ADC alone, 40% for bispecific antibody, and 43.8% for exposure to ≥2 BCMA-directed agents (P = 0.22).
Patients with HRCAs had a lower ORR compared to standard-risk and unknown cytogenetic risk patients (60.8 vs 72.1% vs 70.7%; P = 0.30), but the differences did not reach statistical significance. There was no significant difference in ORR for patients with CrCl ≤30 ml/min compared to patients with CrCl >30 ml/min at teclistamab initiation (66.7% vs 67.1%; P = 1.00). Among the 126 patients who had data on EMD evaluation, there was no significant difference in ORR for patients with EMD compared to patients without EMD (67.7 vs 74.1%; P = 0.14). When comparing ORR based on penta-drug refractory status, ORR was significantly higher for patients who were not penta-refractory (not penta-refractory: 80.2% vs penta-refractory: 56.9% vs unknown status: 57.1%; P = 0.003). Additionally, when comparing subgroups based on select MajesTEC-1 eligibility criteria, the MajesTEC-1 ineligible subgroup had an ORR of 59.4% compared to the MajesTEC-1 eligible subgroup with an ORR of 89.4% (OR 5.74, 95% CI 2.32-17.4; P < 0.001).
Univariable and multivariable analyses were performed to determine factors associated with ORR (Table 2). On multivariate analysis, factors associated with inferior ORR included ECOG PS 3 (OR 0.14, 95% CI 0.02–0.87; P = 0.04), platelet count <50 × 109/L at the start of teclistamab (OR 0.31, 95% CI 0.12–0.78; P = 0.01), prior BCMA-directed therapy (OR 0.47, 95% CI 0.24–0.94; P = 0.03), and penta-drug refractoriness (OR 0.38, 95% CI 0.16–0.86; P = 0.02).
Duration of response, progression-free survival, and overall survival
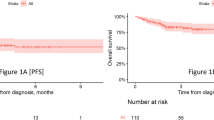
With a median follow-up of 5.3 (range 0.2–16.3) months, the 6-month PFS and OS rates were 53% (95% CI, 46–61%) and 73% (95% CI, 67–80%), respectively (Fig. 2). Median DOR among responders was estimated to be 11 (95% CI 11-NR) months.

A Progression-free survival (PFS). B Overall survival (OS). C Duration of response (DoR).
In subgroup analysis based on BCMA-directed therapy exposure, the 6-month PFS for the cohort with prior BCMA-directed therapy and anti-BCMA naïve cohort was 43% (95% CI, 33–55%) and 63% (54–73%), respectively (logrank P = 0.004) (Fig. 3A). Among patients who received prior BCMA-directed therapy, the 6-month PFS was 46% (95% CI, 33–65%) for CAR T-cell alone, 53% (95% CI, 36–78%) for ADC alone, 17% (95% CI, 2.8–100%) for bispecific antibody alone, and 28% (95% CI, 13–62%) for ≥2 BCMA-directed agents (logrank P = 0.078) (Fig. 3B). The 6-month OS rate was 75% (95% CI, 66–84%) in the prior BCMA treated group and 72% (95% CI, 63–81%) in the anti-BCMA naïve group (logrank P = 0.8) (Fig. 4A). The 6-month OS based on prior BCMA-directed agent was 78% (95% CI, 66–92%) for CAR T-cell alone, 78% (95% CI, 62–97%) for ADC alone, 50% (95% CI, 22–100%) for bispecific antibody alone, and 70% (95% CI, 51–96%) for ≥2 BCMA-directed agents (logrank P = 0.23) (Fig. 4B).

A PFS according to prior BCMA exposure. B PFS according to the type of prior BCMA-directed agent. C PFS according to cytogenetic risk. D PFS according to creatinine clearance. E PFS according to penta-drug refractoriness. F PFS according to MajesTEC-1 eligibility.

A OS according to prior BCMA exposure. B OS according to the type of prior BCMA-directed agent. C OS according to cytogenetic risk. D OS according to creatinine clearance. E OS according to penta-drug refractoriness. F OS according to MajesTEC-1 eligibility.
In subgroup analysis based on cytogenetic risk, the 6-month PFS for the HRCA subgroup was lower than standard-risk and unknown-risk subgroups (46% [95% CI 36–59%] vs 60% [95% CI 49–72%] vs 54% [95% CI 40–72%]), but the differences did not reach statistical significance (logrank P = 0.09) (Fig. 3C). The 6-month OS rate for the HRCA group was 66% (95% CI, 56–77%) compared to 81% (95% CI, 72–90%) in the standard-risk group and 72% in the unknown-risk group (logrank P = 0.009) (Fig. 4C). For patients with CrCl ≤30 ml/min compared with patients with CrCl >30 ml/min, there was no statistically significant difference in 6-month PFS (62% [95% CI, 44–86%] vs 52% [95% CI, 45–60%]; logrank P = 0.94) and OS (80% [95% CI, 66–97%] vs 72% [95% CI, 65–79%]; logrank P = 0.67) (Figs. 3D, 4D). The 6-month PFS rates for penta-drug refractory, not penta-refractory, and unknown penta-refractory status were 49% (95% CI, 38–62%), 61% (50–73%), and 46% (33–63%) (logrank P = 0.07) (Fig. 3E). The respective 6-month OS rates were 65% (95% CI, 55–78%), 80% (71–90%), and 72% (60–86%) (logrank P = 0.08) (Fig. 4E). Although the differences were not statistically significance, there was a trend toward improved PFS and OS associated with not being penta-drug refractory compared to being penta-drug refractory and unknown refractory status. In subgroup analysis based on select MajesTEC-1 eligibility criteria, the 6-month PFS for the MajesTEC-1 ineligible group was 45% (95% CI, 37–54%) compared to 77% (95% CI, 65–91%) for MajesTEC-1 eligible patients; for patients with unknown eligibility status, 6-month PFS was 55% (95% CI, 32–94%) (logrank P < 0.01) (Fig. 3F). The 6-month OS rate for MajesTEC-1 ineligible, eligible, unknown eligibility status groups were 70% (95% CI, 62–78%), 83% (73–96%), and 73% (51–100%) respectively (logrank P = 0.03).
Univariable and multivariable analyses were performed to determine characteristics associated with PFS and OS (Tables 3, 4). On multivariable analysis, receiving prior BCMA-directed therapy (HR 1.82, 95% CI, 1.19–2.79; P = 0.006), platelet count <50 × 109/L at the start of teclistamab (HR 3.18, 95% CI, 1.90–5.35; P < 0.001), and ECOG PS 3 (HR 3.70, 95% CI, 1.23–11.13; P = 0.02) were significant predictors for PFS (Table 3).
Safety
CRS
During the step-up dosing phase of teclistamab, 161 (77.0%) patients were treated in an inpatient setting and 48 (23.0%) initiated therapy in an outpatient setting; one patient did not have the site of initial administration available. CRS occurred in 113/209 (54%) patients with available data. After step-up dose 1, 61 (29%) patients developed CRS with 50 (23.9%) having grade 1 and 10 (4.8%) with grade 2; one patient did not have the CRS grade available. Among these patients, 31 received tocilizumab, and 6 received steroids. After step-up dose 2, 55 (26%) patients had CRS events, including 43 (20.6%) with grade 1 and 12 (5.7%) with grade 2. There were 24 patients who received tocilizumab and 16 received steroids. After the first full dose of teclistamab, 22 (10.5%) patients had a CRS event, including 17 (8.1%) with grade 1, 4 (1.9%) with grade 2, and 1 (0.5%) with grade 4. The grade 4 CRS event was treated with tocilizumab and steroids with resolution. Among these patients, 11 received tocilizumab and 3 received steroids. One patient developed CRS during cycle 1 (second full dose), which resolved with steroids, after completing step-up dosing without having reported CRS. Among patients who received teclistamab step-up dosing in an outpatient setting, 19 (39.6%) developed CRS, which were predominantly grade 1–2, except for the grade 4 event detailed above.
ICANS
ICANS was reported in 15 (7.3%) out of 205 patients with available data. The median time to onset was 2.5 (range 1–55) days. Among these patients, 7 had grade 1, 3 had grade 2, 2 had grade 3, and 2 had grade 4 ICANS. Seven patients had concurrent CRS and ICANS during step-up dosing. All ICANS events resolved with a median duration of 2 (range 0–25) days, including the two grade 4 events. Among patients who developed ICANS, 10 (4.9%) received steroids, 7 (3.4%) received tocilizumab, and 2 (1%) received anakinra.
Infections
Infectious disease prophylaxis was per institutional guidelines, and patients uniformly received herpes simplex virus/varicella zoster virus (HSV/VZV) and Pneumocystis jirovecii (PJP) prophylaxis. Data regarding antibacterial and antifungal prophylaxis was not formally collected as part of this study. Overall, infections occurred in 118 (56.2%) patients during teclistamab therapy, with 46 (21.9%) patients having grade ≥3 infections, including 8 (4%) patients who died from infection. Among the 118 patients, 54 (45.8%) had ≥2 infections while receiving teclistamab. The median time to first onset of infection was 33 (range 0–459) days. The most common infectious pathogens reported within our real-world cohort were viral infections (including COVID-19, CMV reactivation, rhinovirus, RSV, norovirus, and parainfluenza) and bacterial infections (including C. difficile, Pseudomonas, E. coli, Enterococcus, Klebsiella). The most common infections for the 118 patients were pneumonia (22.4%), upper respiratory tract infections (19.0%), and viremia (41%), with asymptomatic/symptomatic CMV reactivation (40%) being the most common.
Among 173 patients with available data, 119 (69%) patients had evidence of hypogammaglobulinemia, a known sequelae of BCMA-targeted therapy. To mitigate this complication, 99 (62%) out of 160 patients were receiving intravenous immunoglobulin (IVIG) at the time of data cut-off. The use of IVIG replacement, (i.e., primary and/or secondary infection prophylaxis) was according to the participating center’s institutional practice.
Cytopenias
Grade ≥3 neutropenia was reported in 83 (39.5%) patients, with 32 (15.2%) patients having persistent neutropenia lasting > 7 days and 46 (21.9%) patients receiving growth factor support. Grade ≥3 thrombocytopenia was reported in 60 (28.6%) patient,s including 10 (5.0%) with significant bleeding and 35 (16.7%) patients requiring platelet transfusion support.
Discussion
In this real-world study conducted through the IMWG Immunotherapy Working Group Committee involving multiple international centers, we report outcomes for 210 patients with RRMM treated with teclistamab. Compared to MajesTEC-1, our real-world cohort was comparable in median age, time since diagnosis, and prior lines of therapy but was enriched with high-risk features, including the presence of EMD (29%) and ISS Stage III disease (30.6%) as well as the presence of HRCA (50%) although the definition in this study includes the presence of +1q. Additionally, our patient population was heavily pretreated, with 83% being triple-class refractory and 44% being penta-drug refractory compared to 78% and 30% in MajesTEC-1, respectively, and also included patients treated with prior BCMA-directed therapy and/or T-cell redirecting therapy (TCR). The ORR of 67.0% and ≥VGPR rate (54.8%) in our real-world cohort were similar to those seen in the MajesTEC-1 trial and responses reported in various real-world studies with ORR ranging from 59.3 to 66% [1, 6,7,8]. Responses may deepen over time, as seen in MajesTEC-1. The 6-month estimated PFS was 53% (95% CI, 46–61%) and 6-month OS rate was 73% (95% CI, 67–80%). In the real-world study by Mohan and colleagues with 110 patients treated with teclistamab, 6-month PFS and OS were comparable with our findings at 52% and 80%, respectively, after a median follow-up of 3.5 months [6]. In another US-based real-world study, Dima and colleagues reported an ORR of 66% with a median PFS of 5.4 months (95% CI, 3.4-NR) and 6-month OS of 70% for their cohort of 106 patients after a median follow-up of 3.8 months [7]. In the Germany-based real-world study of 123 patients, the ORR was 59.3% and the median PFS was 8.7 months after a median follow-up of 5.5 months [8].
The majority of the patients in this real-world study would not have met key MajesTEC-1 eligibility criteria, including 92 (44%) patients with prior BCMA-directed therapy. Encouragingly, 58.3% of patients exposed to prior BCMA-directed therapy were able to achieve at least a partial response, although this ORR was significantly lower than in the anti-BCMA naïve subgroup with an ORR of 74.0% (P = 0.03). In MajesTEC-1, cohort C included 40 patients with prior exposure to BCMA-directed therapy (CAR T-cell therapy and/or ADC) and reported an ORR of 52.5%, including sCR/CR in 30.0% and ≥VGPR in 47.5%. Efficacy was similar in patients who received prior anti-BCMA ADC alone (ORR 55.2%) and CAR T-cell therapy (ORR 53.5%). At a median follow-up of 28.0 months, the median DOR was 14.8 months, the median PFS was 4.5 months, and the median OS was 15.5 months [12]. Dima et al. reported an ORR of 59% for the 56 (53%) patients who had BCMA-directed therapy prior to teclistamab [7]. Mohan et al. reported an ORR of 59% for patients with prior BCMA-directed therapy [6]. In the Germany-based real-world study, ORR for the prior BCMA-exposed cohort was 54.8%, including 33.3% after idecabtagene vicleucel and 73.9% for patients with prior ADC therapy [8]. We saw similar findings of ORR being higher for patients who received anti-BCMA ADC therapy alone compared to BCMA-directed TCR (CAR T and bispecific antibody), although we did not detect a statistically significant difference in our patient population. Loss of BCMA expression and/or mutational events on BCMA have been described as mechanisms for primary resistance to teclistamab and other BCMA-directed bispecific antibodies after prior BCMA-directed TCR [13]. Additionally, T-cell exhaustion, low T-cell count and fitness, and an immunosuppressive microenvironment can result in resistance to bispecific antibodies, which may have contributed to lower ORR and inferior 6-month PFS associated with teclistamab in our patients who were treated with prior bispecific antibody therapy [14,15,16]. In contrast, in DREAMM-1 and DREAMM-2 trials, belantamab mafodotin did not appear to negatively impact lymphocyte numbers and cause systemic immune impairment [17], although loss of BCMA expression has been described in patients treated with belantamab [18]. Future studies in this specific patient cohort with larger sample size and longer follow-up are needed to further characterize patients who may benefit from teclistamab or other anti-BCMA-bispecific antibodies as BCMA-directed therapeutic agents become more commonly used in the management of RRMM, including in earlier lines of therapy. Additionally, methodologies, including antigen expression testing or the use of soluble BCMA (sBCMA) measurements need prospective validation to improve stratification and selection of patients for BCMA-directed therapy and aid in sequencing of therapeutic agents.
In addition to prior BCMA-directed therapy, platelet count <50 × 109/L at the start of teclistamab and ECOG PS 3 were associated with inferior outcomes on multivariate analysis. Both variables are important markers of disease burden and frailty, respectively. Lee and colleagues recently demonstrated that high tumor burden and high sBCMA levels contributed to primary refractoriness to anti-BCMA-bispecific antibodies [14]. Clinicians may need to be cautious when treating patients with these characteristics and consider additional supportive therapies and proactive mitigation or prophylactic strategies for toxicity management in frail patients and in patients with significant disease burden.
In our study, a small subgroup of patients had CrCl ≤30 ml/min prior to receiving teclistamab, and we did not find statistically significant differences in ORR, PFS, and OS for this subgroup when compared with patients with CrCl >30 ml/min. Teclistamab pharmacokinetics were not significantly impacted by mild or moderate renal impairment, but patients with CrCl <40 ml/min were excluded from MajesTEC-1 [19]. Consistent with a single institution’s experience, our findings suggest that patients with significant renal impairment have similar outcomes to patients with CrCl >30 ml/min, although a larger cohort, including those with end-stage renal disease, would be needed to further investigate outcomes and safety in this subgroup [20].
Interestingly, certain high-risk features, including EMD and HRCA, were not associated with statistically significant differences in ORR in our study, although ORR was numerically higher in patients without EMD and in patients with standard-risk cytogenetics. Mohan et al. similarly did not find differences in ORR based on the presence of EMD or HRCA [6]. In contrast, MajesTEC-1 and other real-world teclistamab studies demonstrated decreased efficacy of teclistamab in patients with EMD [1, 7, 8]. As seen in MajesTEC-1 and the Germany-based study, there were no statistically significant differences in PFS based on the presence of HRCAs, although in our study there is a trend in improved PFS associated with standard-risk cytogenetics. There was a significant difference in OS based on cytogenetic risk. However, the median follow-up time was short in this study, and we must acknowledge that there is likely underreporting of HRCAs and EMD in this real-world patient population since bone marrow biopsies and imaging studies were not required as part of routine clinical practice at the start of teclistamab.
The safety profile of teclistamab in this real-world study was comparable to that observed in MajesTEC-1 and other real-world studies. In real-world studies, the incidence of CRS and ICANS was 56–64% and 7.3–14%, respectively, of which the majority were grade 1–2 in severity [8]. CRS occurred in 54% of patients, with most events being grade 1–2; ICANS occurred in 7.3% of patients and were mostly grade ≤2. However, there were a few clinically significant grade ≥3 CRS and ICANS events that occurred in our patient population, alerting to the possibility that high-grade CRS and ICANS may occur more frequently in the real-world setting than as noted in clinical trials. All but one patient in our cohort were able to resume teclistamab dosing following grade ≥3 CRS or ICANS event. In the German study, the incidence of CRS and ICANS was 58.5 and 7.3%, respectively [8]. In the two US-based studies, the incidence of CRS ranged from 56–64% of which more than 90% of events were grade 1–2, and ICANS events occurred in 11–14% of which most were grade 1–2 [6, 7]. In our patient population, 48 (23%) were able to initiate step-up dosing in an outpatient setting with low rates of CRS, suggesting the feasibility of this strategy. Using specific patient eligibility criteria and remote patient monitoring, Sandahl and colleagues demonstrated the feasibility and safety of outpatient teclistamab step-up dosing at the Mayo Clinic [21]. Additionally, other strategies have been implemented or are currently being investigated to allow for safe outpatient administration of teclistamab step-up dosing, including prophylactic tocilizumab and pocket dexamethasone [22, 23].
In trials and real-world studies of teclistamab, infections were frequent adverse events; the incidence of infections was >50%, with >20% being grade ≥3 [3, 6,7,8]. In our study, any grade infection was reported in 56.2% of patients, and 21.9% had grade ≥3 infections, including 8 patients who died from infection. Due to the substantial burden of infections associated with BCMA-directed bispecific antibodies, clinicians and patients should be vigilant for various types of infection to facilitate prompt and early intervention. Additionally, active surveillance and prophylactic strategies should be implemented in patients receiving teclistamab, including the use of growth factors, prophylactic antimicrobials, and vaccinations to avoid serious infectious complications [24, 25]. Hypogammaglobulinemia was reported in 69.8% of patients and 61.9% of patients with available data were receiving IVIG. These data support proactive preventative strategies for infection, including early use of IVIG. In a study by van de Donk and colleagues, primary prophylaxis with IVIG supplementation resulted in a log-fold reduction in the incidence of infections in patients receiving teclistamab, strongly supporting the use of IVIG [26].
Strengths of the study include a large sample size from a diverse international population. This real-world study design allowed for the inclusion of patients with various comorbidities reflective of the general population, who are frailer than those treated in clinical trials. Limitations include a retrospective study design and a short median follow-up of 5.3 months, making it difficult to compare survival outcomes with MajesTEC-1. There is the potential for underreporting of adverse events and heterogeneity in patient selection and institutional practices for treatment schedules and toxicity management, which likely introduced uncompensated impact on the analysis, although this is reflective of real-world practice patterns. To overcome some of these biases, we completed a sensitivity analysis to address potential confounding site effect and stratified disease- and patient-related variables by study sites (Supplement Table 1). Additionally, we included sites as a variable in multivariate analyses (Supplement Table 2). Moreover, bone marrow biopsies and imaging studies were not routinely performed prior to initiation of teclistamab and during follow-up resulting in certain missing data and inability to assess for CR, minimal residual disease (MRD) status, and EMD in all of the included patients as expected in the real-world setting. We treated missing data as a separate category rather than performing a complete case analysis, which allowed us to retain all available data while accounting for potential informative missingness and avoiding bias in assessing associations.
In summary, this study is the largest real-world experience with teclistamab in RRMM given on-label at multiple international academic centers. We demonstrated that teclistamab can lead to rapid responses in the majority of heavily pretreated patients enriched with high-risk disease features, comparable to results seen in MajesTEC-1. Encouragingly, even patients with prior exposure to BCMA-directed therapies were able to achieve deep responses, although less than in patients who were naïve to anti-BCMA agents. The durability of these responses needs to be determined with longer follow-ups. The incidence of CRS and ICANS was comparable to findings reported from MajesTEC-1, and a subgroup of patients was able to receive teclistamab step-up dosing in the outpatient setting. Prevention and management of infections remain a challenge and require close monitoring of these patients. Investigation into reduced frequency dosing schedules, limited or finite, response-adapted or fixed-duration therapy, and infection prophylactic strategies are warranted to lower infection-related morbidity and mortality while maintaining efficacy.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387:495–505.
Garfall, Nooka AL, Donk AK, NWCJvd, Moreau P, Bhutani M, et al. Long-term follow-up from the phase 1/2 MajesTEC-1 trial of teclistamab in patients with relapsed/refractory multiple myeloma. J Clin Oncol. 2024;42:7540.
Nooka AK, Rodriguez C, Mateos MV, Manier S, Chastain K, Banerjee A, et al. Incidence, timing, and management of infections in patients receiving teclistamab for the treatment of relapsed/refractory multiple myeloma in the MajesTEC-1 study. Cancer. 2024;130:886–900.
Chari A, Romanus D, Palumbo A, Blazer M, Farrelly E, Raju A, et al. Randomized clinical trial representativeness and outcomes in real-world patients: comparison of 6 hallmark randomized clinical trials of relapsed/refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2020;20:8–17.e6.
Shah JJ, Abonour R, Gasparetto C, Hardin JW, Toomey K, Narang M, et al. Analysis of common eligibility criteria of randomized controlled trials in newly diagnosed multiple myeloma patients and extrapolating outcomes. Clin Lymphoma Myeloma Leuk. 2017;17:575–83.e2.
Mohan M, Monge J, Shah N, Luan D, Forsberg M, Bhatlapenumarthi V, et al. Teclistamab in relapsed refractory multiple myeloma: multi-institutional real-world study. Blood Cancer J. 2024;14:35.
Dima D, Davis JA, Ahmed N, Jia X, Sannareddy A, Shaikh H, et al. Safety and efficacy of teclistamab in patients with relapsed/refractory multiple myeloma: a real-world experience. Transplant Cell Ther. 2024;30:308.e1–e13.
Riedhammer C, Bassermann F, Besemer B, Bewarder M, Brunner F, Carpinteiro A, et al. Real-world analysis of teclistamab in 123 RRMM patients from Germany. Leukemia. 2024;38:365–71.
Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e46.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transpl. 2019;25:625–38.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing 2023.
Touzeau C, Krishnan AY, Moreau P, Perrot A, Usmani SZ, Manier S, et al. Efficacy and safety of teclistamab in patients with relapsed/refractory multiple myeloma after BCMA-targeting therapies. Blood. 2024;144:2375–88.
Lee H, Ahn S, Maity R, Leblay N, Ziccheddu B, Truger M, et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med. 2023;29:2295–306.
Lee H, Durante M, Skerget S, Vishwamitra D, Benaoudia S, Ahn S, et al. Impact of soluble BCMA and non–T-cell factors on refractoriness to BCMA-targeting T-cell engagers in multiple myeloma. Blood. 2024;144:2637–51.
Verkleij CPM, O’Neill CA, Broekmans MEC, Frerichs KA, Bruins WSC, Duetz C, et al. T-cell characteristics impact response and resistance to T-cell–redirecting bispecific antibodies in multiple myeloma. Clin Cancer Res. 2024;30:3006–22.
Philipp N, Kazerani M, Nicholls A, Vick B, Wulf J, Straub T, et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood. 2022;140:1104–18.
Lowther DE, Houseman EA, Han G, Kleanthous E, Knoblock D, Zhou X, et al. No evidence of BCMA expression loss or systemic immune impairment after treatment with the BCMA-targeted antibody-drug conjugate (ADC) belantamab mafodotin (Belamaf) in the DREAMM-1 and DREAMM-2 trials of patients with relapsed/refractory multiple myeloma (RRMM). Blood. 2022;140:611–3.
Firestone RS, Socci ND, Shekarkhand T, Zhu M, Qin WG, Hultcrantz M, et al. Antigen escape as a shared mechanism of resistance to BCMA-directed therapies in multiple myeloma. Blood. 2024;144:402–7.
Miao X, Wu LS, Lin SXW, Xu Y, Chen Y, Iwaki Y, et al. Population pharmacokinetics and exposure-response with teclistamab in patients with relapsed/refractory multiple myeloma: results from MajesTEC-1. Target Oncol. 2023;18:667–84.
Joiner L, Bal S, Godby KN, Costa LJ. Teclistamab in patients with multiple myeloma and impaired renal function. Am J Hematol. 2023;98:E322–E4.
Sandahl TB, Soefje SA, Fonseca R, Ailawadhi S, Parrondo R, Lin D, et al. Real-world safety and health care resource utilization of teclistamab under an outpatient model for step-up dosing administration. JCO Oncol Pract. 2024;0:OP-24-00489.
Scott SA, Marin EM, Maples KT, Joseph NS, Hofmeister CC, Gupta VA, et al. Prophylactic tocilizumab to prevent cytokine release syndrome (CRS) with teclistamab: a single-center experience. Blood Cancer J. 2023;13:191.
van de Donk NWCJ, Garfall AL, Benboubker L, Uttervall K, Groen K, Rosiñol L, et al. Longer-term follow-up of patients (pts) receiving prophylactic tocilizumab (toci) for the reduction of cytokine release syndrome (CRS) in the phase 1/2 MajesTEC-1 study of teclistamab in relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. 2024;42:7517.
Raje N, Anderson K, Einsele H, Efebera Y, Gay F, Hammond SP, et al. Monitoring, prophylaxis, and treatment of infections in patients with MM receiving bispecific antibody therapy: consensus recommendations from an expert panel. Blood Cancer J. 2023;13:116.
Rodriguez-Otero P, Usmani S, Cohen AD, van de Donk NWCJ, Leleu X, Gállego Pérez-Larraya J, et al. International Myeloma Working Group immunotherapy committee consensus guidelines and recommendations for optimal use of T-cell-engaging bispecific antibodies in multiple myeloma. Lancet Oncol. 2024;25:e205–e16.
Frerichs KA, Verkleij CPM, Mateos MV, Martin TG, Rodriguez C, Nooka A, et al. Teclistamab impairs humoral immunity in patients with heavily pretreated myeloma: importance of immunoglobulin supplementation. Blood Adv. 2023;8:194–206.
Acknowledgements
CRT is supported by the Memorial Sloan Kettering NIH/NCI Cancer Center Support Grant (P30 CA008748). RP is supported by the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre.
Author information
Authors and Affiliations
Contributions
CRT, SA, TGM, and YL conceived and designed the study. CRT, SA, LB, RP, EK, JM-L, RB, ADMSC, SC, RP, SA, DF, MAD, KY, CM, CL, MC, AJSM, HM, SZU, TGM, and YL contributed to data collection and curation. CRT, SA, CH, and TGM analyzed, verified, and interpreted the data. CRT wrote the first draft of the manuscript. CRT, SA, CH, LB, RP, EK, JM-L, RB, ADMSC, SC, RP, SA, DF, MAD, KY, CM, CL, MC, AJSM, HM, BGMD, SZU, TGM, and YL reviewed and edited the manuscript. All authors had full access to the data in the study and had final responsibility for the decision to submit for publication.
Corresponding authors
Ethics declarations
Competing interests
CRT reports research funding from Janssen and Takeda, personal fees from MJH Life Sciences, and has received honoraria for consultancy/participated in the advisory boards for Janssen and Sanofi. RK reports research funding from Pfizer and GSK, consultancy for Roche and GSK, and has received honoraria from J&J, AbbVie, BMS, Pfizer, and GSK. EK has received honoraria from Janssen/J&J, GSK, Sanofi, and Pfizer and reports institutional research funding from Janssen/J&J, GSK, and Pfizer. JM-L reports research funding from Janssen, Pfizer, Incity, consultancy for Amgen, Janssen, BMS, Pfizer, Kite, Roche, and has received honoraria from Amgen, Janssen, BMS, Pfizer, Kite, Roche. RB reports travel support from Kite Pharma. SB reports honoraria from Sanofi, Sobi, Ascentage Pharma and institutional research funding from AbbVie, C4 therapeutics, Carsgen, Takeda, and Johnson & Johnson. RP reports research funding from GSK and Bristol-Myers Squibb foundation, and consultancy for Astra Zeneca and Sanofi Aventis. SA reports institutional research funding from GSK, BMS, Pharmacyclics, Amgen, Sanofi, Janssen, Cellectar, Xencor, AbbVie, and Ascentage, and consultancy for GSK, Sanofi, BMS, Takeda, Beigene, Regeneron, Pharmacyclics, Amgen, and Janssen. DF has received honoraria from Janssen and Sanofi. MAD has received honoraria from Amgen, Takeda, Janssen-Cilag, Bristol-Myers Squibb, Beigene, and Sanofi, and reports consulting or advisory roles for Amgen, Janssen-Cilag, Takeda, Bristol-Myers Squibb, Beigene, Sanofi. HM reports research funding from Janssen and Pfizer and has received honoraria/participated in advisory boards for Janssen/J&J, GSK, Sanofi, Pfizer, Takeda, and AbbVie. SZU received research funding from Amgen, AbbVie, Array Biopharma, BMS, Celgene, Gilead, GSK, Janssen, Merck, Pharmacyclics, Sanofi, Seattle Genetics, SkylineDX, and Takeda, and is a consultant to AbbVie, Amgen, BMS, Celgene, EdoPharma, Genentech, Gilead, GSK, Gracell, Janssen, Oncopeptides, Pfizer, Sanofi, Seattle Genetics, SecuraBio, SkylineDX, Takeda, TeneoBio. TGM reports research funding from Sanofi, Janssen, Amgen, and BMS and is a consultant for GSK, Roche, and Pfizer. YL reports institutional research funding from Janssen and BMS, has participated in advisory boards for Janssen, Sanofi, BMS, Regeneron, Genentech, Tessera, and Legend, has participated in Steering Committees for Janssen and Kite/Gilead, and has participated in scientific advisory boards for NexImmune, Caribou, and has participated in a Data Safety Monitoring Board for Pfizer.
Consent to participate
Individual informed consent was obtained per each participating center’s institutional requirements and guidelines.
Ethical approval
This study received approval from the Institutional Review Boards of the respective participating institutions, including Memorial Sloan Kettering Cancer Center IRB (#24-044). The research was performed in compliance with the terms of the Declaration of Helsinki, and all methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tan, C.R., Asoori, S., Huang, CY. et al. Real-world evaluation of teclistamab for the treatment of relapsed/refractory multiple myeloma (RRMM): an International Myeloma Working Group Study. Blood Cancer J. 15, 53 (2025). https://doi.org/10.1038/s41408-025-01259-z
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-025-01259-z
This article is cited by
-
Outcomes of relapse after teclistamab therapy in multiple myeloma
Blood Cancer Journal (2025)
-
Outcomes of elranatamab in relapsed/refractory multiple myeloma: prognostic impact of monocyte count, MyCARe, and R2-ISS
International Journal of Hematology (2025)
-
De-escalated Teclistamab dosing in relapsed/refractory multiple myeloma: Czech myeloma group real-world evidence analysis
Annals of Hematology (2025)
-
Results of delayed or salvage autologous hematopoietic stem cell transplantation for multiple myeloma
Bone Marrow Transplantation (2025)
