
Abstract
Acute myeloid leukemia (AML) and DNMT3A mutations (DNMT3Amut) are considered to carry intermediate risk under the 2022 European LeukemiaNet (ELN-2022) classification in the absence of other co-mutations or cytogenetic abnormalities. However, this group is highly heterogeneous. In this study, the genomic and transcriptomic features influencing outcomes in DNMT3A-mutated AML were examined in a cohort of 884 patients with AML receiving standard chemotherapy. Stratification by NPM1 and FLT3-ITD status revealed worse survival among patients with NPM1 mutations and wild-type FLT3-ITD (NPM1mut/FLT3-ITDwt) than patients in the ELN-2022 favorable risk group. The other three subgroups (NPM1mut/FLT3-ITDmut, NPM1wt/FLT3-ITDmut, and NPM1wt/FLT3-ITDwt) exhibited worse prognoses than patients in the ELN-2022 intermediate risk group. Additionally, the presence of TET2mut in patients with AML and DNMT3Amut/NPM1mut/FLT3-ITDwt led to reclassification from favorable risk to intermediate risk in the ELN-2022. RNA-sequencing analysis revealed a distinct transcriptomic profile in patients with TET2mut, highlighting the enrichment of leukemic stem cell signatures and dendritic cell migration, with MMP14, CD200, and CT45A5 identified as key differentially expressed genes. In conclusion, co-mutation patterns strongly affected AML outcomes in patients with DNMT3Amut. Patients with TET2mut constituted a unique subgroup within the ELN-2022 favorable DNMT3Amut/NPM1mut/FLT3-ITDwt group, characterized by distinct transcriptomic features and an unfavorable prognosis.
Similar content being viewed by others

Introduction
DNMT3A encodes the DNA methyltransferase 3 alpha enzyme, which adds methyl groups to cytosine residues in CpG dinucleotides [1]. This process is essential for gene expression regulation and the maintenance of genomic stability. Mutations in DNMT3A (DNMT3Amut) have been identified in approximately 14–22% of patients with acute myeloid leukemia (AML) [1,2,3,4,5], with a higher prevalence of 29–34% observed in those with normal karyotype AML [6, 7]. Numerous studies have demonstrated a correlation between DNMT3Amut and poorer survival outcomes [1, 2, 8,9,10,11]. However, the prognostic implications of DNMT3Amut remain uncertain, because these mutations frequently exist alongside other genetic alterations, hindering identification of their independent effects on clinical outcomes. Notably, DNMT3Amut is not listed as a major prognostic factor in the 2022 European Leukemia Net (ELN-2022) classification [12].
Most DNMT3Amut appear alongside other molecular abnormalities, such as FLT3-ITD, NPM1, and IDH1, underscoring the clinical heterogeneity among individuals with these mutations. NPM1 mutations (NPM1mut) have exhibited a strong association with favorable outcomes, whereas FLT3-ITD mutations (FLT3-ITDmut) have been linked to poorer outcomes [13, 14]. However, the prognostic implications of other concurrent mutations in patients with AML and DNMT3Amut require further clarification. Moreover, the transcriptomic features that affect survival in patients with DNMT3Amut remain poorly understood, indicating a need for additional research. Dysregulated pathways that function independently of specific mutational backgrounds should be elucidated to uncover new opportunities for therapeutic intervention. In this study, we performed a detailed subgroup analysis of clinical heterogeneity in patients with AML and DNMT3Amut based on comprehensive genomic and transcriptomic data. Our findings were also thoroughly validated in several external cohorts.
Methods and materials
Sample
A total of 884 newly diagnosed patients with de novo non-M3 AML who received standard chemotherapy at National Taiwan University Hospital (NTUH) were enrolled in this study. AML diagnosis and classification were performed according to the International Consensus Classification [15] and the 5th World Health Organization Classification [16]. Detailed patient characteristics and treatment regimens are provided in the supplementary data [17].
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. Live vertebrates were not used in this project. The study was approved by the NTUH institutional review board, and written informed consent was obtained from all participants in accordance with the Declaration of Helsinki (approval number 201709072RINC and 202109078RINB).
Cytogenetics
Chromosomal analyses were performed according to a procedure described in a previous study [18]. Karyotypes were classified on the basis of risk groups defined by the Medical Research Council [19].
Mutation analysis
We performed targeted next generation sequencing (NGS) with a TruSight myeloid sequencing panel (Illumina, San Diego, CA, USA) to analyze mutations in 15 full exon genes and 39 oncogenic hotspot genes. A HiSeq platform (Illumina, San Diego, CA, USA) with a median reading depth of 12,000× was used for sequencing. Due to the suboptimal sequencing sensitivity of this method, CEBPA mutations were confirmed through Sanger sequencing. FLT3-ITD was analyzed through polymerase chain reaction (PCR) followed by fluorescence capillary electrophoresis. KMT2A-PTD was analyzed through PCR followed by Sanger sequencing [20].
RNA sequencing analysis
Bone marrow (BM) mononuclear cells were obtained from 264 patients with AML, including 80 with DNMT3Amut and 184 with wild-type DNMT3A (DNMT3Awt) and normal karyotypes. The BM samples were then analyzed through RNA sequencing (RNA-seq) per the method of a previous study [21]. RNA was processed into sequencing libraries with the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina NovaSeq 6000 sequencing system (150 bp paired-end). Reads were trimmed with Cutadapt (v2.3), aligned to GRCh38.p12 using STAR (v2.6.1a) [22], and quantified with Gencode annotations (v28) [23]. Read counts were normalized across all samples according to the trimmed mean of M-values with the calcNormFactors function of the edgeR package in R software [24]. Gene expression was calculated in terms of log2(CPM + 1) (counts per million reads) for further analysis. We used the limma package in R to identify differentially expressed genes (DEGs) and determined log2 fold changes (logFC) and false discovery rate (FDR)-adjusted P values using the Benjamini–Hochberg method.
We conducted analyses of gene ontology to investigate the enrichments of expression profiles associated with DNMT3Amut in gene sets curated in the Molecular Signatures Database (MSigDB) [25]. The statistical significance of the degree of enrichment was assessed through a random permutation test with 1000 repetitions [26].
External datasets
We downloaded AML RNA-seq data from the Cancer Genome Atlas (TCGA) generated with the Illumina HiSeq 2000 platform (n = 173) and Beat-AML RNA-seq data for validation [27, 28]. The prognostic relevance of concurrent mutations was further validated on the basis of patient data with molecular annotations from the German–Austrian Acute Myeloid Leukemia Study Group (AMLSG, n = 1540) and the UK National Cancer Research Institute (UK-NCRI) trials (n = 2113) [4, 29].
Statistical analysis
Chi-square tests were performed for comparisons of discrete variables. If the expected contingency table values were below 5, Fisher’s exact test was performed. Mann–Whitney U tests were performed to compare continuous variables and the medians of distributions. Overall survival (OS) was defined as the time from initial diagnosis to the date of last follow-up or death from any cause. Event-free survival (EFS) was defined as the time from diagnosis to the first instance of the following: treatment failure, relapse after first complete remission (CR1), or death from any cause. Disease-free survival (DFS) was defined as the time from diagnosis to either relapse or death from any cause [30]. A P value < 0.05 indicated statistical significance. The effect of allogeneic hematopoietic stem cell transplantation (allo-HSCT) at CR1 on outcomes in patients with DNMT3Amut was assessed according to the Mantel–Byar approach, and results were visualized in Simon–Makuch plots [31]. Statistical analyses were performed with SPSS 23 (SPSS, Chicago, IL, USA) and R software 4.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Patient characteristics and clinical outcomes
DNMT3Amut was identified in 16.2% of the 884 patients in our cohort (143) (Supplementary Table 1). The median age of patients with DNMT3Amut was 53 years (range: 17–82 years), significantly higher than that of patients with DNMT3Awt. Patients with DNMT3Amut also had significantly higher white blood cell counts, platelet counts, and hemoglobin levels at diagnosis. Cytogenetic analysis revealed a significantly higher proportion of patients with DNMT3Amut had intermediate risk profiles than those with DNMT3Awt (93.0% vs. 72.1%, respectively, P < 0.001). Patients with DNMT3Amut had a higher incidence of NPM1mut but a lower incidence of AML with core-binding factor positivity, CEBPA mutations, and KMT2A rearrangements (Table 1).
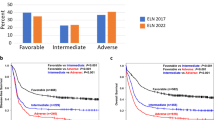
CR1 rates were comparable between patients with DNMT3Amut and DNMT3Awt (74.1 vs. 75.8%, respectively, P = 0.662). Among patients who did not receive allo-HSCT at CR1, relapse rates remained comparable between both groups (40.6% vs. 32.0%, P = 0.102). Patients with DNMT3Amut exhibited similar OS and EFS as those in the ELN-2022 intermediate risk group (Fig. 1A). Patients in this cohort with DNMT3Awt had significantly better OS and EFS than those with DNMT3Amut after a median follow-up time of 7.3 years (median OS 30.1 vs. 21.3 months, respectively, P = 0.005; median EFS 12.6 vs. 7.4 months, P = 0.009) (Fig. 1B).

A Comparison of Kaplan–Meier survival curves for OS and EFS among patients with AML stratified according to ELN-2022 risk groups (favorable, intermediate, adverse) and DNMT3A mutation status. OS and EFS for the DNMT3Amut group closely aligned with those of the ELN-2022 intermediate risk group. B DNMT3Amut were associated with significantly worse OS and EFS than DNMT3Awt.
Frequency and prognostic relevance of concurrent mutations in DNMT3A mut
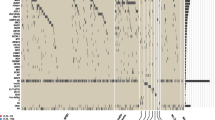
At least one concurrent mutation was identified in most patients with DNMT3Amut (97.2%) (Fig. 2A). The most frequently mutated genes were NPM1 (52.8%), FLT3-ITD (30.6%), IDH2 (26.4%), FLT3-TKD (15.3%), IDH1 (10.4%), and PTPN11 (9.7%) (Fig. 2B, Supplementary Table 2). Given the well-established prognostic significance of NPM1mut and FLT3-ITDmut, we stratified patients with DNMT3Amut into four subgroups by whether these two genetic alterations were present. To further strengthen risk stratification, we also assessed the prognostic effects of concurrent mutations within each subgroup. Patients with DNMT3Amut and different NPM1 and FLT3-ITD mutation statuses differed significantly in OS (P = 0.004) and EFS (P = 0.009) (Fig. 2C). Outcomes were significantly better among DNMT3Amut patients with NPM1mut and wild-type FLT3-ITD (FLT3-ITDwt) than in patients with NPM1mut/FLT3-ITDmut, NPM1wt/FLT3-ITDmut, or NPM1wt/FLT3-ITDwt, in which prognoses were similarly poor. Furthermore, outcomes were significantly worse in the NPM1mut/FLT3-ITDwt subgroup than in the ELN-2022 favorable risk group (median OS, 97.8 months vs. not reached, P = 0.047; median EFS, 18.0 vs. 138.1 months, P = 0.036). The other three subgroups all exhibited worse outcomes than the ELN-2022 intermediate risk group (Supplementary Fig. 1A–D). These results indicate that outcomes among all patients with DNMT3Amut were inferior to those predicted under the current ELN-2022 risk categories.

A Oncoprint of recurrent genetic mutations in patients with DNMT3Amut and AML. Colored bars represent mutations, categorized according to functional gene classes. B Prevalence of concurrent mutations in patients with DNMT3Amut and DNMT3Awt. Mutations observed in at least 1% of patients in one subgroup are displayed. Significant differences between DNMT3Amut and DNMT3Awt groups are marked as follows: *P < 0.05, **P < 0.01, ***P < 0.001. C OS and EFS of patients with DNMT3Amut stratified according to NPM1 and FLT3-ITD status. Patients with NPM1mut/FLT3-ITDwt exhibited significantly better OS and EFS than those in other groups. D OS and EFS in the DNMT3Amut/NPM1mut/FLT3-ITDwt group, stratified according to concurrent genetic mutations: TET2, IDH2, signaling pathway mutations (PTPN11, FLT3-TKD, and NRAS), and total subgroup patients. The worst outcomes were observed in the DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2mut subgroup. E OS and EFS comparison between the DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2mut subgroup and ELN-2022 favorable risk group. Worse outcomes were observed in the DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2mut subgroup. F OS and EFS comparison between the DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2wt subgroup and ELN-2022 intermediate risk group. Better outcomes were observed in the DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2wt subgroup.
The highest frequency of co-mutations was observed in the NPM1mut/FLT3-ITDwt group, including FLT3-TKD (25.5%), IDH2 (19.1%), PTPN11 (17.0%), NRAS (14.9%), and TET2 (14.9%). Stratification according to these co-mutations revealed that TET2 mutations (TET2mut) were associated with the lowest survival rates (Fig. 2D). Notably, in our analysis of patients based on ELN-2022 risk stratification, those with TET2mut exhibited significantly worse OS and a trend towards inferior EFS than patients in the ELN-2022 favorable risk group (median OS, 22.9 months vs. not reached, P = 0.008; median EFS, 11.8 vs. 138.2 months, P = 0.054) (Fig. 2E). Patients in our cohort with TET2mut shared similar OS and EFS with the ELN-2022 intermediate risk group (Supplementary Fig. 2A). By contrast, among patients without TET2mut, OS and EFS were higher relative to patients in the ELN-2022 intermediate risk group (median OS, 98.2 vs. 24.2 months, P = 0.013; median EFS, 18.7 vs. 10.7 months, P = 0.028) (Fig. 2F) and similar relative to patients in the ELN-2022 favorable risk group (Supplementary Fig. 2B). The DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2mut group exhibited a trend towards worse OS comparing with the DNMT3Amut/NPM1mut/FLT3-ITDwt/wild-type TET2 (TET2wt) group (median, 22.9 vs. 98.2 months, respectively, P = 0.097) (Supplementary Fig. 2C). In patients with DNMT3Amut/NPM1mut/FLT3-ITDwt/PTPN11wt/IDH2wt, TET2mut were associated with significantly worse OS (TET2mut, median, 1.3 months vs. not reached, P = 0.024) and a trend toward worse EFS (TET2mut, median, 0.001 vs. 33.2 months, P = 0.090) (Supplementary Fig. 2D).
In the NPM1mut/FLT3-ITDmut group, frequent co-mutations were observed in myeloid dysplasia related genes (MDS-R, 16.6%), including ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, and ZRSR2, and in signaling pathways (13.3%). MDS-R genes were most frequently detected in the NPM1wt/FLT3-ITDmut group (9.3%), whereas the proportion of other co-mutations remained low. Notably, no significant differences in OS or EFS were observed within each group when stratified according to the type of co-mutations (Supplementary Fig. 2E, F). A high frequency of co-mutations was observed in the NPM1wt/FLT3-ITDwt group, including IDH2 (48.1%), MDS-R genes (44.4%), and NRAS (16.7%). Patients with MDS-R genes were classified within the ELN-2022 unfavorable group and had a 5-year survival rate of approximately 25–30% [23]. Even patients with DNMT3Amut/NPM1wt/FLT3-ITDwt who lacked MDS-R gene mutations were associated with poor survival outcomes that were similar to those of patients with MDS-R gene mutations, indicating the negative prognostic effects of DNMT3Amut (Supplementary Fig. 2G). Patients with IDH2 mutations were excluded for further stratification of the NPM1wt/FLT3-ITDwt/IDH2wt group according to NRAS mutation status, which revealed that patients with NRAS mutations had better OS (median, not reached vs. 12.4 months, P = 0.045) (Supplementary Fig. 2H).
Effects of Allo-HSCT at CR1 in patients with DNMT3A mut
Outcomes among patients with DNMT3Amut, irrespective of their NPM1 and FLT3-ITD mutation status, were inferior to those predicted under the current ELN-2022 risk categories. We also examined whether allo-HSCT at CR1 alleviated the poor survival of patients with DNMT3Amut. Among these patients, 64 (44.8%) underwent allo-HSCT, with 29 (20.3%) receiving allo-HSCT at CR1. Among patients who received allo-HSCT at CR1, 12 had NPM1 mutations and 10 had available NPM1 PCR data before HSCT. NPM1 was detected in 5 (50%) of these patients before the transplant (Supplementary table 3). To accurately evaluate the effects of allo-HSCT at CR1, we performed a Mantel–Byar analysis and constructed Simon–Makuch plots. Significantly better OS (median, 209.1 vs. 23.1 months, P = 0.001) and DFS (median, not reached vs. 10.2 months, P < 0.001) was observed among patients who received allo-HSCT at CR1 (Supplementary Fig. 3A). Among patients in the ELN-2022 favorable group with DNMT3Amut/NPM1mut/FLT3-ITDwt, allo-HSCT at CR1 was associated with significantly improved DFS (median, not reached vs. 93.9 months, P = 0.037) but a nonsignificant difference in OS (median, not reached vs. 241.8 months, P = 0.196) compared with post-remission chemotherapy (Supplementary Fig. 3B).
External validation of survival outcomes stratified according to co-mutations in patients with AML and DNMT3A mut
We integrated data from the AMLSG and UK-NCRI trial cohorts to analyze the prognostic effects of co-mutations (Supplementary Table 4). In the DNMT3Amut/NPM1mut/FLT3-ITDwt/PTPN11wt/IDH2wt subgroup, TET2mut were associated with significantly worse OS (TET2mut, median, 2.9 years vs. not reached, P = 0.020) (Supplementary Fig. 4), consistent with findings from our cohort.
Characteristics and clinical outcomes of patients with DNMT3A mut in relapse
Among patients with DNMT3Amut, 63 (44.1%) experienced a relapse, and 34 (54.0%) of all relapsed patients achieved a second complete remission (CR2) after salvage reinduction chemotherapy (Supplementary Table 5). FLT3-ITD and WT1 mutations were associated with a significantly lower likelihood of CR2 (FLT3-ITD, 38% vs. 72%, P = 0.045; WT1, 0% vs. 100%, P = 0.012). The median OS from the time of relapse was 10.5 months. Subsequent allo-HSCT was administered to 30 patients, including 12 at CR2. Patients who received allo-HSCT after relapse had significantly better OS than those without allo-HSCT treatment (median, 14.0 vs. 5.6 months, P = 0.038) (Supplementary Fig. 5).
Comparison of transcriptional signatures between patients with DNMT3A mut and DNMT3A wt
Gene expression profiles were compared between 80 patients with DNMT3Amut and 184 patients with DNMT3Awt and normal karyotypes. Hierarchical clustering was performed for the top DEGs to increase the clustering resolution (Fig. 3A). A total of 818 upregulated and 454 downregulated DEGs were identified between patients with DNMT3Amut and DNMT3Awt on the basis of a cutoff of |logFC|> 0.585 (absolute fold change >1.5 or <0.67) and P < 0.05. Several Hox family genes were among the top upregulated DEGs (Fig. 3B). Using the MsigDB Hallmark gene sets, we observed that the DNMT3Amut transcriptome was enriched for TNFα signaling (normalized enrichment score ([NES] 3.89, FDR 3.85E−10), complement system (NES 3.34, FDR 3.85E−10), inflammatory response (NES 3.27, FDR 3.85E−10), and IL6/JAK/STAT3 signaling (NES 2.74, FDR 3.85E−10). The chemical and genetic perturbations (CGP) gene sets revealed the positive enrichment of several AML-related gene clusters, including the VERHAAK NPM1-mutated AML signature (NES 4.53, FDR 1.44E−9) and AML with aberrant cytoplasmic localization of NPM1 (NES 3.80, FDR 1.44E−9) (Fig. 3C–E, Supplementary Table 6). In a subgroup analysis of 63 patients with DNMT3Amut and normal karyotypes, the majority of originally identified DEGs remained significant (1215 of 1,272, 95.5%) (Fig. 3F).

A Unsupervised hierarchical clustering of patients with DNMT3Amut and DNMT3Awt patients, with the major upregulated and downregulated DEGs displayed. Colors represent normalized gene expression values. B Volcano plot comparison of all RNA-seq genes between patients with DNMT3Amut and DNMT3Awt. Positive (logFC > 0) and negative (logFC < 0) fold changes indicate upregulated and downregulated genes, respectively, in patients with DNMT3Amut. C, D Bubble plots illustrating a comparison of enriched and depleted MSigDB CGP and Hallmark gene sets between patients with DNMT3Amut and DNMT3Awt. Bubble size represents gene counts, and colors indicate statistical significance. E Gene Set Enrichment Analysis plots displaying overrepresentation of immune gene set signatures within the DNMT3Amut transcriptome. F Venn diagram of DEGs identified in initial and updated comparisons between patients with DNMT3Amut and DNMT3Awt. An initial analysis included RNA-seq data from 80 patients with DNMT3Amut and 184 with DNMT3Awt and normal karyotypes. In an updated analysis, a subset of 63 DNMT3Amut patients with normal karyotypes were compared with the original 184 patients with DNMT3Awt.
To validate our RNA-seq data, we compared the transcriptomes of 40 patients with DNMT3Amut and 59 patients with DNMT3Awt and normal karyotypes in the TCGA cohort. Hallmark gene sets for TNFα signaling, the complement system, inflammatory response, and shared AML-related gene clusters in the CGP gene sets were among the primary enriched pathways. Further validation within the Beat-AML cohort revealed comparable results. These findings confirm the alignment between our RNA-seq data and that previously reported (Supplementary Figs. 6 and 7) and underscore the unique immune microenvironment in these patients.
Distinct transcriptomic pathways in patients with DNMT3A mut/NPM1 mut/FLT3-ITDwt/TET2 mut
We observed a correlation between TET2mut and unfavorable outcomes in patients with DNMT3Amut/NPM1mut/FLT3-ITDwt and further investigated whether these patients exhibited overlapping transcriptomic profiles. We compared the RNA-seq data between patients with TET2mut (n = 4) and those with TET2wt (n = 25) within the DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup. The TET2mut group was identified as a distinct cluster through hierarchical clustering (Fig. 4A). Additionally, the principal component analysis plot effectively distinguished patients with TET2mut from those with TET2wt. By contrast, patients with FLT3-ITD, NRAS, and PTPN11 mutations could not be clustered separately from those without these mutations (Supplementary Fig. 8A).

A Unsupervised hierarchical clustering of TET2mut and TET2wt patients within the DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup, displaying the top upregulated and downregulated DEGs. Colors represented normalized gene expression values. B Number of upregulated and downregulated DEGs identified through comparison of transcriptomes in patients with AML with and without TET2, FLT3-TKD, IDH2, NRAS or any other co-mutations. Bubble plots illustrating enriched and depleted MSigDB Hallmark (C) and CGP and Gene Ontology Biological Processes gene sets (D) between patients with TET2mut and TET2wt in the DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup. Bubble size represents gene counts, and color indicates statistical significance. E Gene Set Enrichment Analysis plots indicating enrichment of LSC and immune gene set signatures within the TET2mut transcriptome. F, G Volcano plot of DEGs with genes downregulated (logFC < 0) and upregulated (logFC > 0) in patients with DNMT3Amut/NPM1mut/FLT3-ITDwt based on TET2 mutation status. Key DEGs are highlighted in a heat map, with colors representing normalized gene expression values.
A total of 3801 DEGs were identified between the TET2mut and TET2wt groups (Fig. 4B and Supplementary Table 7). The number of DEGs between the TET2mut and TET2wt groups was notably higher than that between other mutated subgroups. Leukemic stem cell (LSC) signatures were enriched in the TET2mut group (NES 2.42, FDR 1.47E-4), and genes downregulated in LSC were negatively enriched. An analysis of the MsigDB Hallmark and Gene Ontology Biological Process gene sets revealed significant negative enrichment of IL6/JAK/STAT3 signaling (NES −1.60, FDR 8.42E−3), pathways associated with dendritic cell migration (NES − 2.20, FDR 8.80E-5), and lymphocyte chemotaxis (NES −1.95, FDR 2.08E−3) (Fig. 4C–E and Supplementary Fig. 8B). The top DEGs included MMP14 (logFC = 1.89), CD200 (logFC = 1.39), CT45A5 (logFC = –2.60), and CT45A8 (logFC = –1.71) (Fig. 4F–G). MMP14, derived from mesenchymal stem cells, promotes AML progression and chemoresistance [32]. CD200 is a novel LSC marker that can capture the entire LSC compartment from samples provided by patients with AML, including those with NPM1 mutations [33]. The CT45 gene family consists of 10 distinct but highly similar genes that are nearly identical at the protein level (CT45A1–CT45A10). CT45 peptides can induce CD8+ T cell activation through intracellular IFNγ staining [34].
To validate these results and strengthen the robustness of our data, we analyzed RNA-seq data from 40 patients with DNMT3Amut in the TCGA AML cohort, 6 of whom had TET2mut. Selected AML gene clusters, LSC signatures, and pathways related to dendritic cell migration were significantly enriched in patients with TET2mut (Supplementary Fig. 9), in alignment with our findings. Additionally, MMP14 and CD200 were identified as DEGs in the TCGA cohort.
Discussion
Our findings underscore the complexity of AML in patients with DNMT3Amut and the prognostic implications of its associated molecular landscape. Patients with DNMT3Amut were stratified into four subgroups on the basis of their NPM1 and FLT3-ITD mutation status. All subgroups exhibited a poorer prognosis than that predicted under each designated ELN-2022 risk category. Additionally, within the DNMT3Amut/NPM1mut/FLT3-ITDwt group, TET2 mutations may have characterized a unique subgroup with distinct transcriptomic features and an unfavorable prognosis.
In previous studies, patients with DNMT3Amut have exhibited inferior OS and EFS [1, 2, 35]. In our study, patients with DNMT3Amut were stratified on the basis of their NPM1 and FLT3-ITD mutation status, and the results indicated the profound prognostic implications of these co-mutations. The DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup trended toward worse outcomes than the ELN-2022 favorable risk group. These findings suggest that despite the traditional classification of the NPM1mut/FLT3-ITDwt subgroup as favorable, patients in this group with DNMT3Amut may fall under a distinct intermediate risk category. The remaining three subgroups, NPM1mut/FLT3-ITDmut, NPM1wt/FLT3-ITDmut, and NPM1wt/FLT3-ITDwt, all exhibited poorer prognoses than the ELN-2022 intermediate risk group, aligning more closely with the ELN-2022 unfavorable risk criteria. These results highlight the heterogeneity within the DNMT3Amut population and the need for more precise risk stratification. Our findings further underscore the critical role of allo-HSCT at CR1 in improving survival outcomes for patients with AML and DNMT3Amut. Among all patients with AML and DNMT3Amut, significantly better OS and DFS was observed in those who received allo-HSCT at CR1 than in those who received post-remission chemotherapy. Within the DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup, allo-HSCT at CR1 improved DFS, whereas OS remained comparable with other groups. Although we conducted Mantel–Byar analysis and constructed Simon–Makuch plots to address immortal time bias, further randomized studies are necessary to clarify the role of allo-HSCT in this patient population.
To better understand why worse outcomes were observed in the DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup than in ELN-2022 favorable risk patients, we investigated the effects of co-mutations within this subgroup. Combined DNMT3Amut and TET2mut in patients with AML and NPM1mut/FLT3-ITDwt resulted in a risk reclassification from ELN-favorable to intermediate, whereas patients without TET2mut had survival more similar to patients in the ELN favorable risk group. Our study presents the novel finding that TET2mut may adversely affect prognosis in patients with DNMT3Amut/NPM1mut/FLT3-ITDwt. We refined the subgroup to exclude PTPN11 and IDH2 mutations, which further underscored the deleterious effects of TET2mut on survival and disease progression. This finding was sufficiently validated in external cohorts. Among all patients with AML, TET2mut have been associated with adverse prognosis [36,37,38,39]. In a recent study, two epigenetic modifiers, TET2 and DNMT3A, synergistically induced leukemogenesis [40]. In a DNMT3A and TET2 double-knockout mouse model, dysregulated self-renewal of hematopoietic stem cells may have caused hematological abnormalities. Both DNMT3A and TET2 play key roles in repressing lineage-specific transcription factors in hematopoietic stem cells. The concurrent loss of both genes causes notable upregulation of downstream transcription factors, which eventually leads to malignant transformation [40]. In our study, patients with AML and TET2mut formed a separated transcriptomic cluster within the DNMT3Amut/NPM1mut/FLT3-ITDwt subgroup and exhibited the largest number of DEGs compared with patients with TET2wt. Enrichment of LSC signatures may partly explain the poor outcomes observed in this group, and these results were validated in the TCGA cohort. Based on the distinct transcriptomic signatures, prognostic implications, reproducibility across cohorts, and functional evidence, DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2mut AML can be redefined as a distinct subgroup to improve ELN classification systems and risk stratification.
Our study has several limitations. First, we relied on bulk BM aspirates for RNA-seq analysis. Further research incorporating single-cell RNA-seq or mass cytometry can elucidate the primary contributor among cell components. Second, although we validated the poor outcomes in patients with DNMT3Amut/NPM1mut/FLT3-ITDwt/TET2mut AML with external cohort data, this finding should be interpreted with caution due to the small size of the TET2mut subgroup. Further studies remain necessary to ensure the reproducibility of our results. Third, all patients in our study received intensive chemotherapy, and the interaction of specific mutation groups with venetoclax and hypomethylating agents was not examined [41, 42]. Furthermore, most patients with FLT3-ITD did not receive FLT3 inhibitor treatment before and after HSCT, potentially affecting their prognosis.
Our findings underscore the necessity of integrating genetic and transcriptomic analyses to comprehensively understand the heterogeneity of DNMT3A-mutated AML. Irrespective of NPM1 and FLT3-ITD mutation status, poorer prognoses were observed among patients with AML and DNMT3Amut than anticipated under the assigned ELN-2022 risk category. Patients with TET2mut comprised a unique subgroup within the ELN-2022 favorable DNMT3Amut/NPM1mut/FLT3-ITDwt group, characterized by distinct transcriptomic features and an unfavorable prognosis. This study provides a robust and comprehensive dataset of patient demographic characteristics, clinical outcomes, NGS, and RNA-seq information, offering valuable resources for future AML research.
Data availability
Our RNA-seq data can be accessed online from the National Center for Biotechnology Information’s Gene Expression Omnibus under GSE297413. Clinical and mutation data and computer codes with nonidentifiable patient information are accessible on reasonable request. The corresponding author (F.-M.T.) will evaluate the appropriateness of such requests.
References
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A Mutations in Acute Myeloid Leukemia. N Engl J Med. 2010;363:2424–33.
Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119:559–68.
Gaidzik VI, Schlenk RF, Paschka P, Stölzle A, Späth D, Kuendgen A, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG). Blood. 2013;121:4769–77.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374:2209–21.
Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, et al. Incidence and Prognostic Influence of DNMT3A Mutations in Acute Myeloid Leukemia. J Clin Oncol. 2011;29:2889–96.
Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, et al. Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia. 2012;26:1247–54.
Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrózek K, et al. Age-Related Prognostic Impact of Different Types of DNMT3A Mutations in Adults With Primary Cytogenetically Normal Acute Myeloid Leukemia. J Clin Oncol. 2012;30:742–50.
Ribeiro AFT, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119:5824–31.
Bezerra MF, Lima AS, Piqué-Borràs MR, Silveira DR, Coelho-Silva JL, Pereira-Martins DA, et al. Co-occurrence of DNMT3A, NPM1, FLT3 mutations identifies a subset of acute myeloid leukemia with adverse prognosis. Blood. 2020;135:870–5.
Loghavi S, Zuo Z, Ravandi F, Kantarjian HM, Bueso-Ramos C, Zhang L, et al. Clinical features of De Novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J Hematol OncolJ Hematol Oncol. 2014;7:74.
Elrhman HAEA, El-Meligui YM, Elalawi SM. Prognostic Impact of Concurrent DNMT3A, FLT3 and NPM1 Gene Mutations in Acute Myeloid Leukemia Patients. Clin Lymphoma Myeloma Leuk. 2021;21:e960–9.
Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–77.
Wakita S, Marumo A, Morita K, Kako S, Toya T, Najima Y, et al. Mutational analysis of DNMT3A improves the prognostic stratification of patients with acute myeloid leukemia. Cancer Sci. 2023;114:1297–308.
Ostronoff F, Othus M, Ho PA, Kutny M, Geraghty DE, Petersdorf SH, et al. Mutations in the DNMT3A exon 23 independently predict poor outcome in older patients with acute myeloid leukemia: a SWOG report. Leukemia. 2013;27:238–41.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–19.
Lo M, Tsai XC, Lin C, Tien F, Kuo Y, Lee W, et al. Validation of the prognostic significance of the 2022 European LeukemiaNet risk stratification system in intensive chemotherapy treated aged 18 to 65 years patients with de novo acute myeloid leukemia. Am J Hematol. 2023;98:760–9.
Hou HA, Tsai CH, Lin CC, Chou WC, Kuo YY, Liu CY, et al. Incorporation of mutations in five genes in the revised International Prognostic Scoring System can improve risk stratification in the patients with myelodysplastic syndrome. Blood Cancer J. 2018;8:39.
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The Importance of Diagnostic Cytogenetics on Outcome in AML: Analysis of 1,612 Patients Entered Into the MRC AML 10 Trial. Blood. 1998;92:2322–33.
Lee W, Lin C, Tsai C, Tseng M, Kuo Y, Liu M, et al. Effect of mutation allele frequency on the risk stratification of myelodysplastic syndrome patients. Am J Hematol. 2022;97:1589–98.
Tien FM, Yao CY, Tsai XCH, Lo MY, Chen CY, Lee WH, et al. Dysregulated immune and metabolic pathways are associated with poor survival in adult acute myeloid leukemia with CEBPA bZIP in-frame mutations. Blood Cancer J. 2024;14:15.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–73.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–50.
Tien FM, Hou HA, Tsai CH, Tang JL, Chiu YC, Chen CY, et al. GATA2 zinc finger 1 mutations are associated with distinct clinico-biological features and outcomes different from GATA2 zinc finger 2 mutations in adult acute myeloid leukemia. Blood Cancer J. 2018;8:87.
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–31.
The Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013;368:2059–74.
Tazi Y, Arango-Ossa JE, Zhou Y, Bernard E, Thomas I, Gilkes A, et al. Unified classification and risk-stratification in Acute Myeloid Leukemia. Nat Commun. 2022;13:4622.
Hou HA, Lin CC, Chou WC, Liu CY, Chen CY, Tang JL, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014;28:50–8.
Simon R, Makuch RW. A non-parametric graphical representation of the relationship between survival and the occurrence of an event: application to responder versus non-responder bias. Stat Med. 1984;3:35–44.
Wu J, Liu X, Li X, Wang Q, Zhang N, Zhou F. Mesenchymal Stromal Cell-Derived MMP14 Facilitates Leukemia Progression and Chemotherapy Resistance. Blood. 2023;142:5993.
Ho JM, Dobson SM, Voisin V, McLeod J, Kennedy JA, Mitchell A, et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020;4:5402–13.
Coscia F, Lengyel E, Duraiswamy J, Ashcroft B, Bassani-Sternberg M, Wierer M, et al. Multi-level Proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy Target in Ovarian Cancer. Cell. 2018;175:159–170.e16.
Park DJ, Kwon A, Cho BS, Kim HJ, Hwang KA, Kim M, et al. Characteristics of DNMT3A mutations in acute myeloid leukemia. BLOOD Res. 2020;55:17–26.
Delhommeau F, Valle VD, Massé A, Couedic JPL, Lécluse Y, Marzac C, et al. Mutation in TET2 in Myeloid Cancers. N Engl J Med. 2009;360:2289–301.
Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–10.
Pan X, Chang Y, Ruan G, Zhou S, Jiang H, Jiang Q, et al. TET2 mutations contribute to adverse prognosis in acute myeloid leukemia (AML): results from a comprehensive analysis of 502 AML cases and the Beat AML public database. Clin Exp Med. 2024;24:35.
Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, et al. TET2 Mutations Improve the New European LeukemiaNet Risk Classification of Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J Clin Oncol. 2011;29:1373–81.
Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48:1014–23.
Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–52.
Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106–7.
Acknowledgements
We would like to express our thanks to the DNA Sequencing Core of the First Core Laboratory, National Taiwan University College of Medicine for their assistance with the experiments. This manuscript was edited by Wallace Academic Editing.
Funding
This work was partially sponsored by grants from the Ministry of Science and Technology (Taiwan) (MOST 104–2314-B-002–128-MY4, 106-2314-B-002-226-MY3, 108-2628-B-002-015, 109-2314-B-002-213, 111-2314-B-002-279 12-2314-B-002-197, and 113-2314-B-002-103), the Ministry of Health and Welfare (Taiwan) (MOHW 107-TDU-B-211-114009 and 111-TDU-B-221-114001).
Author information
Authors and Affiliations
Contributions
Contribution: S-CN was responsible for study design, literature collection, data management and interpretation, statistical analysis and manuscript writing; F-MT, Y-CY and X-CT were responsible for bioinformatics analysis; Y-YK, M-HT and L-IL were responsible for mutation analysis and interpretation; M-YL, C-YC, W-HL, C-CL, Y-SW, B-SK, M-Y, J-LT and W-CC contributed patient samples and clinical data; Y-LP, M-HT and M-CL performed the gene mutation and chromosomal studies. H-AH and H-FT coordinated the study and wrote and revised the manuscript. F-MT planned, designed, conceived and coordinated the study, analyzes the statistics and wrote and revised the manuscript. All authors provided final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ni, SC., Yao, CY., Tsai, X.CH. et al. Genomic and transcriptomic determinants of clinical outcomes in patients with AML and DNMT3A mutations. Blood Cancer J. 15, 97 (2025). https://doi.org/10.1038/s41408-025-01287-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-025-01287-9
