
Abstract
For over two decades, the EBMT has updated recommendations on indications for haematopoietic cell transplantation (HCT) practice based on clinical and scientific developments in the field. This is the ninth special EBMT report on indications for HCT for haematological diseases, solid tumours and immune disorders. Our aim is to provide guidance on HCT indications according to prevailing clinical practice in EBMT countries and centres. In order to inform patient decisions, these recommendations must be considered with the risk of the disease, risk of HCT procedure and non-HCT strategies, including evolving cellular therapies, and their availability on site. HCT techniques are constantly evolving and we make no specific recommendations, but encourage harmonisation of practice, where possible, to ensure experience across indications can be meaningfully aggregated via registry outputs. We also recommend working according to JACIE certification standards to maintain quality in clinical and laboratory practice, including benchmarking of survival outcomes [1,2,3]. Since the last edition, innovative cellular and gene therapies have entered in activity across indications affecting clinical decision making. As the number and type of regulatory authority-approved cellular therapies grow, recommendations for best practice and quality of patient care were developed to support clinicians and will be regularly updated.
Similar content being viewed by others
Introduction
This is the ninth report from the EBMT covering indications for haematopoietic cell transplantation (HCT) according to prevailing clinical practice in EBMT countries and centres. For over two decades, EBMT has considered changes in HCT practice alongside developments in non-transplant treatments. As in previous editions, these 2025 recommendations are based upon clinical trials, registry data, and the opinion of EBMT experts from the board, scientific council and relevant working parties, but not upon a formal extensive or systematic review of the literature. They aim to provide general guidance on transplant indications to inform individual patient decisions by the multidisciplinary team.
They must be considered in conjunction with the risk of the disease status, the likelihood of the successful outcome of HCT, assessment of patient co-morbidities and estimation of non-relapse mortality (NRM) risk alongside the results of non-transplant strategies and their availability on site. Besides potential survival benefits, assessment must include quality of life, patient-reported outcomes and late effects. The recommendations are not intended to be used to choose a particular transplant protocol, conditioning regimen or stem cell source, but we encourage harmonisation of practice, where possible, to ensure meaningfully aggregated experience across indications via registry outputs.
Over 30 years numbers of HCT continue to rise [4], alongside a marked growth in chimeric antigen receptor (CAR)-T cell therapies in recent years. Despite a ‘dip’ related to the COVID pandemic, current registry numbers show a complete recovery from the pandemic dip with increased cellular therapy [5, 6].
Transplant categorisation, definitions and factors
Haematopoietic cell transplant (HCT)
HCT refers to any procedure where haematopoietic stem cells (HSC) of any donor type and source are given to a recipient with the intention of repopulating and replacing the haematopoietic system in total or in part. HSC for HCT can be derived from bone marrow (BM), peripheral blood (PB) or cord blood (CB) [7].
Donor categories and stem cell sources
Donor type is categorised as autologous, syngeneic and allogeneic, the latter being either related or unrelated. Beyond HLA-matched related (i.e. sibling) donors (MSD), a well-matched unrelated donor (MUD) is defined as a 10/10 identical unrelated donor (UD) based on HLA high-resolution typing for class I (HLA-A, -B, -C) and II (HLA-DRB1, -DQB1). A mismatched unrelated donor (MMUD) refers to an unrelated donor mismatched in at least one antigen or allele at HLA-A, -B, -C, -DR or -DQ and a haploidentical donor refers to one haplotype mismatched family donor. Criteria for UD selection have been proposed [8, 9], but are beyond the scope of these recommendations and incorporation into clinical practice will depend on the effort from donor registries and transplant centres balanced against strategies to incorporate mismatched alternative donors (MMAD), including haploidentical donors, into practice.
In this current version, we continue to combine the recommendations for MMAD, incorporating CB, haploidentical and MMUD into a single category separated from well-matched related and UD. Beyond this general approach, the relative value of the various modalities is described and addressed in more detail below in sections for the relevant indications.
Since the last update, innovative cellular therapies and gene therapies have entered in activity across indications affecting clinical decision making. Increasingly numbers of advanced therapy medicinal products (ATMPs) are available, including CAR-T for haematological cancers [10] and autoimmune diseases (ADs) [11, 12]: gene therapies are available for rare genetic disorders and significant progress has been made in gene therapy for the hemoglobinopathies [13]. The focus is continually shifting from developing these therapies and proving they can work to refining their effectiveness, reducing side effects and expanding their indications [14, 15]. While this guidance does not primarily intend to cover cellular therapies, it makes reference to CAR-T cells and gene therapies as treatment options alongside HCT where appropriate.
Categorisation of the type of indication for transplant procedures
EBMT indications are classified into four categories (Supplementary information Table), to describe the levels of evidence and recommendations for types of transplants and different indications. For certain indications, specific haploidentical categorisations are detailed in the footnote (*). Although we broadly categorise according to adult and paediatric (with age cut-offs also influenced by EBMT registry definitions), the indications should be interpreted with flexibility, particularly in the teenage and young adult age group and some ‘paediatric’ and teenage and young adult indications may occasionally extend into older adult age groups.
Transplant indications in adults
The updated 2025 classification of HCT procedures in adults is shown in Table 1.
Acute myeloid leukaemia (AML)
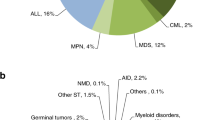
As per EBMT’s last activity survey, AML accounts for 40% of allo-HCT transplants [5]. The decision to perform allo-HCT in AML patients is guided by the risk/benefit ratio that accounts for both risk of relapse and the predicted NRM of the procedure. Genetic risk factors form the basis of modern prognostication for predicting relapse including cytogenetics [16,17,18], molecular mutations [19, 20], response to initial therapy and post-induction measurable residual disease (MRD) status [21,22,23]. Several risk scores are available for evaluating the risk of disease recurrence and mortality of allo-HCT. The risk of NRM is usually predicted by the haematopoietic cell transplantation-specific co-morbidity index (HCT-CI) which scores patients’ comorbidities [24] while disease-risk index (DRI) predicts the risk of disease relapse independently of comorbidities [25]. Other factors as donor and transplant characteristics should be considered as well and data-mining-based machine-learning algorithms that include all these variables may improve the prediction of day +100 mortality [26].
Overall, allo-HCT in first complete remission (CR1) is recommended for adverse-risk AML and the majority of intermediate-risk AML defined by the European Leukaemia Net (ELN) risk stratification based on cytogenetics and mutational profile of the disease and the Clinical Practice Guidelines in Oncology (NCCN Guidelines®) [7, 16, 27]. On the contrary, allo-HCT is not recommended for favourable-risk AML, unless there is inadequate clearance of MRD [28]. Specifically, allo-HCT in CR1 is recommended for AML patients with adverse cytogenetics [16] and patients with normal cytogenetics with either both wild-type or both mutated NPM1 and FLT3-ITD or those harbouring ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, ZRSF2 or TP53 mutations [16, 28]. However, allo-HCT is not recommended in CR1 AML patients with core-binding factor (CBF) that are associated with translocations t(8;21), inv (16) or t(16;16), NPM1 mutation plus wild-type FLT3-ITD and mutated CEPBA with normal karyotype [16]. In patients over 60 years, allo-HCT is recommended for patients with high-risk and intermediate-risk AML who are fit enough and willing to undergo intensive induction chemotherapy [16]. Allo-HCT remains a potentially curative treatment modality in fit patients with primary refractory AML (failure to achieve a morphological CR after two courses of intensive induction chemotherapy) providing a suitable donor can be identified rapidly and the predicted TRM is acceptable [28]. Allo- HCT may also be effective in patients with relapsed AML [29, 30], especially in those achieving CR2 [31]. However, patients with resistant disease should be considered for clinical trials. MSD or MUD are the preferred donor options in AML patients but the use of PTCy as GVHD prophylaxis in mismatched unrelated and haplo-identical transplants with encouraging results has widened the donor pool [32, 33] and it may change other aspects of allo-HCT, including the intensity of the conditioning as well as the risk of relapse post-transplantation [28, 34]. Although prospective data are not consistent it is generally considered that reduced intensity conditioning (RIC) regimens decrease NRM, while myeloablative conditioning (MAC) reduce the risk of relapse, with the most benefit seen in MRD-positive patients [35,36,37].
In favourable prognosis AML, low levels of CBF fusion gene transcripts may persist after treatment without impacting survival. However, failure to achieve a 3-log reduction in CBF fusion transcript after two cycles of chemotherapy is associated with a high risk of relapse and these patients may benefit from allo-HCT [38, 39]. Similarly, younger adults with NPM1-mutated AML who remain MRD-positive after two cycles of chemotherapy have higher relapse rates and seem to benefit from allo-HCT [40, 41]. Although not widely used, auto-HCT may be considered for intermediate-risk AML patients and AML-M3 patients achieving CR2 and MRD negativity [7].
Acute lymphoblastic leukaemia (ALL)
As per EBMT’s latest activity survey, ALL is the second most common indication for allo-HCT, accounting for 16% of allo-HCT transplants [5]. While allo-HCT represents the standard of care in Ph+ALL, its role in Ph-ALL has been evaluated in prospective studies considering the availability of a MSD and before the era of systematic MRD monitoring [42]. However, the results of allo-HCT from unrelated donors are constantly improving including those from non-T-cell-depleted haploidentical donors, as alternatives for transplant-eligible patients lacking a MSD [43, 44]. On the other hand, the evaluation of MRD is today considered as the most important prognostic factor in Ph-B-ALL and most international study groups build the indication for allo-HCT on routine assessment of MRD. This usually refers to adult patients up to 60 years, while there is no consensus regarding older patients [45]. MRD-based indication for allo-HCT requires standardised methodology for MRD detection in an experienced laboratory [45]. Overall, patients with MRD > 0.01% after 3 blocks of standard therapy are considered to have an indication to proceed to allo-HCT [45]. Allo-HCT should also be considered in ALL with high-risk cytogenetics at diagnosis even in MRD-negative patients, at least until additional studies are available [46,47,48]. Adverse cytogenetics in Ph-ALL include, but, are not limited to, low hypodiploidy, KMT2A (previously MLL) translocations, t(8;14), complex karyotype (≥5 chromosomal abnormalities). With respect to auto-HCT, in the donor versus no donor studies and subsequent meta-analyses, no beneficial effect was observed [49,50,51]. The question as to whether auto-HCT should be revisited for MRD-negative patients has been discussed but is not routinely considered [52].
In Ph+ ALL treated with imatinib and chemotherapy allo-HCT improves overall survival (OS) and is still indicated in CR1 [53]. However, emerging data show that indications may be restricted in patients treated with third generation TKIs in combination with novel monoclonal antibodies (mAbs) and achieving deep MRD negativity [54, 55]. Allo-HCT is indicated for ‘Ph-like’ ALL as well, due to the inadequate MRD response but also due to poor outcomes in MRD negative patients as well [56]. Finally, allo-HCT is indicated for relapsed/refractory ALL patients in CR2 or beyond [57] and may rescue a subset of patients with resistant disease responding to novel agents [58].
Regarding novel agents, the treatment paradigm for Ph relapsed/refractory B-ALL has been revolutionised in recent years with the introduction of immunotherapy: a first-in-class bispecific T-cell engager anti-CD19 monoclonal antibody (mAb) blinatumomab and the drug-conjugated anti-CD22-calicheamicin mAb inotuzumab ozogamicin. In addition, anti-CD19 CAR-T cell therapy was approved for patients up to 25 years of age based on the ELIANA study [59], and for adult ALL based on the ZUMA-3 trial [60] and FELIX trial [61]. Most trials of CAR-T cells in relapsed/refractory ALL demonstrate impressive response rates, with >70% of patients achieving CR regardless of cytogenetic background, prior therapies, or age [62]. Prognostic factors associated with higher remission rates and better outcome in adult ALL include lower disease burden (assessed by bone marrow blast count), lower LDH and higher platelet count prior to lymphodepletion [63, 64]. Importantly, conditioning with fludarabine and cyclophosphamide is superior to cyclophosphamide alone [63]. Due to the time delay between detection of relapse and infusion of CAR-T cells, bridging therapy is often necessary, although results may be inferior in patients previously treated with blinatumomab [65] and toxicity may be higher in patients with a previous allo-HCT [64]. Relapse after CAR-T cell therapy occurs in 30–50% of patients (with CD19 negative relapse in up to 40%) [62, 66]. The question of post-CAR-T cell consolidation with allo-HCT is still open and data are controversial. In any case, patients with molecular MRD positivity following CAR-T cell therapy, loss of B-cell aplasia and without a previous HCT are candidates for consolidative allo-HCT post CAR-T cell infusion [66].
It is also recommended to change therapy in patients with persistent or recurrent MRD before proceeding to allo-HCT [45]. Blinatumomab is so far the only agent evaluated in MRD Ph-B-ALL patients which leads to a significant MRD negativity (78%), survival benefit and can effectively serve as bridge to allo-HCT [67, 68]. The choice of conditioning regimen can also affect outcome of allo-HCT, and there is a clear benefit of 12 Gy TBI over chemotherapy-based regimens in terms of reducing relapse in paediatric ALL [69]. Thiotepa and busulfan-based conditioning are an alternative in case of unavailability or intolerance of myeloablative TBI [70, 71].
In contrast to B-ALL, salvage options in T-ALL are limited and consolidation with allo-HCT in CR1 should be considered in high-risk patients. Similarly to B-ALL, MRD is a key predictor of relapse [72]. Other high-risk features include early T-precursor immunophenotype [73] and adverse cytogenetics [74, 75]. Complex cytogenetics are associated with poor outcomes, while NOTCH1 and/or FBXW7 mutations are associated with improved outcomes [72, 75].
Chronic myeloid leukaemia (CML)
Tyrosine kinase inhibitors (TKI) remain the backbone of CML therapy [76, 77], indicated in first line and subsequent lines of therapy. Allo-HCT is not recommended in CML front-line treatment. However, preventing progression to advanced phase CML is the main treatment goal, as outcomes following progression remain poor regardless of therapeutic approach [78]. In this respect, allo-HCT should be indicated in CML patients whilst still in chronic phase 1-CML, according to the following recommendations.
In chronic phase-CML patients failing 1st generation-TKI or 2nd generation-TKI, treatment with ≥3rd generation-TKI is associated with a high progression free survival (PFS) and OS [79, 80]. Allo-HCT is indicated in patients treated with ≥3rd generation-TKI with BCR::ABL1 > 1% (International Scale; IS) or in those with BCR::ABL1 > 0.1% with sustained molecular increase. CML patients carrying high-risk features, such as major route additional chromosomal abnormalities [81] or the BCR::ABL kinase domain mutation T315I, must be closely monitored, and allo-HCT is indicated in case of molecular increase under ≥3rd generation-TKI. Additionally, allo-HCT is recommended in those few chronic phase-CML patients intolerant (i.e. persistent cytopenia) to all available TKI. Donor search should be started in chronic phase-CML patients failing first line TKI with high-risk features, in patients treated with at least two TKIs or after front-line 2nd generation-TKI failure.
De novo advanced phase-CML patients should start treatment with 2nd generation-TKI, or ≥3rd generation-TKI if available; and in this setting, allo-HCT should be considered in de novo advanced phase-CML patients failing front-line TKI. Progression to advanced phase-CML from chronic phase-CML should prompt allo-HCT. Donor search should be commenced as soon as signs of progression are observed, if not started before in chronic phase-CML. HLA-typing should be started in patients diagnosed with de novo or progression to blast phase. These patients can be treated with TKI± intensive chemotherapy adapted to the immunophenotype, and undergo allo-HCT in chronic phase 2, without the need to achieve cytogenetic response [82].
Retrospective data suggest that allo-HCT from MMAD in the PTCy setting is a feasible option [83, 84]; hence, a lack of MSD or MUD is not a contraindication to allo-HCT. BM and PB are both valid options as graft source in chronic phase-CML. In advanced phase CML, PB remains the preferred graft source.
Myeloproliferative neoplasms (MPNs)
Allo-HCT remains the only curative option for patients with MPNs. However, polycythaemia vera and essential thrombocythaemia are not allo-HCT indications unless disease transformation to secondary myelofibrosis (MF) or MDS/leukaemia has occurred [85,86,87]. Recently, EBMT/ELN guidelines for allo-HCT in MF have been published [88]. Here, eligible patients with primary MF and an intermediate-2 or high-risk Dynamic International Prognostic Scoring System (DIPSS) score, or a high-risk Mutation-Enhanced International Prognostic Score Systems (MIPSS70 or MIPSS70-plus) score, or MYSEC-PM high/intermediate-2 risk (for secondary MF), and a low-risk or intermediate-risk Myelofibrosis Transplant Scoring System (MTSS) score should be considered candidates for allo-HCT [88, 89]. Ideally, candidates for allo-HCT with splenomegaly >5 cm below the left costal margin should receive spleen-directed therapy prior to allo-HCT, ideally with a JAK inhibitor [88, 90]. As regard younger patients with intermediate risk-1 disease (DIPSS) or intermediate disease (MIPSS70 or MIPSS70-plus) and low MTSS score, allo-HCT could be an option balancing patient preference, donor type, therapeutic options and other risk factors (including multi-hit TP53 mutations) [88]. Experience of allo-HCT in patients with low-risk index disease is limited and remains controversial. Recent EBMT data suggest no disadvantage with pre-transplant exposure to ruxolitinib, and better outcomes after transplant in ruxolitinib-responsive patients compared with patients with no response or loss of response to ruxolitinib [91]. The effect of other JAK inhibitors on allo-HCT outcome, such as fedratinib and momelotinib remains as yet undetermined. Auto-HCT for MPN is not recommended.
Myelodysplastic syndromes (MDS)
Allo-HCT remains the only curative option for patients with higher-risk MDS, but patient-related factors, especially age and co-morbidities, often impact upon feasibility of HCT. The EBMT proposed a transplant-specific risk score for MDS patients [92], which, alongside HCT-CI, may be used to judge the feasibility of allo-HCT and predict outcomes. In addition, disease characteristics that impact the risk of transformation into AML and survival need to be considered.
Both IPSS and the revised IPSS (IPSS-R) have been used to stratify patients and propose transplantation for higher risk eligible patients based on prospective studies [93,94,95,96,97]. Higher risk includes both ‘high’ and ‘very high risk’ according to IPSS-R and ‘intermediate-2’ and ‘high risk’ according to IPSS. Of note, some lower risk cases have also been included in these prospective studies where they represent a minority of the population. The benefit of transplantation in low risk MDS, including patients who are heavily transfused, has not been proven by prospective trials (in contrast to higher risk patients). More recently, a large international study has utilised IPSS-M to stratify patients and determine potential benefits of transplantation [98]. This study confirmed that eligible patients in the ‘moderate-high’, ‘high’ and ‘very high-risk’ categories benefited from immediate transplantation while other lower risk categories did not (but may benefit for a delayed transplantation approach) which remains in line with previous EBMT and other international recommendations [99]. Of note, some lower risk patients may acquire a transplantation indication when progressing or when presenting with specific high-risk features (transfusion refractoriness, marrow fibrosis, acquisition of poor prognostic somatic mutations). Pre-transplantation therapy has not been proven by prospective trial to be required before transplantation. A recent EBMT study demonstrated that down staging IPSS-R with pre-HCT therapy does not convert into an improved outcome [100]. However, in patients who cannot be transplanted upfront due to logistical issues, hypomethylating agents are usually used (alone or in combination) to prevent disease progression. Marrow blast persistence that is stable over time is not a contraindication for transplantation.
Chronic myelomonocytic leukaemia (CMML)
Allo-HCT represents the only potentially curative treatment option in CMML but is applicable to only a minority of patients. High rates of relapse (30–50%) and NRM (>20%) remain the main reasons for allo-HCT failure historically and careful patient selection is mandated [101]. The EBMT recently generated contemporary guidance focused on the allo-HCT decision in CMML [101]. Recent ICC and WHO classification revisions removed the CMML-0 classification whilst maintaining CMML-1 (<5% blasts in peripheral blood and <10% in bone marrow) and CMML-2 (5–19% blasts in peripheral blood and 10-19% in bone marrow and lowered the threshold to a persistent absolute (≥0.5 × 109/ L) and relative (≥10%) peripheral blood monocytosis. There are two variants, ‘dysplastic’ and ‘proliferative’, dependent on the circulating leucocyte count (≤13 × 109/L and >13 × 109/L, respectively). For patient risk stratification, the use of a CMML-specific Prognostic Scoring System (CPSS) is recommended, ideally one including molecular information such as the CPSS-molecular (CPSS-Mol) [102]. Regarding CPSS-Mol high-risk disease, eligible patients with a suitable donor should proceed directly to allo-HCT [102]. For intermediate-2 risk CPSS-Mol, those with ≥1 additional risk factor (e.g. extramedullary disease, splenomegaly, hyperleukocytosis) should also proceed to upfront transplant [102]. Patients with low and intermediate-1 CPSS-Mol disease should be dynamically monitored closely but not routinely undergo allo-HCT [102]. For those with an allo-HCT indication, upfront allo-HCT is preferred rather than undergoing pre-transplant therapy unless delay in donor availability or in the presence of aggressive disease with rapid kinetics [102]. In general, hypomethylating agent-based regimens are preferred if pre-transplant therapy is required. However, available data for this indication are still limited.
Chronic lymphocytic leukaemia (CLL)
The widespread availability of signalling pathway inhibitors, such as the Bruton’s TKIs, ibrutinib and acalabrutinib, the BCL2- inhibitor venetoclax, and phosphatidylinositol-3-kinase inhibitors has significantly changed management algorithms and HCT indications across all lines of therapy in CLL. These drugs are frequently used either alone or in combination based on disease risk stratification, licensing and reimbursement status as per each country. Of note, the combination of ibrutinib and venetoclax in the Phase III FLAIR (ISRCTN01844152) trial was associated with improved PFS and OS in untreated CLL patients compared to combination chemoimmunotherapy, with duration guided by MRD response [103]. Eligible patients with relapsed/ refractory CLL who have exhausted their main pharmacological therapeutic options (including sequencing of both covalent and/ or non-covalent Bruton’s TKIs and venetoclax-based regimens) may be considered for cellular therapies, including allo-HCT or CAR-T cells, or clinical trials. Such truly ‘double-refractory patients’ have an estimated survival of 4-6 months without intervention [104]. Cellular and molecular therapies are not mutually exclusive and could be used synergistically to exploit their full potential. Patients with CLL and a concomitant MDS and those with clonally related aggressive transformation of CLL should be considered for allo-HCT regardless of the treatment stage of their CLL [105]. Auto-HCT is generally not recommended in CLL. However, it can still be considered a clinical option in patients with a histological transformation clonally unrelated to CLL [106].
Lymphomas
Since December 2015, lymphoma patients are reported to the EBMT Registry as being in ‘true’ CR1 (first CR directly by standard first-line treatment), or as being in ‘first’ CR (achieved by one or more salvage attempts after primary induction failure), clearly segregating patients with different prognoses. Therefore, the recommendations for lymphoma (Table 1) refer to ‘true’ CR1 if CR1 is mentioned, and CR1 after prior refractoriness is included as CR > 1, and, in some lymphoma types, transplant indications in CR1 are more restrictive than in previous editions of these recommendations. In most types of lymphoma, retrospective analyses now show comparable results for MSD, MUD and haploidentical donor transplantation with PTCy (as cord blood and other alternative donors are used quite rarely in this setting and are not covered by these statements). Therefore, we now have consistency across current lymphoma classifications. While general recommendations and supporting data are discussed in the text below, Table 1 outlines specific transplant indications along with the corresponding strength of evidence for each disease status.
Large B-cell lymphoma (LBCL)
LBCL is defined as any entity behaving clinically similar to diffuse large B-cell lymphoma (DLBCL) with respect to therapeutic options within the context of this guideline, including high-grade B-cell lymphoma NOS (not otherwise specified), double-hit and triple-hit lymphomas and peripheral mediastinal B-cell lymphoma.
Anti-CD19 CAR-Ts, axicabtagene ciloleucel (axi-cel) and lisocaptagene maraleucel (liso-cel), demonstrated superior PFS compared to standard of care, including salvage immunochemotherapy followed by auto-HCT in responders, as shown in two phase III clinical trials in patients with primary refractory disease or early relapses within 12 months following first-line therapy [107, 108]. For axi-cel a significant OS benefit was shown in the 5-year follow-up [109]. For patients who achieve a CR after salvage treatment prior to cell therapy, a retrospective analysis found better PFS if patients were consolidated with auto-HCT compared to CAR-T cell treatment [110]. Of note, no patients treated with CAR-T cells in second line were included in this analysis and more than half of the patients had received tisagenlecleucel, which is probably less efficient compared to axi-cel [111]. Still, in patients with early chemosensitive relapse, we consider high-dose therapy/auto-HCT to be a valid clinical option. For patients with primary refractory disease or early relapse with untested or chemorefractory disease, treatment with axi-cel or liso-cel represents the current standard of care.
Patients with late relapses face reasonable outcomes with auto-HCT and this standard has not been challenged by a clinical trial as of today. For the treatment of relapses following high-dose therapy/auto-HCT, CAR-T cell therapy can still lead to good responses [112]. In case of failure of second-line cell therapy, allo-HCT remains a clinical option for eligible patients [113,114,115,116,117,118,119].
The benefit of auto-HCT as part of the first-line treatment for patients with double-hit lymphomas is questionable and not recommended as routine practice [120]. For patients with high-risk disease defined by inadequate response on the interim PET-CT, auto-HCT as consolidation in CR1 can be considered. Still, superiority of this approach needs to be confirmed by comparative studies [121].
For patients with primary CNS lymphoma, auto-HCT in first remission after first-line therapy remains a standard approach in eligible patients, supported by several randomised trials and shown to be feasible even in fit older patients [122,123,124,125,126]. For patients with primary CNS lymphoma experiencing relapse or refractory disease, auto-HCT can also represent a promising option [127, 128]. In addition, there is growing evidence that CAR-T cell treatment has the potential to offer similar outcomes to those observed in other relapsed/refractory LBCL with an acceptable response duration in this setting [129,130,131] and is therefore considered to be the clinical option in second line.
Follicular lymphoma (FL)
Most advanced-stage FL patients benefit from a long PFS after induction and maintenance and should not be offered any cellular therapy in first remission [132]. Previously untreated FL patients with high-grade transformation should be treated as DLBCL. Auto-HCT is a clinical option in patients with transformed FL, if they had received prior chemoimmunotherapy. Patients with untransformed FL in 2nd remission or beyond and patients with POD24 should be considered for auto-HCT [133]. The FL treatment algorithm is changing. Bispecific antibodies are conditionally approved by EMA from 3rd line. [134,135,136] Two CAR-T cell products are EMA-approved for FL based on Phase 2 non-comparative trials: axi-cel [137] from 4th line and tisa-cel [138] from 3rd line. Liso-cel also shows promising results in FL [139]. CAR-T cells can be used in accordance with the EMA-label in countries where they are available. Allo-HCT is recommended for relapses after auto-HCT, if CAR-T is not available [140,141,142].
Waldenström’s macroglobulinemia (lymphoplasmacytic lymphoma with IgM gammopathy; WM)
The treatment advances for patients with WM using immunotherapy with anti-CD20 monoclonal antibody, chemoimmunotherapy [143, 144], purine analogues [145], proteosome inhibitors [146,147,148], Bruton’s TKIs [149, 150] and most recently bcl2 inhibitors have achieved deep and durable responses. Therefore, auto-HCT should not be considered as first-line clinical option. Auto-HCT should be considered as clinical option at subsequent disease relapses depending on the type of prior treatment, duration of response, age, performance status and comorbidities. Allo-HCT might be considered for younger patients with multiple relapses, those not achieving durable responses, or patients with refractory disease to immunochemotherapy, proteosome inhibitors-based treatment and/or Bruton’s TKIs and bcl-2 inhibitors.
Mantle cell lymphoma (MCL)
Studies testing Bruton’s TKIs as part of first-line therapy are questioning the benefit of 1st-line auto-HCT consolidation in MCL [151]. In 2nd line, auto-HCT is an option in HCT-naïve patients responding to salvage chemoimmunotherapy.
Available evidence does not suggest benefit of allo-HCT in MCL in CR1. In the salvage setting, allo-HCT is an option in patients without access to CAR-T or who have failed CAR-T [152, 153].
CAR-T should not be used as 1st-line treatment of MCL outside of clinical trials. The labelled indication of CAR-T in MCL is patients having failed at least two prior therapies including Bruton’s TKIs [153]. With Bruton’s TKIs moving to 1st-line treatment, CAR-T has become a clinical option as 2nd-line therapy in patients with primary Bruton’s TKIs failure.
T-cell lymphomas (TCL)
Peripheral TCLs (PTCL) usually carry a poor prognosis. Allo-HCT is the only curative option for transplant-eligible patients with primary refractory PTCL or those who are in relapse. While allo-HCT is not an indication for major PTCL entities as first-line consolidation (T Follicular Helper Lymphoma (TFH-L), Peripheral TCL, Not Otherwise Specified (PTCL-nos) or Anaplastic Large Cell Lymphoma (ALCL)), auto-HCT is a recommended strategy in CR1 [154, 155]. In Enteropathy-Associated T-Cell Lymphoma (EATL) auto-HCT is also recommended in CR1. In Hepatosplenic TCL (HSTL), Monomorphic Epitheliotropic Intestinal T-Cell Lymphoma (MEITL), Extranodal NK/TCL (ENKTL), and Adult T-Cell Leukaemia/Lymphoma (ATLL), allo-HCT is strongly recommended in first-line therapy. While patients undergoing transplant in CR and PR show superior survival, patients with stable disease or progression should be considered for allo-HCT because other treatments do not typically result in long-term remission.
Primary cutaneous TCL (CTCL) in early stage have an excellent outcome, and HCT is generally not recommended. However, patients with EORTC/ISCL advanced stages (IIB to IV) have a dismal prognosis with conventional therapy [156]. Allo-HCT offers these patients a clinically relevant and persistent graft-versus-lymphoma effect [157,158,159].
A prospective matched controlled study in patients with advanced CTCL [160] demonstrated superior PFS of patients treated with allo-HCT as compared to conventional therapy. Therefore, allo-HCT is recommended for all patients with poor-risk CTCL stage IIB-IV, refractory or relapse after first-line therapy, large-cell transformation, nodal or visceral involvement, as soon as they reach CR, very good partial response (VGPR) or stable disease with minimal residual disease. Because of high relapse rates, auto-HCT is generally not recommended.
Hodgkin lymphoma (HL)
Targeted agents such as brentuximab vedotin (BV) and checkpoint inhibitors (CPIs) have already modified results of both auto-HCT and allo-HCT in patients with relapsed/refractory cHL. For now, as in previous recommendations, auto-HCT remains standard of care for patients with relapsed HL chemosensitive to standard therapy, and allo-HCT in those after a failed prior autograft [161,162,163,164,165].
The combination of BV and CPIs with salvage therapeutic strategies increases the percentage of metabolic complete remissions (mCR) before auto-HCT, thus potentially improving results of the procedure and the number of potential candidates for high-dose therapy [166,167,168,169,170,171]. Consolidation strategies after auto-HCT in high-risk of relapse patients have also improved long-term outcomes of auto-HCT. In the absence of prospective randomised clinical trials, allo-HCT is generally considered in patients who fail both BV and CPIs. CAR-Ts have been tested in the setting of prospective clinical trials; the few available results do not show convincingly better results than other treatment modalities to date [172]. Nodular lymphocyte predominant HL comprises only 5% of all patients being diagnosed with HL; HCT is restricted to those high-risk patients with relapsed disease [173].
Multiple myeloma (MM)
The development over the last decade of new agents such as anti-CD38 monoclonal antibodies and bispecific antibodies targeting BCMA and GPRC5D among other antigens, represent major advances. The incorporation of these agents into induction regimens may ultimately displace high-dose melphalan and upfront auto-HCT from its current central role in the treatment of younger, fitter patients. At present, however, first-line auto-HCT remains the standard of care for younger MM patients [174,175,176]. Age should be considered in conjunction with general health and fitness [177]. TBI should not be used in the conditioning regimen due to increased toxicity without appreciable benefit, and the addition of bortezomib or lenalidomide to conditioning regimens was not found to improve patient outcomes [178]. Double or ‘tandem’ autografting was found to be superior to a single auto-HCT in historical studies, although the benefit of the second transplant procedure in patients receiving contemporary quadruplet induction is not clear. Immunomodulatory drugs and bortezomib patients with high risk myeloma [179,180,181,182].
Two trials randomised patients who relapsed late after an autologous transplant to either a second salvage auto-HCT or a chemotherapy-only approach [183, 184]. Cook et al. reported a superior median time to progression (19 vs 11 months) in the salvage auto-HCT group without post-transplant maintenance [184]. In the randomised GMMG phase III ReLApsE trial, a post-hoc landmark analysis beginning at the time of HCT suggested an OS benefit for HCT (hazard ratio [HR] 0.56; 95% CI 0.32-0.99). In general, more durable remissions have been reported in patients whose time to progression was >24 months after the first auto-HCT [185].
Allo-HCT is a treatment with curative potential but is associated with considerable NRM and is generally reserved for selected high-risk patients in experienced centres [186]. The combination of auto-HCT followed by RIC allograft (‘auto-allo’) has shown a survival benefit in selected high-risk patients, albeit inconsistently in various clinical trials [187,188,189,190]. Recently, allo-HCT with PTCy has been shown to be feasible in MM, but relapse remains a major challenge [191]. Overall, there has been a gradual decline in the use of allo-HCT in patients with myeloma in EBMT centres over the last decade. There were approximately 200 allo-HCT reported to the EBMT in 2023.
CAR-T has shown promising results in patients with refractory/relapsed MM [192,193,194]. Idecabtagene vicleucel is the first cell-based gene therapy approved by the FDA for adults with relapsed/refractory MM after four or more lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 mAb. The second anti-BCMA CAR-T therapy, ciltacabtagene autoleucel, is indicated for the treatment of adult patients with relapsed and refractory MM, who have received at least one prior therapy, including an immunomodulatory agent and a proteasome inhibitor who have demonstrated disease progression on the last therapy, and are refractory to lenalidomide. The 27-month PFS and OS rates in patients with advanced disease were 54.9% (95% CI, 44.0 to 64.6) and 70.4% (95% CI, 60.1 to 78.6), respectively [195]. There are several trials evaluating the efficacy and tolerability of anti-BCMA CAR-T therapy at earlier disease stages though logistic and cost constraints have limited access to these agents so far.
AL amyloidosis
Patients with systemic immunoglobulin-light-chain (AL) amyloidosis without severe heart failure may benefit from auto-HCT [196]. The Mayo Clinic Prognostic Staging System for Light Chain Amyloidosis Incorporating cardiac biomarkers may aid in patient selection [197]. The benefit of auto-HCT was not confirmed in a prospective randomised trial that included patients with advanced cardiac amyloidosis [198]. Many recently published studies have reported improved early mortality after an appropriate risk assessment and consistently good haematologic responses and impressive long-term survival [199, 200]. Cytogenetic aberrations, as t(11;14), can also guide therapy [201]. The recently reported efficacy of bispecific antibodies in this disease is likely to reduce the use of auto-HCT.
Acquired severe aplastic anaemia (SAA)
HLA-identical sibling allo-HCT is considered the standard of care for adult patients with SAA; outcomes decline depending on the age, with increased mortality which is meaningful above 40 years, and unacceptable above 50 years [202,203,204,205]. Thus, in addition to age, careful assessment of co-morbidities prior to HCT is essential to assess the risk of mortality and morbidity for upfront HCT, especially in border-line age groups (i.e. 35–50 years). To reduce the risk of chronic GVHD, all patients should receive in vivo lymphodepletion with ATG or alemtuzumab, and bone marrow is the recommended source of stem cells [206,207,208]. The choice of conditioning regimen also depends on age; patients <30 years old should receive high-dose cyclophosphamide (200 mg/kg), and those aged 30–40 years old, a fludarabine-based (120 mg/m2) regimen with lower dose cyclophosphamide (120 mg/kg). There is no indication for using radiation in the conditioning for MSD HCT. MSD HCT is also the preferred treatment option for all patients reasonably young (up to 70 years) and fit who have failed (refractory or relapsed) an initial non-transplant treatment, which is immunosuppressive therapy (IST), now based on the triple therapy ATG (preferably horse ATG), ciclosporin and eltrombopag [209, 210]. Failure of IST also includes progression/transformation into a myeloid malignancy (MDS or AML), somehow defined as ‘clonal evolution; however, while allo-HCT is recommended in case of proven MDS or AML, the presence/emergence of somatic mutations alone does not qualify as an indication to allo-HCT [211].
MUD HCT can be considered as a first-line choice in young patients aged <18 years based on the excellent outcome compared to historical matched controls [212], provided transplant is feasible within the first two months after diagnosis. Alemtuzumab-based conditioning is also recommended in this situation [213]. If the interval to find a suitable MUD and proceed to HCT is predicted to be longer, IST with triple therapy is recommended as first-line treatment as in adults.
Otherwise, MUD HCT is indicated in young and adult patients after failure to respond to one course of IST (again, triple therapy), normally assessed at 3–6 months. Age of recipient is also an issue for MUD HCT, and along with assessment of co-morbidities and other patient and transplant characteristics (e.g. CMV status, source of cells, use of ATG, interval from diagnosis to transplant, HLA matching degree), should help in evaluating patients who would benefit most from the procedure. On the other hand, since IST with triple therapy has maximised the likelihood of response to IST, allo-HCT is now considered the preferred treatment option for all patients who have failed initial IST, as defined above. In this setting, recipient age, HLA disparity and interval from diagnosis to transplant (within 1 year) impact on transplant outcome. A non-transplant approach is still recommended in elderly (>60–70 years), and it can be considered also in patients who had an initial response to triple therapy (e.g. second course of IST, or reintroduction of eltrombopag), as well as in other clinical settings (e.g. eltrombopag if not given first line, androgens, second course of immunosuppressive therapy) [206,207,208, 212,213,214,215,216]. Here, the conditioning regimen is usually based on the standard association of fludarabine and cyclophosphamide (120 mg/m2 and 120 mg/kg, respectively), associated with lymphodepletion with ATG or alemtuzumab. As in MSD HCT, bone marrow is the recommended stem cell source for MUD HCT in SAA, especially for ATG-based conditioning regimens, while alemtuzumab-based conditioning may result in excellent outcome even using peripheral blood as stem cell source. Ongoing studies are exploring the impact of PTCY as an alternative GVHD prophylaxis in the context of MUD allo-HCT in SAA.
Alternative donors for allo-HCT (e.g. MMUD, haploidentical or cord blood) may be considered after failure to respond to IST. Excellent results have been reported especially in young patients up to 20 years of age in the absence of MSD or MUD [217,218,219,220,221,222,223]. Registry studies have documented that the outcome of MMUD allo-HCT for SAA is worse than that of MUD, but still acceptable; recent data suggest that the survival difference may be somehow mitigated or even overcome with the introduction of PTCY as GVHD prophylaxis [224]. This approach of GVHD prophylaxis has drastically changed the field, with very promising data even in older patients [217, 225]. Haplo-HCT in SAA should follow the so-called ‘DeZern’ protocol (fludarabine, cyclophosphamide, ATG and low-dose TBI, followed by PTCY, CNI and MMF). This regimen, albeit in small studies, resulted in excellent outcomes, making this strategy a clinical option for refractory/relapsed patients [226, 227]. Haplo-HCT is now under investigation also as possible initial therapy of SAA [228]. Although the treatment algorithm of SAA is under debate [229, 230], it is too early to recommend haplo-HCT as initial treatment for SAA patients [231]. Similarly, the question whether this promising conditioning regimen (which requires TBI, even at the dose of 400 cGy as upfront therapy) might be extended to other donor types [232] requires further investigation.
Cord blood HCT is also an option for refractory/relapsed SAA patients. Even if results from small studies are promising, this strategy remains experimental and should be delivered through specific trials or protocols (a SAAWP-recommended protocol is available), always seeking advice from a SAA specialist centre.
Constitutional SAA and bone marrow failure (BMF) syndromes, including Fanconi anaemia (FA), dyskeratosis congenita (DKC) and other telomere diseases, may not only be present in childhood but also in adults, often with more subtle clinical features. All cases of SAA being considered for HCT should have an appropriate diagnostic workup, including molecular genetics, to establish or exclude constitutional BMF syndromes, as it has important consequences for donor choice and conditioning regimen (see relevant sections below). On the other hand, additional testing may be useful to investigate clonal evolution even in possible (or confirmed) inherited BMF, since several factors (e.g. disease penetrance, concurrent mutations and further somatic mutations—including possible somatic rescue) may impact on disease phenotype, possibly impacting both the indication for allo-HCT, as well as the specific HCT procedure, including donor selection.
Paroxysmal nocturnal hemoglobinuria (PNH)
The introduction of anti-complement therapy with eculizumab changed the natural history of the disease, and allo-HCT became generally not recommended for patients with classical PNH (haemolysis ± thrombosis) for whom eculizumab is available. In recent years, additional complement inhibitors have been developed aiming to improve the haematological benefit and/or patients’ convenience (e.g. long-lasting agents and oral agents) [233,234,235,236]. They have further limited the room for allo-HCT in patients with suboptimal response to complement inhibitors. Potential indications remain dependent on the individual clinical manifestations: (i) severe bi- or tri-lineage cytopenia due to severe BM failure (the so-called AA/PNH syndrome), using the same criteria for SAA above (i.e. age, disease severity, and failure to respond to one course of immunosuppressive therapy in case of donor other than MSD) and (ii) evolution/transformation of PNH to a myeloid malignancy (MDS or AML) [237, 238]. Given the improved outcome of allo-HCT for PNH [239], allo-HCT remains a potential treatment option when anti-complement treatment is not available [240]. Given the pleiomorphic clinical presentation of PNH, expert advice should be sought from a PNH specialist centre.
Details about these indications as well as the specific treatment regimens and challenges in AA patients are summarised in the most recent guidelines on the management of AA from the British Society of Haematology [241].
Solid tumours
Currently, the EBMT Registry includes >70,000 HCT procedures in >50,000 patients, of whom >40,000 are adults with solid tumours. Over 7500 procedures have been performed in adults in the last 5 years. However, with the possible exception of patients with germ cell tumours, and highly selected patients with breast cancer, sarcoma and medulloblastoma, HCT is generally not recommended or remains developmental for most indications in solid tumours [7]. With limited evidence from prospective studies published recently, the new recommendations in 2025 have changed little compared to prior indications.
Despite the encouraging role of immune surveillance and immune responses against several solid tumours [242,243,244] recommendations for allo-HCT would require further prospective trials, which are not a priority for medical oncology.
CAR-T, often associated with adjunctive agents, and other forms of adaptive immune effector therapies [245,246,247,248,249,250] are currently an area of active clinical research with data from Phase I/II studies suggesting a potential role of these approaches in several solid tumours [251, 252].
The role of auto-HCT in breast cancer at high risk of recurrence and metastatic disease has been assessed by several randomised trials and meta-analyses of individual patient data [253, 254]. As discussed in more detail in previous reports [255], the overall conclusion is that auto-HCT in breast cancer improves PFS but not OS in most studies. However, auto-HCT may still represent a clinical option for selected patients with specific biological characteristics and/or having gross involvement of axillary nodes (adjuvant setting) or highly chemosensitive disease (advanced setting) [255,256,257,258].
In germ cell tumours, auto-HCT is a standard of care for patients with disease refractory to platinum-based chemotherapy or with a second or further relapse, a clinical option as a second line in high-risk patients, and generally not recommended as first-line therapy [259,260,261], with the possible exception of primary mediastinal non-seminoma germ cell tumours [262]. Finally, auto-HCT can be considered a potential clinical option in selected patients with Ewing’s and soft tissue sarcomas [263, 264], and medulloblastoma [265].
Auto-HCT, being per se capable of inducing marked and rapid tumour regression, may still represent a treatment modality for selected chemosensitive solid tumours and is worthy of further study in combination with effective target agents, including immunotherapy. While awaiting results of further prospective trials, the EBMT registry remains an important source to survey indications, outcome and clinical risk factors in patients with solid tumours treated with auto-, allo-HCT and cellular therapies.
Autoimmune diseases (AD)
Autologous and, less frequently, allo-HCT represent promising therapeutic approaches for many severe AD cases that are resistant to state-of-the-art therapies, following careful consideration of the associated risks and benefits [266]. Worldwide, most transplant procedures for AD have been performed for multiple sclerosis (MS), followed by systemic sclerosis (SSc), currently representing the two standard indications for auto-HCT in this population [7].
Evidence is continually evolving for auto-HCT, predominantly in MS [267,268,269,270,271], and SSc [272,273,274,275,276], for which auto-HCT is regarded as a standard of care [277]. In addition, there is evidence to support treatment of carefully selected patients with Crohn’s disease [278, 279], systemic lupus erythematosus (SLE) [280,281,282], neuromyelitis optica [283, 284], chronic inflammatory demyelinating polyradiculoneuropathy [285], myasthenia gravis [286], stiff person syndrome [287, 288], systemic vasculitis (ANCA positive [289], Takayasu [290], Behcȩt’s disease [291] and refractory coeliac disease [292]. Collectively, the available data support the consideration of auto-HCT as an efficacious, on-off intensive therapeutic procedure for severe and refractory AD. Indeed, in the last decade, better outcomes have been obtained with auto-HCT, owing to a growing centre experience in selecting the most appropriate patients to transplant paralleled by advances in conditioning and supportive care regimens, certification and national socioeconomic factors [293]. Health economic reports have supported cost-effectiveness in some AD [269, 294, 295].
Despite improved survival over time, allo-HCT has remained predominantly used in younger patients [293]. According to recent EBMT registry data, this strategy can potentially provide long-term disease control in refractory AD, warranting further investigations mainly in younger patients [296]. The EBMT and other professional societies have published multidisciplinary guidelines and recommendations for both autologous and allo-HCT across various ADs, providing support to clinicians, scientists, patients, and caregivers [277, 293, 297,298,299,300,301,302,303]. Recently, the EBMT has also provided updated recommendations for the best practice of HCT in AD during the COVID-19 pandemic [304].
Recently, CAR-T cell therapy has been successfully used to treat patients with refractory autoantibody-driven ADs [305, 306]. Evidence for CAR-T therapies initially derived from small pilot studies [11] or single cases using autologous CD19 CAR-T in SLE [307,308,309], SSc [310] and idiopathic inflammatory myopathies (IIM) [311,312,313], followed by positive results from the first phase I/II trial of the BCMA-CD19 compound CAR-T in SLE [314]. Positive results were also obtained for refractory cases of rheumatoid arthritis [315, 316]. Meanwhile, first experiences have been obtained for CD19 CAR-T cell therapy for neurologic indications, including refractory cases of myasthenia gravis [315, 317], MS [318] and SPS [319]. More recently, positive results have been reported from phase 1 trials using BCMA CAR-T therapy in NMOSD [320] and autologous RNA CAR-T therapy in MG [321] with an acceptable safety profile.
Although representing a unique and promising therapeutic approach, further studies are required to position CAR-T cell therapies in the current treatment landscape of AD. EBMT recommendations for the use of CAR-T cells in ADs have been published [12]. A multidisciplinary team is strongly recommended to ensure appropriate patient selection and to carefully monitor the efficacy and toxicity of CAR-T cells in ADs.
Inherited diseases in adults
While allo-HCT in inherited diseases is predominantly performed in childhood, adult patients with inherited diseases, including haemoglobinopathies, constitutional BMF syndromes and inborn errors of metabolism (IEM) and immunity (IEI) are increasingly considered for HCT [322]. Given the complexity of these cases, it is advisable to seek expert guidance from specialised centres. The indications are the same as for inherited paediatric diseases (covered below), although presentation in adults may differ, including age of onset, course and prognosis. In some cases, access to HCT during childhood has been limited by health service resource and other non-clinical factors, necessitating consideration of HCT in adulthood.
As for the paediatric patients, there are differences between Transfusion dependent Thalassaemia (TDT) and Sickle Cell Disease (SCD).
In TDT, limited experience has been registered in patients older than 14 years over the last years, with success rates, although improved, still not exceeding 80% [323]. However, because it has been clearly demonstrated that clinical condition and iron toxicity long-life-control is a major determinant of transplant outcome [324], improved clinical care and management of TDT makes transplant a today rationale option in patients beyond the above defined threshold [325].
In SCD, the EBMT hemoglobinopathies registry data have recently demonstrated that age is no longer a factor determining adverse outcomes [326] with success rates over 90% in adults as in children.
The recommendation to perform transplant for hemoglobinopathies in disease highly experienced centres already expressed for children is even more stringent for adult patient [325]
Transplant indications in children and adolescents
Allo-HCT in children and adolescents accounts for over 20% of overall allo-HCT activity, with particular use in congenital and non-malignant diseases, many of which are rare. Transplant complications in paediatric patients are compounded by the vulnerabilities of the developing child, including organ dysfunction related to development, such as infertility, delayed hormonal development, growth retardation, and a high risk of malignancies in congenital disorders with chromosomal breakage syndromes.
Advances in high-resolution HLA matching for UDs, conditioning regimens, supportive care for both infectious and non-infectious complications and the improvements in haplo transplants especially in non-malignant diseases (e.g. PIDs), have progressively reduced mortality. These developments have encouraged the use of allo-HCT, particularly for non-malignant indications, at earlier stages of disease progression, with patients in better performance status, rather than as a ‘last chance for cure’.
GVHD remains a major limitation for patients without optimally matched donors. Emerging allo-HCT strategies aim to improve outcomes for patients with MMAD. The updated 2025 classification of HCT procedures in children and adolescents is presented in Table 2.
Acute myeloid leukaemia (AML)
Childhood AML is a rare and heterogeneous disease, with increasing cure and survival rates achieved through intensive chemotherapy, particularly for patients with favourable prognostic markers. As a result, allo-HCT is not recommended as frontline therapy for low-risk patients but remains the standard of care for those in CR1 with high- or very-high-risk disease who have a well-matched donor [327,328,329,330].
Alternative donors, particularly haploidentical family members, are playing an increasingly significant role in treating high- and very-high-risk childhood AML and patients beyond CR1 [331, 332]. Children who relapse and achieve a second CR are also candidates for allo-HCT from the best available donor.
Two randomised studies are currently underway to evaluate conditioning regimens in children with AML: one comparing BuCyMel versus TreoFluTT (AML-AIEOP BFM 2020) and another comparing BuCyMel versus BuFluClo (SCRIPT). Auto-HCT in this context is generally not recommended [333].
Acute lymphoblastic leukaemia (ALL)
Allo-HCT from MSD and MUD is the standard of care for high-risk ALL patients in CR1, as well as for those in CR2 or later [334,335,336,337,338]. Classical risk factors include molecular markers, chromosomal abnormalities, biological factors, and resistance to initial chemotherapy [339]. However, minimal residual disease (MRD) has now emerged as the most critical prognostic factor to distinguish high- from very-high-risk ALL groups [340,341,342].
When an MSD or MUD is unavailable, mismatched alternative donors (MMAD), such as cord blood (CB), mismatched unrelated donors (MMUD), or haploidentical family donors, are viable clinical options [343]. Unlike in adults, PBSCs offer no significant advantage over BM in terms of engraftment or relapse incidence. Therefore, BM is the preferred stem cell source for children undergoing MSD-HCT [344].
The FORUM trial (EudraCT: 2012-003032-22; ClinicalTrials.gov: NCT01949129) recently evaluated the role of total body irradiation (TBI) in a non-inferiority study involving 417 patients. In the intention-to-treat population, the 2-year OS was significantly higher following TBI compared to chemotherapy-only conditioning. The 2-year cumulative incidence of relapse and treatment-related mortality (TRM) was 0.12 and 0.02 for TBI, versus 0.33 and 0.09 for chemotherapy-only conditioning, respectively.
Regrettably, the trial demonstrated that improved OS and lower relapse rates were achieved with TBI plus etoposide compared to chemotherapy-only conditioning. At present, TBI plus etoposide is recommended for patients >4 years old with high-risk ALL undergoing allo-HCT [345].
In regions without access to TBI or for children with contraindications, chemotherapy regimens such as BuFluTT or TreoFluTT remain valuable alternatives [346].
Tisagenlecleucel is a CD19-specific chimeric antigen receptor T cell therapy, EMA- and US FDA-approved for children, adolescents, and young adults with relapsed and/or refractory B-ALL. The registration for tisagenlecleucel was based on a CR rate of 81%, 12-month OS of 76%, and EFS of 50% [59].
Chronic myeloid leukaemia (CML)
As mentioned earlier, for adult patients, with the advent of TKIs, allo-HCT is no longer recommended as a first-line treatment for CML in children and adolescents. However, it remains a standard option for patients experiencing treatment failure, recurrence after receiving salvage second-generation TKI therapy, and advanced-phase CML [347,348,349].
For paediatric patients, the decision to proceed with allo-HCT requires careful individual consideration to balance the well-established long-term complications of HCT with the potential adverse effects of prolonged TKI treatment, which may include growth failure, hepatic, and cardiac complications [350,351,352]. Stronger evidence from prospective cooperative studies is needed to better understand disease progression after TKI discontinuation and to address issues specific to paediatric patients with CML [348, 353].
A recent paper by Pichler et al. recommends FluMelTT as the optimal conditioning protocol for children with CML [354].
Myelodysplastic Syndromes (MDS) and Juvenile Myelomonocytic Leukaemia (JMML)
Allo-HCT from an MSD or MUD is the treatment of choice for children with primary MDS, including JMML, as well as secondary AML [355,356,357]. Auto-HCT is not recommended outside clinical trials.
Lymphoma
Nearly all children and adolescents with HL and NHL are cured with multidrug chemotherapy. Only a small subset of paediatric patients are eligible for HCT (Table 2) [358,359,360,361,362]. These include patients with residual disease after re-induction therapy with contemporary chemotherapy protocols, patients with early relapses of NHL, and those with an inadequate response or relapse of ALK-positive ALCL. All other approaches should be discussed with experts in frontline chemotherapy trials.
Solid tumours
Although published results have not yet demonstrated an unequivocal benefit for most indications, children and adolescents with solid tumours may undergo auto-HCT following high-dose chemotherapy as a clinical option or as part of research protocols, preferably within first-line treatment strategies (Table 2). Neuroblastoma (stage 4 in children older than 1 year, or with high-risk factors in lower stages) remains the only indication where the benefit of auto-HCT has been proven by randomised trials [363, 364]. The use of GD2-CART01 cells was feasible and safe in treating high-risk neuroblastoma in a single-centre study. Treatment-related toxic effects developed, and the activation of the suicide gene controlled side effects. GD2-CART01 cells may have a sustained antitumor effect [365].
In general, allo-HCT for children with solid tumours should only be explored within prospective clinical trials conducted at highly experienced centres.
Acquired severe aplastic anaemia
Allo-HCT from MSD is the standard front-line therapy for children and adolescents (up to 18–20 years) with acquired SAA. In patients without MSD, a well-matched (10/10) unrelated HCT is now also considered a standard front-line therapy in many patients if the donor is readily available and the transplant feasible within the first two months after diagnosis. The search should in any case be initiated before starting any immunosuppressive therapy [366,367,368,369,370]. For those who lack a well-matched donor (i.e. MMUD and Haplo) allo-HCT is currently not recommended as front-line treatment, even if recent data using an haplo donor may be promising [220, 221, 223,224,225,226,227,228]. MMUD and haplo-HCT is considered standard in patients failing their first course of immunosuppression, being preferred over a second course of immunosuppression.
Constitutional bone marrow failure syndromes
Allo-HCT is the only treatment able to restore normal haematopoiesis in these patients. Transfusion-dependent FA patients with a suitably well-matched family or UD should be transplanted while in the phase of moderate cytopenia with no poor-risk clonal abnormalities and no MDS/AML [371,372,373,374]. Haplo donor HCT offers very good results when either in vivo or ex vivo TCD is included [371,372,373] but for patients who lack a well-matched donor, HCT from MMAD should be better considered as a clinical option in the context of a clinical protocol. Although outcomes are reported to be better at age <10 years, this is not the only criterion for decision-making. Details on transplant conditioning for specific indications are beyond the scope of these recommendations, but it is important that standard doses of chemotherapy and/or irradiation are absolutely avoided in HCT for FA due to the underlying defect in DNA repair. Although radiation-free regimens including busulfan, cyclophosphamide, fludarabine, ATG with the infusion of a T-cell-depleted graft provide excellent outcomes in HCT from allogeneic donors other than MSD [373], the addition of low-dose TBI may be indicated for those patients with clonal evolution or receiving transplantation from a UD due to a higher risk of graft rejection. In addition, MSD must be tested for chromosomal fragility, given that some FA subjects can have nearly normal somatic and haematological phenotype. BM is recommended above PB as HSC source, as PB is an independent risk factor for second malignancies.
Patients with DKC and other inherited BMF syndromes should be transplanted if they have an MSD or a MUD [375,376,377]. A recent large retrospective SAAWP study on allo-HCT for DKC and other telomeropathies showed that pre-transplant organ damage (lung and liver) was associated with poorer outcome [375], supporting thorough organ assessment before HCT. RIC regimens incorporating fludarabine are currently recommended [376, 377]. Potential sibling donors should be tested for telomere length and for mutations of gene of the telomerase-shelterin complex to rule out alterations despite normal somatic and haematologic phenotype. Patients with Blackfan–Diamond anaemia with a MSD should be transplanted if they do not respond to steroids. If an MSD is not available, allo-HCT may be performed with a MUD in experienced centres [378]. Discussion with a specialist centre is advised regarding possible HCT in patients with constitutional BMF.
Autoimmune diseases (AD)
Autologous and allo-HCT may be considered clinical options for children and adolescents with AD [266, 293, 296, 379]. Overall, given the overlap between autoimmune, autoinflammatory conditions, and inborn errors of immunity (IEI) in the paediatric age group, appropriate specialist expertise in diagnostics is essential (such as NGS, WGS/exome sequencing) and in evaluating alternative treatment options when selecting patients for HCT. Special consideration should be given to AD that remains refractory to multiple lines of conventional and disease-modifying treatments, for which allo-HCT might represent the final opportunity for disease control and cure [296].
Auto-HCT may be considered for carefully selected subpopulations of patients with juvenile inflammatory arthritis (e.g. polyarticular or systemic course or onset, inadequate response and/or intolerance to prednisone or disease-modifying antirheumatic drugs) and other ADs including SSc, SLE, vasculitis and polymyositis-dermatomyositis. Paediatric MS is a rare indication for auto-HCT, and long- term responses have been reported [368].
Crohn’s disease is a potential indication for auto-HCT. However, there should be careful consideration of monogenic forms of inflammatory bowel disease (e.g. IL-10 signalling defects, immunodysregulation polyendocrinopathy enteropathy X-linked—IPEX [369]—syndrome, chronic granulomatous disease or increasingly X-linked inhibitor of apoptosis—XIAP–deficiency [300], which are IEI for which allo-HCT is appropriate. Auto-HCT and allo-HCT have both been performed in severe autoimmune cytopenias, with similar outcomes [380]. Allo-HCT may also result in long-term responses in severe juvenile inflammatory arthritis [381]. A recent retrospective EBMT study reported the long-term outcome of allo-HCT in various haematological and non-haematological severe AD, including also paediatric patients [368]. Better transplant outcomes have been reported for patients <18 years and in more recent years of transplant.
Inherited diseases
Allo-HCT for inherited conditions is most commonly used in the paediatric and teenage and young adult populations, though an increased number of older patients require assessment and treatment. Genetically modified auto-HCT has become available for few diseases and is being explored in others with promising results.
Inborn errors of immunity (IEI)
IEIs are an expanding group of over 500 genetic disorders often involving susceptibility to infections, immune dysregulation related manifestations such as autoimmunity and autoinflammation [382]. Patients may experience chronic lymphoproliferation or cancers. Allo-HCT offers curative options for adaptive immune defects and in specific innate immune deficiencies. The decision to proceed to HCT depends on factors like immunological parameters, disease severity, organ damage, HLA matching, and family willingness [383]. Early intervention is critical to prevent damage from the disease or treatments [384,385,386]. A thorough, multidisciplinary evaluation is essential for optimal treatment planning.
T-cell immunity defects are among the most serious conditions, often making allo-HCT necessary. Severe combined immunodeficiency (SCID) represents the most critical forms, typically leading to an early mortality unless HCT, or in some instances, gene therapy is initiated during infancy. Non-SCID-T cell deficiencies are varied, resulting in a broad spectrum of symptoms and severity. For the most severe conditions, such as MHC class II deficiency [387], Wiskott-Aldrich syndrome, DOCK8 deficiencies [388], CD40 ligand deficiency [389] among others, allo-HCT has to be strongly considered early in life, particularly when a suitable HLA-identical donor is available.
Advances in haploidentical transplants, including selective in vitro graft depletion (e.g. TCRab/CD19) [390,391,392] or T-replete graft with in vivo depletion with PTCy have further broaden the indications for transplantation [393, 394]. Feasibility has been shown with both approaches in the field of IEIs [386].
Primary immune regulation disorders have been increasingly recognised and are characterised by diverse auto-immune and auto-inflammatory features. While allo-HCT can be an option, decision to go to transplant, and determining the appropriate timing remain complex. Control of the inflammation as much as possible prior to the procedure is crucial. The place of emerging biotherapies and targeted immunosuppressive drugs will need to be clarified, either as an alternative to transplantation or as a bridge to the procedure [386, 395,396,397].
Primary HLH, regardless of the underlying genetic cause is a clear indication for allo-HCT, including alternative donors. Achieving disease remission prior to allo-HCT plays a critical role in improving overall survival. In certain cases, pre-emptive HCT, before the onset of any HLH-related symptoms, can be considered [398, 399].
Among innate immune deficiencies, complete leucocyte adhesion deficiency is a straightforward indication of HCT [400]. In chronic granulomatous disease, the improved outcomes following HCT have expanded the HCT indication, particularly when an HLA-matched donor is available [401].
Stem cell gene therapy has been pioneered in X-linked and ADA-SCID, leading to EMA licensing of the gammaretroviral product Strimvelis® [402], while excellent results have recently been reported with a lentivirus-based approach [402, 403]. The most recent lentiviral gene therapy studies in other IEI have provided encouraging results and positioning of this alternative curative treatment option is expected in the next years [404,405,406].
However, accessibility to these innovative therapies for rare or ultra-rare conditions remains challenging [407, 408].
Inborn errors of metabolism (IEM)
Allo-HCT is effective in selected patients with peroxisomal diseases (PSD), lysosomal storage diseases (LSD), and some other IEMs [409]. Among LSD, the most successful application of this approach is seen in in type I mucopolysaccharidosis (or Hurler’s syndrome), a severe, multi-system disorder that progressively affects neurocognitive function. Engraftment of donor-derived myeloid cells, including microglia, provides the missing enzyme to the recipient. Early intervention (before the age of 2) with a wild type donor, and a tailored busulfan-based conditioning regimen to reach full donor chimerism achieve the best results. Cord blood and bone marrow are both eligible as HSC source. HCT can mitigate disease progression but does not completely eliminate long-term disease manifestations.
In metachromatic leukodystrophy, a disorder characterised by central and peripheral demyelination, allo-HCT is ineffective in early infantile disease but can have a beneficial impact when applied in juvenile or adult forms, provided the intervention occurs early in the disease’s progression. Allo-HCT also has a role in other LSD, such as MPSII, MPSVII and Wolman disease, but given the rarity of these later conditions, a thorough assessment by an expert multidisciplinary team is crucial.
Among PSD, allo-HCT is effective in preventing disease progression in childhood, (or in rare instances adult) forms of X-linked adrenoleukodystrophy (X-ALD), a cerebral inflammatory disease. Allo-HCT is not recommended for individuals with advanced disease but transplant is offered at the earliest sign of disease.
Gene therapy using gene-corrected autologous HSC holds considerable promise in treating IEMs by facilitating supra-physiological enzyme production [410]. This approach is now an established treatment for late-infantile metachromatic leukodystrophy [411]. A phase I-II trial has been completed in MPS IH, with very promising results in the short and medium term [412, 413] while this approach is currently being investigated in MPS II and MPS IIIA. Lentiviral GT product has also been approved by the EMA for X-ALD [414] but the risk of this product needs to be carefully weighted since a significant risk of MDS has been reported, related to clonal vector insertions within oncogenes [415].
Osteopetrosis (OP)
OP is a heterogeneous genetic disease related to several gene defects that impair bone resorption. Urgent HCT is indicated after exclusion of neurodegenerative and osteoclast-extrinsic defects. Atypical, delayed disease may occur, for whom HCT may be considered in case of haematological insufficiency or imminent visual impairment [416].
Haemoglobinopathies
Haemoglobinopathies are non-malignant diseases and have prolonged survival with modern medical therapy, albeit life-long, complex and expensive and is ‘standard-of-care’ only in limited parts of the world, different to where hemoglobinopathies are most prevalent. Importantly, allo-HCT for haemoglobinopathies should be performed early in life to reduce disease related complications like irreversible damage due to iron overload in patients with transfusion dependent thalassaemia (TDT) and systemic vasculopathy in patients with Sickle cell Disease (SCD).
Moreover, since TDT and SCD present specific clinical characteristics potentially leading to unexpected and disease-specific complications, transplantation (particularly experimental approaches) should only be performed in highly experienced centres [325, 417].
Transfusion-dependent thalassaemia (TDT)
TDT can today benefit from optimal transfusion and iron chelation therapy permitting, is consistent long-life, a prolonged survival until adulthood. However, transfusion consequences and iron toxicity are not completely avoidable [418].
For patients without an MSD, a transplant from a MUD can obtain similar results and is therefore a practicable clinical option [323, 325, 419, 420].
HCT in TDT from haploidentical related donors is now increasingly performed as a clinical option in experienced centres with outstanding results [421,422,423].
Sickle cell disease (SCD)
Although SCD standard clinical care improved over recent years, current management relies on lifelong use of palliative short-term and long-term therapies and survival and quality of life remain significantly reduced. For this reason, allo-HCT from a MSD is standard of care and should be offered prior to the emergence of serious complications [325, 424].
Recently, hemoglobinopathies registry data demonstrated improved results in the last years with a probability of success clearly exceeding 90% of cases [326]. The general theme that MSD and MUD produce overlapping OS and event-free survival has not been reproduced in SCD, where HCT from MUD is inferior to MSD with a significant decline in outcome [425]. Recent reports demonstrated successful outcomes with haploidentical transplantation [417, 426] and alternative family donors are considered a clinical option using either PTCy or T-cell-depleted strategies.
Gene therapy in haemaoglobinopathies
CRISPR-Cas9 gene-editing approaches have been recently approved by FDA and EMA and are currently available in few EU countries for TDT [427] and SCD [428]. A lentiviral-based gene therapy approach for TDT [429] and SCD [430] has received market authorisation but is not available in Europe [431].
Conclusions
For over two decades, the EBMT indications reports have incorporated developments in HCT practice based on scientific and technical developments in HCT. We encourage harmonisation of practice, where possible, to ensure meaningfully aggregated experience across indications via registry outputs. We also recommend working according to JACIE certification standards and participation in benchmarking of survival outcomes to maintain quality in HCT and other cellular therapy practice [1,2,3].
Reporting in the EBMT registry allows the collection of crucial real-world data also in long-term follow up, alongside basic reporting of activity data that is published in EBMT Survey reports [4, 5]. The EBMT is increasingly engaging with the patient community and considering Patient Reported Outcomes as part of data collection [432].
Moving forward, all treatment decisions, whether HCT or non-HCT, need to be considered in conjunction with the risk of the disease, risk of HCT procedure and non-HCT strategies, and impact on both quantity and quality of life. HCTs are evolving in complexity alongside health economic costs associated with their delivery. Local delivery of HCT should be balanced against working in close partnership (‘shared care‘) with more distant HCT centres with more developed facilities and expertise is also an important consideration. The increasing application of CAR-T and other genetically modified cell therapies will require ongoing consideration in clinical decision-making in HCT across the indications.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
References
Saccardi R, Putter H, Eikema DJ, Busto MP, McGrath E, Middelkoop B, et al. Benchmarking of survival outcomes following Haematopoietic Stem Cell Transplantation (HSCT): an update of the ongoing project of the European Society for Blood and Marrow Transplantation (EBMT) and Joint Accreditation Committee of ISCT and EBMT (JACIE). Bone Marrow Transpl. 2023;58:659–66.
Saccardi R, Rintala T, McGrath E, Snowden JA. JACIE Accreditation of HCT Programs. 2024 Apr 11. In: Sureda A, Corbacioglu S, Greco R, Kröger N, Carreras E, editors. The EBMT handbook: hematopoietic cell transplantation and cellular therapies [Internet]. 8th edition. Cham (CH): Springer; 2024. Chapter 5.PMID: 39437072 Free Books & Documents. Review.
Snowden JA, Saccardi R, Orchard K, Ljungman P, Duarte RF, Labopin M, et al. Benchmarking of survival outcomes following haematopoietic stem cell transplantation: A review of existing processes and the introduction of an international system from the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Bone Marrow Transpl. 2020;55:681–94.
Passweg JR, Baldomero H, Chabannon C, Basak GW, De La Cámara R, Corbacioglu S, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transpl. 2021;56:1651–64.
Passweg JR, Baldomero H, Atlija M, Kleovoulou I, Witaszek A, Alexander T, et al. The 2023 EBMT report on hematopoietic cell transplantation and cellular therapies. Increased use of allogeneic HCT for myeloid malignancies and of CAR-T at the expense of autologous HCT. Bone Marrow Transplant [Internet]. 12 de febrero de 2025 [citado 13 de febrero de 2025]; Disponible en: https://www.nature.com/articles/s41409-025-02524-2.
Baldomero H, Neumann D, Hamad N, Atsuta Y, Sureda A, Iida M, et al. The role of registries in hematological disorders. Best Pract Res Clin Haematol. 2024;37:101556.
Snowden JA, Sánchez-Ortega I, Corbacioglu S, Basak GW, Chabannon C, De La Camara R, et al. Indications for haematopoietic cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2022. Bone Marrow Transpl. 2022;57:1217–39
Weisdorf D, Spellman S, Haagenson M, Horowitz M, Lee S, Anasetti C, et al. Classification of HLA-matching for retrospective analysis of unrelated donor transplantation: revised definitions to predict survival. Biol Blood Marrow Transpl. 2008;14:748–58.
Weisdorf D, Cooley S, Wang T, Trachtenberg E, Haagenson MD, Vierra-Green C, et al. KIR donor selection: feasibility in identifying better donors. Biol Blood Marrow Transpl. 2019;25:e28–e32.
Chabannon C, Ruggeri A, Montoto S, Van Biezen A, Van Der Werf S, Markslag A, et al. Celebrating the registration of 9.000 patients treated with CAR T cells in the EBMT registry: Collection of real-world data in the context of hematopoietic cellular therapies. Best Pract Res Clin Haematol. 2024;37:101557.
Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-cell therapy in autoimmune disease—a case series with follow-up. N Engl J Med. 2024;390:687–700.
Greco R, Alexander T, Del Papa N, Muller F, Saccardi R, Sanchez-Guijo F, et al. Innovative cellular therapies for autoimmune diseases: expert-based position statement and clinical practice recommendations from the EBMT practice harmonization and guidelines committee. EClinicalMedicine. 2024;69:102476.
Locatelli F, Cavazzana M, Frangoul H, Fuente JDL, Algeri M, Meisel R. Autologous gene therapy for hemoglobinopathies: from bench to patient’s bedside. Mol Ther. 2024;32:1202–18. mayo de
Penack O, Peczynski C, Boreland W, Wolff D, Moiseev I, Schoemans H, et al. Management of complications of chimeric antigen receptor T-cell therapy: a report by the European Society of Blood and Marrow Transplantation. haematol [Internet]. 30 de mayo de 2024 [citado 4 de marzo de 2025]; Disponible en: https://haematologica.org/article/view/haematol.2023.284810.
Levine BL, Pasquini MC, Connolly JE, Porter DL, Gustafson MP, Boelens JJ, et al. Unanswered questions following reports of secondary malignancies after CAR-T cell therapy. Nat Med 2024;30:338–41. febrero de
Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H. at al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–77.
Canaani J, Labopin M, Itälä-Remes M, Blaise D, Socié G, Forcade E, et al. Prog- nostic significance of recurring chromosomal abnormalities in transplanted patients with acute myeloid leukemia. Leukemia. 2019;33:1944–52.
Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood. 2016;127:62–70.
Moarii M, Papaemmanuil E. Classification and risk assessment in AML: integrating cytogenetics and molecular profiling. Hematol Am Soc Hematol Educ Program 2017;2017:37–44.
Kantarjian H, Kadia T, DiNardo C, Daver N, Borthakur G, Jabbour E, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11:41.
Araki D, Wood BL, Othus M, Radich JP, Halpern AB, Zhou Y, et al. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: time to move toward a minimal residual disease-based definition of complete remission? J Clin Oncol. 2016;34:329–36.
Kim HJ, Kim Y, Kang D, Kim HS, Lee JM, Kim M, et al. Prognostic value of measurable residual disease monitoring by next-generation sequencing before and after allogeneic hematopoietic cell transplantation in acute myeloid leukemia. Blood Cancer J. 2021;11:109.
Fenwarth L, Thomas X, de Botton S, Duployez N, Bourhis JH, Lesieur A, et al. A personalized approach to guide allogeneic stem cell transplantation in younger adults with acute myeloid leukemia. Blood. 2021;137:524–32.
Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allo-HCT. Blood. 2005;106:2912–9.
Armand P, Gibson CJ, Cutler C, Ho VT, Koreth J, Alyea EP, et al. A disease risk index for patients undergoing allogeneic stem cell transplantation. Blood. 2012;120:905–1. Jul
Shouval R, Labopin M, Bondi O, Mishan-Shamay H, Shimoni A, Ciceri F, et al. Prediction of allogeneic hematopoietic stem-cell transplantation mortality 100 days after transplantation using a machine learning algorithm: a European Group for Blood and Marrow Transplantation Acute Leukemia Working Party Retrospective Data Mining Study. J Clin Oncol. 2015;33:3144–51.
NCCN Guidelines Version 2.2025 Acute Myeloid Leukemia. National Comprehensive Cancer Network (NCCN). Available at: https://www.nccn.org.
Versluis J, Cornelissen JJ, Craddock C, Sanz MÁ, Nagler A. AML in adults. In: Sureda A, Corbacioglu S, Greco R, Kröger N, Carreras E, editors. The EBMT handbook: hematopoietic cell transplantation and cellular therapies [Internet]. 8th edition. Cham (CH): Springer, 2024.
Nagler A, Savani BN, Labopin M, Polge E, Passweg J, Finke J, et al. Outcomes after use of two standard ablative regimens in patients with refractory acute myeloid leukaemia: a retrospective, multicentre, registry analysis. Lancet Haematol. 2015;2:e384–92.
Heinicke T, Krahl R, Kahl C, Cross M, Scholl S, Wolf HH, et al. Allogeneic hematopoietic stem cell transplantation improves long-term outcome for relapsed AML patients across all ages: results from two East German Study Group Hematology and Oncology (OSHO) trials. Ann Hematol. 2021;100:2387–98. https://doi.org/10.1007/s00277-021-04565-1. Sep
Gilleece MH, Labopin M, Savani BN, Yakoub-Agha I, Socié G, Gedde-Dahl T, et al. Allogeneic haemopoietic transplantation for acute myeloid leukaemia in second complete remission: a registry report by the Acute Leukaemia Working Party of the EBMT. Leukemia. 2020;34:87–99.
Gooptu M, Romee R, St Martin A, Arora M, Al Malki M, Antin JH, et al. HLA-haploidentical vs matched unrelated donor transplants with posttransplant cyclophosphamide-based prophylaxis. Blood 2021;138:273–82. Jul
Shaw BE, Jimenez-Jimenez AM, Burns LJ, Logan BR, Khimani F, Shaffer BC, et al. J. National Marrow Donor Program-Sponsored Multicenter, Phase II Trial of HLA-mismatched unrelated donor bone marrow transplantation using post-transplant cyclophosphamide. J Clin Oncol. 2021;39:1971–82. Jun
Bolaños-Meade J, Hamadani M, Wu J, Al Malki MM, Martens MJ, Runaas L, et al. Post-transplantation cyclophosphamide-based graft-versus-host disease prophylaxis. N Engl J Med. 2023;388:2338–48. de junio de
Scott BL, Pasquini MC, Logan BR, Wu J, Devine SM, Porter DL, et al. Myeloablative versus reduced-intensity hematopoietic cell transplantation for acute myeloid leukemia and myelodysplastic syndromes. J Clin Oncol. 2017;35:1154–61. Apr
Scott BL, Pasquini MC, Fei M, Fraser R, Wu J, Devine SM, et al. Myeloablative versus reduced-intensity conditioning for hematopoietic cell transplantation in acute myelogenous leukemia and myelodysplastic syndromes-long-term follow-up of the BMT CTN 0901 clinical trial. Transpl Cell Ther. 2021;27:483.e1–483.e6. Jun
Paras G, Morsink LM, Othus M, Milano F, Sandmaier BM, Zarling LC, et al. Conditioning intensity and peritransplant flow cytometric MRD dynamics in adult AML. Blood. 2022;139:1694–706. Mar
Schetelig J, Schaich M, Schäfer-Eckart K, Hänel M, Aulitzky WE, Einsele H, et al. Hematopoietic cell transplantation in patients with intermediate and high-risk AML: results from the randomized Study Alliance Leukemia (SAL) AML 2003 trial. Leukemia. 2015;29:1060–8.
Rücker FG, Agrawal M, Corbacioglu A, Weber D, Kapp-Schwoerer S, Gaidzik VI, et al. Measurable residual disease monitoring in acute myeloid leukemia with t (8;21)(q22;q22.1): results from the AML Study Group. Blood 2019;134:1608–18.
Jourdan E, Boissel N, Chevret S, Delabesse E, Renneville A, Cornillet P, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121:2213–23.
Ivey A, Hills RK, Simpson MA, Jovanovic JV, Gilkes A, Grech A, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374:422–33.
Hoelzer D, Bassan R, Dombret H, Fielding A, Ribera JM, Buske C, ESMO Guidelines Committee. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27:v69–v82. Sep
Giebel S, Labopin M, Socié G, Beelen D, Browne P, Volin L, et al. Improving results of allogeneic hematopoietic cell transplantation for adults with acute lymphoblastic leukemia in first complete remission: an analysis from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. 2017;102:139–49.
Nagler A, Labopin M, Koc Y, Angelucci E, Tischer J, Arat M, et al. Outcome of T-cell-replete haploidentical stem cell transplantation improves with time in adults with acute lymphoblastic leukemia: a study from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Cancer. 2021;127:2507–14.
Gökbuget N, Boissel N, Chiaretti S, Dombret H, Doubek M, Fielding A, et al. Management of ALL in adults: 2024 ELN recommendations from a European expert panel. Blood 2024;143: 1903–30.
Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189–97.
Pullarkat V, Slovak ML, Kopecky KJ, Forman SJ, Appelbaum FR. Impact of cytogenetics on the outcome of adult acute lymphoblastic leukemia: results of Southwest Oncology Group 9400 study. Blood. 2008;111:2563–72.
Lazaryan A, Dolan M, Zhang MJ, Wang HL, Kharfan-Dabaja MA, Marks DI, et al. Impact of cytogenetic abnormalities on outcomes of adult Philadelphia-negative acute lymphoblastic leukemia after allogeneic hematopoietic stem cell transplantation: a study by the Acute Leukemia Working Committee of the Center for International Blood and Marrow Transplant Research. Haematologica. 2021;106:2295–6.
Giebel S, Marks DI, Boissel N, Baron F, Chiaretti S, Ciceri F, et al. Hematopoietic stem cell transplantation for adults with Philadelphia chromosome-negative acute lymphoblastic leukemia in first remission: a position statement of the European Working Group for Adult Acute Lymphoblastic Leukemia (EWALL) and the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transpl. 2019;54:798–809.
Gupta V, Richards S, Rowe J. Allogeneic, but not autologous, hematopoietic cell transplantation improves survival only among younger adults with acute lymphoblastic leukemia in first remission: an individual patient data meta-analysis. Blood. 2013;121:339–50.
Pidala J, Djulbegovic B, Anasetti C, Kharfan-Dabaja M, Kumar A. Allogeneic hematopoietic cell transplantation for adult acute lymphoblastic leukemia (ALL) in first complete remission. Cochrane Database Syst Rev. 2011;2011:Cd008818.
Gorin NC, Giebel S, Labopin M, Savani BN, Mohty M, Nagler A. Autologous stem cell transplantation for adult acute leukemia in 2015: time to rethink? Present status and future prospects. Bone Marrow Transpl. 2015;50:1495–502.
Brissot E, Labopin M, Beckers MM, Socié G, Rambaldi A, Volin L, et al. Tyrosine kinase inhibitors improve long-term outcome of allogeneic hematopoietic stem cell transplantation for adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia. Haematologica. 2015;100:392–9.
Jabbour E, Short NJ, Ravandi F, Huang X, Daver N, DiNardo CD, et al. Combi- nation of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018;5:e618–e27.
Foà R, Bassan R, Vitale A, Elia L, Piciocchi A, Puzzolo MC, et al. Dasatinib- blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. 2020;383:1613–23.
Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK, Chen K, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood. 2017;129:572–81.
Brissot E, Labopin M, Russo D, Martin S, Schmid C, Glass B, et al. Alternative donors provide comparable results to matched unrelated donors in patients with acute lymphoblastic leukemia undergoing allogeneic stem cell transplantation in second complete remission: a report from the EBMT Acute Leukemia Working Party. Bone Marrow Transpl. 2020;55:1763–72.
Pavlů J, Labopin M, Zoellner AK, Sakellari I, Stelljes M, Finke J, et al. Allogeneic hematopoietic cell transplantation for primary refractory acute lymphoblastic leukemia: a report from the Acute Leukemia Working Party of the EBMT. Cancer. 2017;123:1965–70.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisa- genlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–48.
Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE- X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398:491–502.
Roddie C, Sandhu KS, Tholouli E, Logan AC, Shaughnessy P, Barba P, et al. Obecabtagene autoleucel in adults with B-cell acute lymphoblastic leukemia. N Engl J Med. 2024;391:2219–30. de diciembre de
Mohty M, Gautier J, Malard F, Aljurf M, Bazarbachi A, Chabannon C, et al. CD19 chimeric antigen receptor-T cells in B-cell leukemia and lymphoma: current status and perspectives. Leukemia. 2019;33:2767–78.
Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–63.
Zhang X, Lu XA, Yang J, Zhang G, Li J, Song L, et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. 2020;4:2325–38.
Pillai V, Muralidharan K, Meng W, Bagashev A, Oldridge DA, Rosenthal J, et al. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv. 2019;3:3539–49.
Dholaria B, Savani BN, Huang XJ, Nagler A, Perales MA, Mohty M. The evolving role of allogeneic haematopoietic cell transplantation in the era of chimaeric antigen receptor T-cell therapy. Br J Haematol. 2021;193:1060–75.
Gökbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131:1522–31. https://doi.org/10.1182/blood-2017-08-798322. Apr
Gökbuget N, Zugmaier G, Dombret H, Stein A, Bonifacio M, Graux C, et al. Curative outcomes following blinatumomab in adults with minimal residual disease B-cell precursor acute lymphoblastic leukemia. Leuk Lymphoma. 2020;61:2665–73. Nov
Peters C, Dalle JH, Locatelli F, Poetschger U, Sedlacek P, Buechner J.IBFM Study Group;; Arend von Stackelberg; IntReALL Study Group; Adriana Balduzzi; I-BFM SCT Study Group; Selim Corbacioglu; EBMT Paediatric Diseases Working Party; Peter Bader et al. Total body irradiation or chemotherapy conditioning in childhood ALL: a multinational, randomized, noninferiority phase III study. J Clin Oncol. 2021;39:295–307. https://doi.org/10.1200/JCO.20.02529. Feb.
Kebriaei P, Anasetti C, Zhang MJ, Wang HL, Aldoss I, de Lima M.Acute Leukemia Committee of the CIBMTR et al. Intravenous busulfan compared with total body irradiation pretransplant conditioning for adults with acute lymphoblastic leukemia. Biol Blood Marrow Transpl. 2018;24:726–33. Apr.
Eder S, Canaani J, Beohou E, Labopin M, Sanz J, Arcese W, et al. Thiotepa-based conditioning versus total body irradiation as myeloablative conditioning prior to allogeneic stem cell transplantation for acute lymphoblastic leukemia: a matched-pair analysis from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Am J Hematol. 2017;92:997–1003. Oct
Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114:5136–45.
Jain N, Lamb AV, O’Brien S, Ravandi F, Konopleva M, Jabbour E, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127:1863–9.
Petit A, Trinquand A, Chevret S, Ballerini P, Cayuela JM, Grardel N, et al. Onco- genetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood. 2018;131:289–300.
Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/ FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113:3918–24.
Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34:966–84. Apr
Shah NP, Bhatia R, Altman JK, Amaya M, Begna KH, Berman E, et al. Chronic Myeloid Leukemia, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw. 2024;22:43–69. Feb
Chalandon Y, Sbianchi G, Gras L, Koster L, Apperley J, Byrne J, et al. Allogeneic hematopoietic cell transplantation in patients with chronic phase chronic myeloid leukemia in the era of third generation tyrosine kinase inhibitors: A retrospective study by the chronic malignancies working party of the EBMT. Am J Hematol. 2023;98:112–21. Jan
Réa D, Mauro MJ, Boquimpani C, Minami Y, Lomaia E, Voloshin S, et al. A phase 3, open-label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood. 2021;138:2031–41. Nov
Cortes J, Apperley J, Lomaia E, Moiraghi B, Undurraga Sutton M, Pavlovsky C, et al. Ponatinib dose-ranging study in chronic-phase chronic myeloid leukemia: a randomized, open-label phase 2 clinical trial. Blood. 2021;138:2042–50. Nov
Wang W, Cortes JE, Tang G, Khoury JD, Wang S, Bueso-Ramos CE, et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood. 2016;127:2742–50. Jun
Brioli A, Lomaia E, Fabisch C, Sacha T, Klamova H, Morozova E, et al. Management and outcome of patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era - analysis of the European LeukemiaNet Blast Phase Registry. Leukemia. 2024;38:1072–80. May
Ortí G, Gras L, Koster L, Kulagin A, Byrne J, Apperley JF, et al. Graft-versus-host disease prophylaxis with post-transplantation cyclophosphamide in chronic myeloid leukemia patients undergoing allogeneic hematopoietic cell transplantation from an unrelated or mismatched related donor: a comparative study from the chronic malignancies working party of the EBMT (CMWP-EBMT). Transpl Cell Ther. 2024;30:93.e1–93.e12. Jan
Onida F, Gras L, Ge J, Koster L, Hamladji RM, Byrne J, et al. Mismatched related donor allogeneic haematopoietic cell transplantation compared to other donor types for Ph+ chronic myeloid leukaemia: a retrospective analysis from the Chronic Malignancies Working Party of the EBMT. Br J Haematol. 2024;204:2365–77. Jun
Ortí G, Gras L, Zinger N, Finazzi MC, Sockel K, Robin M, et al. Outcomes after allogeneic hematopoietic cell transplant in patients diagnosed with blast phase of myeloproliferative neoplasms: A retrospective study from the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Am J Hematol. 2023;98:628–38.
Kröger N, Holler E, Kobbe G, Bornhäuser M, Schwerdtfeger R, Baurmann H, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 2009;114:5264–70.
McLornan DP, Sirait T, Hernández-Boluda JC, Czerw T, Hayden P, Yakoub-Agha I. European-wide survey on allogeneic haematopoietic cell transplantation practice for myelofibrosis on behalf of the EBMT chronic malignancies working party. Curr Res Transl Med. 2021;69:103267.
Kröger N, Bacigalupo A, Barbui T, Ditschkowski M, Gagelmann N, et al. Indication and management of allogeneic haematopoietic stem-cell transplantation in myelofibrosis: updated recommendations by the EBMT/ELN International Working Group. Lancet Haematol. 2024;11:e62–e74. Jan
Gagelmann N, Ditschkowski M, Bogdanov R, Bredin S, Robin M, Cassinat B, et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood. 2019;133:2233–42.
Polverelli N, Mauff K, Kroger N, et al. Impact of spleen size and splenectomy on outcomes of allogeneic hematopoietic cell transplantation for myelofibrosis: a retrospective analysis by the chronic malignancies working party on behalf of European Society for Blood and Marrow Transplantation (EBMT). Am J Hematol. 2021;96:69–79.
Kröger N, Sbianchi G, Sirait T, Wolschke C, Beelen D, Passweg J, et al. Impact of prior JAK-inhibitor therapy with ruxolitinib on outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis: a study of the CMWP of EBMT. Leukemia. 2021;35:3551–60.
Gagelmann N, Eikema DJ, Stelljes M, Beelen D, de Wreede L, Mufti G, et al. Optimized EBMT transplant-specific risk score in myelodysplastic syndromes after allogeneic stem-cell transplantation. Haematologica. 2019;104:929–36.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88. 15 de marzo de
Robin M, Porcher R, Adès L, Raffoux E, Michallet M, François S, et al. HLA-matched allogeneic stem cell transplantation improves outcome of higher risk myelodysplastic syndrome A prospective study on behalf of SFGM-TC and GFM. Leukemia. 2015;29:1496–501. julio de
Kröger N, Sockel K, Wolschke C, Bethge W, Schlenk RF, Wolf D, et al. Comparison between 5-azacytidine treatment and allogeneic stem-cell transplantation in elderly patients with advanced MDS according to donor availability (VidazaAllo study). J Clin Oncol. 2021;39:3318–27. 20 de octubre de
Nakamura R, Saber W, Martens MJ, Ramirez A, Scott B, Oran B, et al. Biologic assignment trial of reduced-intensity hematopoietic cell transplantation based on donor availability in patients 50-75 years of age with advanced myelodysplastic syndrome. J Clin Oncol. 2021;39:3328–39. 20 de octubre de
Tentori CA, Gregorio C, Robin M, Gagelmann N, Gurnari C, Ball S, et al. Clinical and genomic-based decision support system to define the optimal timing of allogeneic hematopoietic stem-cell transplantation in patients with myelodysplastic syndromes. J Clin Oncol. 2024;42:2873–86. 20 de agosto de
DeFilipp Z, Ciurea SO, Cutler C, Robin M, Warlick ED, Nakamura R, et al. Hematopoietic cell transplantation in the management of myelodysplastic syndrome: an evidence-based review from the American Society for Transplantation and Cellular Therapy Committee on Practice Guidelines. Transpl Cell Ther. 2023;29:71–81. febrero de
Scheid C, Eikema DJ, van Gelder M, Salmenniemi U, Maertens J, Passweg J, et al. Does IPSS-R downstaging before transplantation improve the prognosis of patients with myelodysplastic neoplasms? Blood. 2024;144:445–56. de julio de
Onida F, Gagelmann N, Chalandon Y, Kobbe G, Robin M, Symeonidis A, et al. Management of adult patients with CMML undergoing allo-HCT: recommendations from the EBMT PH&G Committee. Blood. 2024;143:2227–44. May
Elena C, Galli A, Such E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128:1408–17.
Munir T, Cairns DA, Bloor A, Allsup D, Cwynarski K, Pettitt A, et al. National cancer research institute chronic lymphocytic leukemia subgroup. chronic lymphocytic leukemia therapy guided by measurable residual disease. N Engl J Med. 2024;390:326–37.
Aronson JH, Skånland SS, Roeker LE, Thompson MC, Mato AR. Approach to a patient with «double refractory» chronic lymphocytic leukemia: «double, double toil and trouble» (Shakespeare). Am J Hematol. 2022;97:S19–S25.
Cwynarski K, van Biezen A, de Wreede L, Stilgenbauer S, Bunjes D, Metzner B, et al. Autologous and allogeneic stem-cell transplantation for transformed chronic lymphocytic leukemia (Richter’s syndrome): a retrospective analysis from the chronic lymphocytic leukemia subcommittee of the chronic leukemia working party and lymphoma working party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2012;30:2211–7.
Gisselbrecht C, Schmitz N, Mounier N, Singh Gill D, Linch DC, Trneny M, et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol. 2012;30:4462–9.
Locke FL, Jacobson C, Perales MA, Kersten MJ, Westin JR. Primary analysis of ZUMA‑7: a phase 3 randomized trial of axicabtagene ciloleucel (Axi-Cel) versus standard‑of‑care therapy in patients with relapsed/refractory large B-cell lymphoma. Blood. 2021;138:2.
Kamdar M, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet. 2022;399:2294–308. Jun
Neelapu SS, Jacobson CA, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. 2023;141:2307–15. May
Shadman M, Ahn KW, Kaur M, Lekakis L, Beitinjaneh A, Iqbal M, et al. Autologous transplant vs. CAR-T therapy in patients with DLBCL treated while in complete remission. Blood Cancer J. 2024;14:108. Jul
Bachy E, Le Gouill S, Di Blasi R, Sesques P, Manson G, Cartron G, et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat Med. 2022;28:2145–54. Oct
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44. https://doi.org/10.1056/NEJMoa1707447. DecEpub 2017 Dec 10
Glass B, Hasenkamp J, Wulf G, Dreger P, Pfreundschuh M, Gramatzki M, et al. Rituximab after lymphoma-directed conditioning and allogeneic stem-cell transplantation for relapsed and refractory aggressive non-Hodgkin lymphoma (DSHNHL R3): an open-label, randomised, phase 2 trial. Lancet Oncol. 2014;15:757–66.
van Kampen RJ, Canals C, Schouten HC, Nagler A, Thomson KJ, Vernant JP, et al. Allogeneic stem-cell transplantation as salvage therapy for patients with diffuse large B-cell non-Hodgkin’s lymphoma relapsing after an autologous stem-cell transplantation: an analysis of the European Group for Blood and Marrow Transplantation Registry. J Clin Oncol. 2011;29:1342–8.
Fenske TS, Ahn KW, Graff TM, DiGilio A, Bashir Q, Kamble RT, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol. 2016;174:235–48.
Dodero A, Patriarca F, Milone G, Sarina B, Miceli R, Iori A, et al. Allogeneic stem cell transplantation for relapsed/refractory B cell lymphomas: results of a multicenter Phase II Prospective Trial including Rituximab in the Reduced-Intensity Conditioning Regimen. Biol Blood Marrow Transpl. 2017;23:1102–9.
Fried S, Shouval R, Walji M, Flynn JR, Yerushalmi R, Shem-Tov N, et al. Allogeneic hematopoietic cell transplantation after chimeric antigen receptor T cell therapy in large B cell lymphoma. Transpl Cell Ther. 2023;29:99–107. Feb
Derigs P, Bethge WA, Krämer I, Holtick U, von Tresckow B, Ayuk F.German Lymphoma Alliance and the German Registry for Stem Cell Transplantation et al. Long-term survivors after failure of chimeric antigen receptor t cell therapy for large b cell lymphoma: a role for allogeneic hematopoietic cell transplantation? a german lymphoma alliance and german registry for stem cell transplantation analysis. Transpl Cell Ther. 2023;29:750–6. Dec.
Zurko J, et al. Allogeneic transplant following CAR T-cell therapy for large B-cell lymphoma. Haematologica. 2023;108:98–109. https://doi.org/10.3324/haematol.2022.281242. Jan
Herrera AF, Mei M, Low L, Kim HT, Griffin GK, Song JY, et al. Relapsed or refractory double-expressor and double-hit lymphomas have inferior progression-free survival after autologous stem-cell transplantation. J Clin Oncol. 2017;35:24–31.
Le Gouill S, et al. Obinutuzumab vs rituximab for advanced DLBCL: a PET-guided and randomized phase 3 study by LYSA. Blood. 2021;137:2307–20. https://doi.org/10.1182/blood.2020008750
Ferreri AJM, Cwynarski K, Pulczynski E, Fox CP, Schorb E, La Rosee P, et al. Wholebrain radiotherapy or autologous stem-cell transplantation as consolidation strategies after high-dose methotrexate-based chemoimmunotherapy in patients with primary CNS lymphoma: results of the second randomisation of the International Extranodal Lymphoma Study Group-32 phase 2 trial. Lancet Haematol. 2017;4:e510–e23.107.
Ferreri AJ, Illerhaus G. The role of autologous stem cell transplantation in primary central nervous system lymphoma. Blood. 2016;127:1642–9.
Houillier C, Taillandier L, Dureau S, Lamy T, Laadhari M, Chinot O, et al. Radiotherapy or autologous stem-cell transplantation for primary CNS lymphoma in patients 60 years of age and younger: results of the intergroup ANOCEFGOELAMS randomized phase II PRECIS study. J Clin Oncol. 2019;37:823–33.
Illerhaus G, Ferreri A, Binder M, Borchmann P, Hasenkamp J, Stilgenbauer S, et al. Consolidative HCT‐ASCT is superior to non‐myeloablative chemo-immunotherapy in newly‐diagnosed pcnsl ‐ updated results of the randomized phase III Matrix/IELSG43 trial. In Proceedings of the 17th ICML, abstract 015, Lugano, 13–17 June 2023.
Schorb E, et al. High-dose chemotherapy and autologous haematopoietic stem-cell transplantation in older, fit patients with primary diffuse large B-cell CNS lymphoma (MARTA): a single-arm, phase 2 trial. Lancet Haematol. 2024;11:e196–e205. https://doi.org/10.1016/S2352-3026(23)00371-X
Soussain C, Suzan F, Hoang-Xuan K, et al. Results of intensive chemotherapy followed by hematopoietic stem-cell rescue in 22 patients with refractory or recurrent primary CNS lymphoma or intraocular lymphoma. J Clin Oncol. 2001;19:742–9.
Soussain C, Hoang-Xuan K, Taillandier L, et al. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: Societe francaise de greffe de moelle osseuse-therapie cellulaire. J Clin Oncol. 2008;26:2512–8.
Choquet S, Soussain C, Azar N, Morel V, Metz C, Ursu R, et al. CAR T-cell therapy induces a high rate of prolonged remission in relapsed primary CNS lymphoma: Real-life results of the LOC network. Am J Hematol. 2024;99:1240–9.
Nayak L, Chukwueke UN, Meehan C, Redd R, Hogan S, Lee EQ, et al. A pilot study of axicabtagene ciloleucel (axi-cel) for relapsed /refractory primary and secondary central nervous system lymphoma (PCNSL and SCNSL) Presentation at: ASCO24, 30 May–03 June, 2024; Chicago, Illinois, & Online. Abstract 2006.
Frigault MJ, Dietrich J, Gallagher K, Roschewski M, Jordan JT, Forst D, et al. Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: a phase 1/2 clinical trial. Blood. 2022;139:2306–15.
Bachy E, Seymour JF, Feugier P, Offner F, López-Guillermo A, Belada D, et al. Sustained progression-free survival benefit of rituximab maintenance in patients with follicular lymphoma: long-term results of the PRIMA study. JCO. 2019;37:2815–24. de noviembre de
Casulo C, Friedberg JW, Ahn KW, Flowers C, DiGilio A, Smith SM, et al. Autologous transplantation in follicular lymphoma with early therapy failure: a national LymphoCare study and center for international blood and marrow transplant research analysis. Biol Blood Marrow Transpl. 2018;24:1163–71. junio de
Kim TM, Taszner M, Novelli S, Cho SG, Villasboas JC, Merli M, et al. Safety and efficacy of odronextamab in patients with relapsed or refractory follicular lymphoma. Annals of Oncology [Internet]. 13 de agosto de 2024 [citado 8 de octubre de 2024];0(0). Disponible en: https://www.annalsofoncology.org/article/S0923-7534(24)03759-1/fulltext
Linton KM, Vitolo U, Jurczak W, Lugtenburg PJ, Gyan E, Sureda A, et al. Epcoritamab monotherapy in patients with relapsed or refractory follicular lymphoma (EPCORE NHL-1): a phase 2 cohort of a single-arm, multicentre study. Lancet Haematol. 2024;11:e593–605. de agosto de
Budde LE, Sehn LH, Matasar M, Schuster SJ, Assouline S, Giri P, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 2022;23:1055–65. de agosto de
Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23:91–103. de enero de
Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med 2022;28:325–32. febrero de
Morschhauser F, Dahiya S, Palomba ML, Garcia-Sancho AM, Ortega JLR, Kuruvilla J, et al. Transcend Fl: phase 2 study results of lisocabtagene maraleucel (liso-Cel) in patients (pts) with relapsed/refractory (r/R) follicular lymphoma (fl). Hematol Oncol. 2023;41:877–80.
Robinson SP, Canals C, Luang JJ, Tilly H, Crawley C, Cahn JY, et al. The outcome of reduced intensity allogeneic stem cell transplantation and autologous stem cell transplantation when performed as a first transplant strategy in relapsed follicular lymphoma: an analysis from the Lymphoma Working Party of the EBMT. Bone Marrow Transpl. 2013;48:1409–14. noviembre de
Sureda A, Zhang MJ, Dreger P, Carreras J, Fenske T, Finel H, et al. Allogeneic hematopoietic stem cell transplantation for relapsed follicular lymphoma: a combined analysis on behalf of the Lymphoma Working Party of the EBMT and the Lymphoma Committee of the CIBMTR. Cancer. 2018;124:1733–42. 15 de abril de
Mussetti A, Kanate AS, Wang T, He M, Hamadani M, Finel H, et al. Haploidentical versus matched unrelated donor transplants using post-transplantation cyclophosphamide for lymphomas. Transpl Cell Ther. 2023;29:184.e1–184.e9. marzo de
Kastritis E, Gavriatopoulou M, Kyrtsonis MC, Roussou M, Hadjiharissi E, Symeonidis A, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: final analysis of a phase 2 study. Blood. 2015;126:1392–4. 10 de septiembre de
Rummel MJ, Niederle N, Maschmeyer G, Banat GA, Von Grünhagen U, Losem C, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381:1203–10. abril de
Rummel M, Kaiser U, Balser C, Stauch M, Brugger W, Welslau M, et al. Bendamustine plus rituximab versus fludarabine plus rituximab for patients with relapsed indolent and mantle-cell lymphomas: a multicentre, randomised, open-label, non-inferiority phase 3 trial. Lancet Oncol. 2016;17:57–66. enero de
Dimopoulos MA, García-Sanz R, Gavriatopoulou M, Morel P, Kyrtsonis MC, Michalis E, et al. Primary therapy of Waldenström macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood. 2013;122:3276–82. de noviembre de
Treon SP, Tripsas CK, Meid K, Kanan S, Sheehy P, Chuma S, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström’s macroglobulinemia. Blood. 2014;124:503–10. de julio de
Castillo JJ, Meid K, Flynn CA, Chen J, Demos MG, Guerrera ML, et al. Ixazomib, dexamethasone, and rituximab in treatment-naive patients with Waldenström macroglobulinemia: long-term follow-up. Blood Adv. 2020;4:3952–9. de agosto de
Castillo JJ, Meid K, Gustine JN, Leventoff C, White T, Flynn CA, et al. Long-term follow-up of ibrutinib monotherapy in treatment-naive patients with Waldenstrom macroglobulinemia. Leukemia. 2022;36:532–9. febrero de
Dimopoulos MA, Opat S, D’Sa S, Jurczak W, Lee HP, Cull G, et al. Zanubrutinib versus ibrutinib in symptomatic waldenström macroglobulinemia: final analysis from the randomized phase III ASPEN study. JCO. 2023;41:5099–106. de noviembre de
Dreyling M, Doorduijn J, Gine E, et al. Ibrutinib combined with immunochemotherapy with or without autologous stem-cell transplantation versus immunochemotherapy and autologous stem-cell transplantation in previously untreated patients with mantle cell lymphoma (TRIANGLE): a three-arm, randomised, open-label, phase 3 superiority trial of the European Mantle Cell Lymphoma Network. Lancet. 2024;403:2293–306.
Munshi PN, Hamadani M, Kumar A, et al. ASTCT, CIBMTR, and EBMT clinical practice recommendations for transplant and cellular therapies in mantle cell lymphoma. Bone Marrow Transpl. 2021;56:2911–21.
Liebers N, Finel H, Edelmann D, et al. A propensity score-matched analysis on the outcomes of brexucabtagene autoleucel from Zuma-2 study and allogeneic stem cell transplantation from the EBMT database in relapsed and refractory Post-BTKi mantle cell lymphoma [abstract]. Blood (ASH Annu Meet Abstr). 2023;142:3494.
Schmitz N, Truemper L, Bouabdallah K, et al. A randomized phase 3 trial of autologous vs allogeneic transplantation as part of first-line therapy in poor-risk peripheral T-NHL. Blood. 2021;137:2646–56.
Tournilhac O, Altmann B, Friedrichs B, et al. Long-term follow-up of the prospective randomized AATT Study (Autologous or Allogeneic Transplantation in Patients With Peripheral T-Cell Lymphoma). J Clin Oncol. 2024;42:3788–94.
Domingo-Domenech E, Duarte RF, Boumedil A, Onida F, Gabriel I, Finel H, et al. Allogeneic hematopoietic stem cell transplantation for advanced mycosis fun- goides and Sézary syndrome. An updated experience of the Lymphoma Working Party of the European Society for Blood and Marrow Transplantation. Bone Marrow Transpl. 2021;56:1391–401.
Trautinger F, Eder J, Assaf C, Bagot M, Cozzio A, Dummer R, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57–74.
Duarte RF, Canals C, Onida F, Gabriel IH, Arranz R, Arcese W, et al. Allogeneic hematopoietic cell transplantation for patients with mycosis fungoides and Sézary syndrome: a retrospective analysis of the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28:4492–9.
Duarte RF, Boumendil A, Onida F, Gabriel I, Arranz R, Arcese W, et al. Long-term outcome of allogeneic hematopoietic cell transplantation for patients with mycosis fungoides and sézary syndrome: a European Society for Blood and Marrow Transplantation Lymphoma Working Party extended analysis. J Clin Oncol. 2014;32:3347–8.
de Masson A, Beylot-Barry M, Ram-Wolff C, Mear JB, Dalle S, d’Incan M, et al. Allogeneic transplantation in advanced cutaneous T-cell lymphomas (CUTALLO): a propensity score matched controlled prospective study. Lancet. 2023;401:1941–50.
Linch DC, Winfield D, Goldstone AH, Moir D, Hancock B, McMillan A, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet. 1993;341:1051–4.
Schmitz N, Pfistner B, Sextro M, Sieber M, Carella AM, Haenel M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet. 2002;359:2065–71.
Sureda A, Canals C, Arranz R, Caballero D, Ribera JM, Brune M, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study—a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante deMedula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica. 2012;97:310–7.
Merryman RW, Castagna L, Giordano L, Ho VT, Corradini P, Guidetti A, et al. Allogeneic transplantation after PD-1 blockade for classic Hodgkin lymphoma. Leukemia. 2021;35:2672–83.
Epperla N, Hamadani M. Double-refractory Hodgkin lymphoma: tackling relapse after brentuximab vedotin and checkpoint inhibitors. Hematol Am Soc Hematol Educ Program. 2021;2021:247–53. Dec
Moskowitz AJ, Shah G, Schöder H, Ganesan N, Drill E, et al. Phase II trial of pembrolizumab plus gemcitabine, vinorelbine, and liposomal doxorubicin as second-line therapy for relapsed or refractory classical Hodgkin lymphoma. J Clin Oncol. 2021;39:3109–17.
Advani RH, Moskowitz AJ, Bartlett NL, Vose JM, Ramchandren R, et al. Brentuximab vedotin in combination with nivolumab in relapsed or refractory Hodgkin lymphoma: 3-year study results. Blood. 2021;138:427–38.
Moskowitz AJ, Schöder H, Yahalom J, McCall SJ, Fox SY, et al. PET-adapted sequential salvage therapy with brentuximab vedotin followed by augmented ifosamide, carboplatin, and etoposide for patients with relapsed and refractory Hodgkin’s lymphoma: a non-randomised, open-label, single-centre, phase 2 study. Lancet Oncol. 2015;16:284–92.
Garcia-Sanz R, Sureda A, de la Cruz F, Canales M, Gonzalez AP, et al. Brentuximab vedotin and ESHAP is highly effective as second-line therapy for Hodgkin lymphoma patients (long-term results of a trial by the Spanish GELTAMO Group). Ann Oncol. 2019;30:612–20.
Driessen J, de Wit F, Herrera AF, Zinzani PL, LaCasce AS, et al. Brentuximab vedotin and chemotherapy in relapsed/refractory Hodgkin lymphoma: a propensity score-matched analysis. Blood Adv. 2024;8:2740–52.
Mei MG, Lee HJ, Palmer JM, Chen R, Tsai NC, et al. Response-adapted anti-PD-1-based salvage therapy for Hodgkin lymphoma with nivolumab alone or in combination with ICE. Blood. 2022;139:3605–316.
Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu MF, Ivanova A, et al. Anti-CD30 CAR-T cell therapy in relapsed and refractory Hodgkin lymphoma. J Clin Oncol. 2020;38:3794–804.
Eichenauer DA, Engert A. Current treatment options for nodular lymphocyte-predominant Hodgkin lymphoma. Curr Opin Oncol. 2021;33:395–9.
Dhakal B, Szabo A, Chhabra S, Hamadani M, D’Souza A, Usmani SZ, et al. Autologous transplantation for newly diagnosed multiple myeloma in the era of novel agent induction: a systematic review and meta-analysis. JAMA Oncol. 2018;4:343–50.
Wiebach H, Gezer D, Brummendorf TH, Crysandt M, Wilop S. Tolerability of high-dose chemotherapy and autologous stem cell transplantation in elderly patients with multiple myeloma: a single-center retrospective analysis. Curr Res Transl Med. 2020;68:139–44.
Mina R, Petrucci MT, Corradini P, Spada S, Patriarca F, Cerrato C, et al. Treatment intensification with autologous stem cell transplantation and lenalidomide maintenance improves survival outcomes of patients with newly diagnosed multiple myeloma in complete response. Clin Lymphoma Myeloma Leuk. 2018;18:533–40.
Swan D, Hayden PJ, Eikema D, Koster L, Sauer S, Blaise D, et al. Trends in autologous stem cell transplantation for newly diagnosed multiple myeloma: Changing demographics and outcomes in European Society for Blood and Marrow Transplantation centres from 1995 to 2019. Br J Haematol. 2022;197:82–96. abril de
Rodriguez TE, Hari P, Stiff PJ, Smith SE, Sterrenberg D, Vesole DH. Busulfan, melphalan, and bortezomib versus high-dose melphalan as a conditioning regimen for autologous hematopoietic stem cell transplantation in multiple myeloma. Biol Blood Marrow Transpl. 2016;22:1391–6.
Cavo M, Petrucci MT, Di Raimondo F, Zamagni E, Gamberi B, Crippa C, et al. Upfront single versus double autologous stem cell transplantation for newly diagnosed multiple myeloma: an intergroup, multicenter, phase III study of the European Myeloma Network (EMN02/HO95 MM Trial). Blood. 2016;128:991.
Cavo M, Beksac M, Dimopoulos MA, Pantani L, Gay F, Hájek R, et al. Intensification therapy with bortezomib-melphalan-prednisone versus autologous stem cell transplantation for newly diagnosed multiple myeloma: an intergroup, multicenter, Phase III Study of the European Myeloma Network (EMN02/HO95 MM Trial). Blood. 2016;128:673.
Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366:1782–91.
McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366:1770–81.
Goldschmidt H, Baertsch MA, Schlenzka J, Becker N, Habermehl C, Hielscher T, et al. Salvage autologous transplant and lenalidomide maintenance vs. lenalidomide/dexamethasone for relapsed multiple myeloma: the randomized GMMG phase III trial ReLApsE. Leukemia 2021;35:1134–44. abril de
Cook G, Williams C, Brown JM, Cairns DA, Cavenagh J, Snowden JA, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:874–85. julio de
Jimenez-Zepeda VH, Mikhael J, Winter A, Franke N, Masih-Khan E, Trudel S, et al. Second autologous stem cell transplantation as salvage therapy for multiple myeloma: impact on progression-free and overall survival. Biol Blood Marrow Transpl. 2012;18:773–9.
Sobh M, Michallet M, Gahrton G, Iacobelli S, van Biezen A, Schönland S, et al. Allogeneic hematopoietic cell transplantation for multiple myeloma in Europe: trends and outcomes over 25 years. A study by the EBMT Chronic Malignancies Working Party. Leukemia. 2016;30:2047–54.
Bruno B, Rotta M, Patriarca F, Mordini N, Allione B, Carnevale-Schianca F, et al. A comparison of allografting with autografting for newly diagnosed myeloma. N Engl J Med. 2007;356:1110–20.
Gahrton G, Iacobelli S, Bjorkstrand B, Hegenbart U, Gruber A, Greinix H, et al. Autologous/reduced-intensity allogeneic stem cell transplantation vs autologous transplantation in multiple myeloma: long-term results of the EBMT- NMAM2000 study. Blood. 2013;121:5055–63.
Rosinol L, Perez-Simon JA, Sureda A, de la Rubia J, de Arriba F, Lahuerta JJ, et al. A prospective PETHEMA study of tandem autologous transplantation versus autograft followed by reduced-intensity conditioning allogeneic transplantation in newly diagnosed multiple myeloma. Blood. 2008;112:3591–3.
Patriarca F, Einsele H, Spina F, Bruno B, Isola M, Nozzoli C, et al. Allogeneic stem cell transplantation in multiple myeloma relapsed after autograft: a multicenter retrospective study based on donor availability. Biol Blood Marrow Transpl. 2012;18:617–26.
Ghosh N, Ye X, Tsai HL, Bolaños-Meade J, Fuchs EJ, Luznik L, et al. Allogeneic blood or marrow transplantation with post-transplantation cyclophosphamide as graft-versus-host disease prophylaxis in multiple myeloma. Biol Blood Marrow Transpl. 2017;23:1903–9.
Beauvais D, Danhof S, Hayden PJ, Einsele H, Yakoub-Agha I. Clinical data, limitations and perspectives on chimeric antigen receptor T-cell therapy in multiple myeloma. Curr Opin Oncol. 2020;32:418–26.
Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398:314–24.
Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384:705–16.
Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene autoleucel, an anti-b-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. JCO. 2023;41:1265–74. de febrero de
Dispenzieri A, Kyle RA, Lacy MQ, Therneau TM, Larson DR, Plevak MF, et al. Superior survival in primary systemic amyloidosis patients undergoing peripheral blood stem cell transplantation: a case-control study. Blood. 2004;103:3960–3.
Muchtar E, Dispenzieri A, Gertz MA, Kumar SK, Buadi FK, Leung N, et al. Treatment of AL amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus statement 2020 update. Mayo Clin Proc. 2021;96:1546–77. junio de
Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357:1083–93.
D’Souza A, Dispenzieri A, Wirk B, Zhang MJ, Huang J, Gertz MA, et al. Improved outcomes after autologous hematopoietic cell transplantation for light chain amyloidosis: a Center for International Blood and Marrow Transplant Research Study. J Clin Oncol. 2015;33:3741–9.
Sidiqi MH, Aljama MA, Buadi FK, Warsame RM, Lacy MQ, Dispenzieri A, et al. Stem cell transplantation for light chain amyloidosis: decreased early mortality over time. J Clin Oncol. 2018;36:1323–9.
Bochtler T, Hegenbart U, Kunz C, Benner A, Kimmich C, Seckinger A, et al. Prognostic impact of cytogenetic aberrations in AL amyloidosis patients after high-dose melphalan: a long-term follow-up study. Blood. 2016;128:594–602.
Marsh JC, Ball SE, Cavenagh J, Derbyshire P, Dokal I, Gordon-Smith EC, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147:43–70.
Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185–96.
Giammarco S, Peffault de Latour R, Sica S, Dufour C, Socie G, Passweg J, et al. Transplant outcome for patients with acquired aplastic anemia over the age of 40: has the outcome improved? Blood. 2018;131:1989–92.
Risitano AM, Eikema DJ, Tuffnell J, Piepenbroek B, Potter V, Sicre De Fontbrune F, et al. Role of age and donor type in 3646 severe aplastic anemia patients undergoing hematopoietic stem cell transplantation in 2011-2020: a retrospective EBMT-Saawp study. Blood. 2024;144:595–595. de noviembre de
Bacigalupo A, Socié G, Schrezenmeier H, Tichelli A, Locasciulli A, Fuehrer M, et al. Bone marrow versus peripheral blood matched sibling transplants, in acquired aplastic anemia: survival advantage for marrow in all age groups. Haematologica. 2012;97:1142–8.
Marsh JC, Gupta V, Lim Z, Ho AY, Ireland RM, Hayden J, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft-versus-host disease after allogeneic stem cell transplantation for acquired aplastic anaemia. Blood. 2011;118:2351–7.
Marsh JC, Pearce RM, Koh MB, Lim Z, Pagliuca A, Mufti GJ, et al. Retrospective study of Alemtuzumab versus ATG-based conditioning without irradiation for unrelated and matched sibling donor transplants in acquired severe aplastic anaemia: a study from the British Society for Blood and Marrow Transplantation (BSBMT). Bone Marrow Transpl. 2014;49:42–48.
Townsley DM, Scheinberg P, Winkler T, Desmond R, Dumitriu B, Rios O, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540–50. de abril de
Peffault De Latour R, Kulasekararaj A, Iacobelli S, Terwel SR, Cook R, Griffin M, et al. Eltrombopag added to immunosuppression in severe aplastic anemia. N Engl J Med. 2022;386:11–23. de enero de
Kaya D, Cook R, Iacobelli S, Kulasekararaj AG, Napolitani G, Gerlevik S, et al. Dynamics of oligoclonal hematopoiesis in severe aplastic anemia patients undergoing immunosuppressive treatment: longitudinal somatic mutation analysis from the EBMT race trial. Blood. 2024;144:33–33. de noviembre de
Bacigalupo A, Marsh JC. Unrelated donor search and unrelated donor trans- plantation in the adult aplastic anaemia patient aged 18-40 years without an HLA-identical sibling and failing immunosuppression. Bone Marrow Transpl. 2013;48:198–200.
de Latour RP. Transplantation for bone marrow failure: current issues. Hematology Am Soc Hematol Educ Program. 2016;2016:90–8.
Olnes MJ, Scheinberg P, Calvo KR, Desmond R, Tang Y, Dumitriu B, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11–19.
Desmond R, Townsley DM, Dumitriu B, Olnes MJ, Scheinberg P, Bevans M, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014;123:1818–25.
Lengline E, Drenou B, Peterlin P, Tournilhac O, Abraham J, Berceanu A, et al. Nationwide survey on the use of eltrombopag in patients with severe aplastic anemia: a report on behalf of the French Reference Center for Aplastic Anemia. Haematologica. 2018;103:212–20.
DeZern AE, Zahurak M, Symons H, Cooke K, Jones RJ, Brodsky RA. Alternative donor transplantation with high-dose post-transplantation cyclophosphamide for refractory severe aplastic anemia. Biol Blood Marrow Transplant. 2017;23:498–504. marzo de
Peffault de Latour R, Purtill D, Ruggeri A, Sanz G, Michel G, Gandemer V, et al. Influence of nucleated cell dose on overall survival of unrelated cord blood transplantation for patients with severe acquired aplastic anaemia. A study by Eurocord and the Aplastic Anaemia Working Party of the European Group for Blood and Marrow Transplantation. Biol Blood Marrow Transpl. 2010;1:78–85.
De Latour RP, Rocha V, Socie G. Cord blood transplantation in aplastic anaemia. Bone Marrow Transpl. 2013;48:201–2.
Ciceri F, Lupo-Stanghellini MT, Korthof ET. Haploidentical transplantation in patients with acquired aplastic anaemia. Bone Marrow Transpl. 2013;48:183–5.
Clay J, Kulasekarara AG, Potter V, Grimaldi F, McLornan D, Raj K, et al. Non- myeloablative peripheral blood haploidentical stem cell transplantation for refractory severe aplastic anemia. Biol Blood Marrow Transpl. 2014;20:1711–6.
Pagliuca S, Peffault de Latour R, Volt F, Locatelli F, Zecca M, Dalle JH, et al. Long-term outcomes of cord blood transplantation from an HLA-identical sibling for patients with bone marrow failure syndromes: a report from Eurocord, Cord Blood Committee and Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation. Biol Blood Marrow Transpl. 2017;23:1939–48.
Bacigalupo A. Alternative donor transplants for severe aplastic anemia. Hematol Am Soc Hematol Educ Program. 2018;2018:467–73.
Montoro J, Eikema DJ, Tuffnell J, Potter V, Kalwak K, Halkes CJM, et al. Alternative donor transplantation for severe aplastic anemia: a comparative study of the SAAWP EBMT. Blood. 2024;144:323–33. 18 de julio de
Prata PH, Eikema DJ, Afansyev B, Bosman P, Smiers F, Diez-Martin JL, et al. Haploidentical transplantation and posttransplant cyclophosphamide for treating aplastic anemia patients: a report from the EBMT Severe Aplastic Anemia Working Party. Bone Marrow Transpl. 2020;55:1050–8.
DeZern AE, Zahurak ML, Symons HJ, Cooke KR, Rosner GL, Gladstone DE, et al. Haploidentical BMT for severe aplastic anemia with intensive GVHD prophylaxis including posttransplant cyclophosphamide. Blood Adv. 2020;4:1770–9. de abril de
DeZern AE, Eapen M, Wu J, Talano JA, Solh M, Dávila Saldaña BJ, et al. Haploidentical bone marrow transplantation in patients with relapsed or refractory severe aplastic anaemia in the USA (BMT CTN 1502): a multicentre, single-arm, phase 2 trial. Lancet Haematol. 2022;9:e660–9. septiembre de
DeZern A, Zahurak ML, Symons HJ, Cooke KR, Huff CA, Jain T, et al. Alternative donor BMT with post-transplant cyclophosphamide as initial therapy for acquired severe aplastic anemia. Blood Journal. de abril de 2023;blood.2023020435.
Babushok DV, DeZern AE, De Castro CM, Rogers ZR, Beenhouwer D, Broder MS, et al. Modified Delphi panel consensus recommendations for management of severe aplastic anemia. Blood Adv. 2024;8:3946–60. de agosto de
Iftikhar R, DeFilipp Z, DeZern AE, Pulsipher MA, Bejanyan N, Burroughs LM, et al. Allogeneic hematopoietic cell transplantation for the treatment of severe aplastic anemia: evidence-based guidelines from the American Society for Transplantation and Cellular Therapy. Transplant Cell Ther. 2024;30:1155–70. diciembre de
Scheinberg P, Kulasekararaj AG. Consensus recommendations for severe aplastic anemia. Blood Adv. 2024;8:5719–20. de noviembre de
DeZern AE, Zahurak M, Jones RJ, Brodsky RA. Uniform conditioning regardless of donor in bone marrow transplantation for severe aplastic anemia. Haematologica2023;109:657–60. de septiembre de
Risitano AM, Peffault De Latour R. How we(’ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol. 2022;196:288–303. enero de
Risitano AM, Peffault De Latour R, Marano L, Frieri C. Discovering C3 targeting therapies for paroxysmal nocturnal hemoglobinuria: achievements and pitfalls. Semin Immunol 2022;59:101618. enero de
Risitano AM, Marotta S, Ricci P, Marano L, Frieri C, Cacace F, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10:1157. de junio de
Risitano AM, Marotta S. Toward complement inhibition 2.0: next generation anticomplement agents for paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2018;93:564–77. agosto de
de Latour PR, Schrezenmeier H, Bacigalupo A, Blaise D, de Souza CA, Vigouroux S, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97:1666–73.
Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124:2804–11.
Frieri C, Eikema DJ, Tuffnell J, Piepenbroek B, Benakli M, Helbig G, et al. Improved outcomes in paroxysmal nocturnal hemoglobinuria patients undergoing allogeneic hematopoietic stem cell transplantation in 2011–2020: a retrospective EBMT-Saawp study. Blood. 2024;144:305–305. de noviembre de
Pellegrini M. Access to expensive drugs in EU countries for very rare hematological blood disorders: the example of paroxysmal nocturnal hemoglobinuria. A survey by ERN-EUROBLOODNET. En EHA 2024;
Kulasekararaj A, Cavenagh J, Dokal I, Foukaneli T, Gandhi S, Garg M, et al. Guidelines for the diagnosis and management of adult aplastic anaemia: A British Society for Haematology Guideline. Br J Haematol. 2024;204:784–804. marzo de
Barkholt L, Bregni M, Remberger M, Blaise D, Peccatori J, Massenkeil G, et al. Allogeneic haematopoietic stem cell transplantation for metastatic renal carcinoma in Europe. Ann Oncol. 2006;17:1134–40.
Carnevale-Schianca F, Cignetti A, Capaldi A, Vitaggio K, Vallario A, Ricchiardi A, et al. Allogeneic nonmyeloablative hematopoietic cell transplantation in metastatic colon cancer: tumor-specific T cells directed to a tumor-associated antigen are generated in vivo during GVHD. Blood. 2006;107:3795–803.
Secondino S, Carrabba MG, Pedrazzoli P, Castagna L, Spina F, Grosso F.European Group for Blood and Marrow Transplantation Solid Tumors Working Party et al. Reduced-intensity stem cell transplantation for advanced soft tissue sarcomas in adults: a retrospective analysis of the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:418–20.
Comoli P, Chabannon C, Koehl U, Lanza F, Urbano-Ispizua A, Hudecek M, et al. Development of adaptive immune effector therapies in solid tumors. Ann Oncol. 2019;30:1740–50.
Comoli P, Pentheroudakis G, Ruggeri A, Koehl U, Lordick F, Mooyaart JE.EBMT Cellular Therapy & Immunobiology Working Party; European Society of Medical Oncology et al. Current strategies of cell and gene therapy for solid tumors: results of the joint international ESMO and CTIWP-EBMT survey. Ann Oncol. 2024;35:404–6.
Haanen JB, Mackensen A, Schultze-Florey C, Alsdorf W, Wagner-Drouet E, Heudobler D, et al. Updated results from BNT211-01 (NCT04503278), an ongoing, first-in-human, phase I study evaluating the safety and efficacy of CLDN6 CAR T cells and a CLDN6-encoding mRNA vaccine in patients with relapsed/refractory CLDN6+ solid tumors. ESMO Congress, Barcelona September 2024, Abstract 611O.
D’Angelo SP, Araujo DM, Abdul Razak AR, Agulnik M, Attia S, Blay JY, et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): an international, open-label, phase 2 trial. Lancet 2024;13:1460–71.
Rosenberg SA. Lymphocytes as a living drug for cancer. Science. 2024;385:25–26.
Secondino S, Pedrazzoli P, Basso S, Bossi P, Bianco A, Imarisio I, et al. Long-lasting responses with chemotherapy followed by T-cell therapy in recurrent or metastatic EBV-related nasopharyngeal carcinoma. Front Immunol. 2023;14:1208475.
Secondino S, Canino C, Alaimo D, Muzzana M, Galli G, Borgetto S, et al. Clinical trials of cellular therapies in solid tumors. Cancers. 2023;15:3667.
Keam SJ. Afamitresgene Autoleucel: first approval. Mol Diagn Ther. 2024;28:861–6.
Berry DA, Ueno NT, Johnson MM, Lei X, Caputo J, Rodenhuis S, et al. High-dose chemotherapy with autologous stem cell support versus standard-dose chemotherapy: overview of individual patient data from 15 randomized adjuvant therapy breast cancer trials. J Clin Oncol. 2011;29:3214–23.
Berry DA, Ueno NT, Johnson MM, Lei X, Caputo J, Smith DA, et al. High-dose chemotherapy with autologous hematopoietic stem-cell transplantation in metastatic breast cancer: overview of six randomized trials. J Clin Oncol. 2011;29:3224–31.
Martino M, Ballestrero A, Zambelli A, Secondino S, Aieta M, Bengala C, et al. Long-term survival in patients with metastatic breast cancer receiving intensified chemotherapy and stem cell rescue: data from the Italian registry. Bone Marrow Transpl. 2013;48:414–8.
Nitz UA, Mohrmann S, Fischer J, Lindemann W, Berdel WE, Jackisch C, et al. West German Study Group. Comparison of rapidly cycled tandem high-dose chemotherapy plus peripheral-blood stem-cell support versus dose-dense conventional chemotherapy for adjuvant treatment of high-risk breast cancer: results of a multicentre phase III trial. Lancet. 2005;366:1935–44.
Martino M, Lanza F, Pavesi L, Öztürk M, Blaise D, Leno Núñez R, et al. High-dose chemotherapy and autologous hematopoietic stem cell transplantation as adjuvant treatment in high-risk breast cancer: data from the European Group for Blood and Marrow Transplantation Registry. Biol Blood Marrow Transpl. 2016;22:475–81.
Steenbruggen TG, Steggink LC, Seynaeve CM, van der Hoeven JJM, Hooning MJ, Jager A, et al. High-dose chemotherapy with hematopoietic stem cell transplant in patients with high-risk breast cancer and 4 or more involved axillary lymph nodes—20-year follow-up of a phase 3 randomized clinical trial. JAMA Oncol. 2020;6:528–34. https://doi.org/10.1001/jamaoncol.2019.6276
Necchi A, Lanza F, Rosti G, Martino M, Farè E, Pedrazzoli P. European Society for Blood and Marrow Transplantation, Solid Tumors Working Party (EBMT- STWP) and the Italian Germ Cell Cancer Group (IGG) High-dose chemotherapy for germ cell tumors: do we have a model? Expert Opin Biol Ther. 2015;15:33–44.
Agrawal V, Abonour R, Abu Zaid M, Althouse SK, Ashkar R, Albany C, et al. Survival outcomes and toxicity in patients 40 years old or older with relapsed metastatic germ cell tumors treated with high-dose chemotherapy and peripheral blood stem cell transplantation. Cancer. 2021;127:3751–60. https://doi.org/10.1002/cncr.33771
Lorch A, Bascoul-Mollevi C, Kramar A, Einhorn L, Necchi A, Massard C, et al. Conventional-dose versus high-dose chemotherapy as first salvage treatment in male patients with metastatic germ cell tumors: evidence from a large international database. J Clin Oncol. 2011;29:2178–84.
Secondino S, Badoglio M, Rosti G, Labopin M, Delaye M, Bokemeyer C, et al. High-dose chemotherapy with autologous stem cell transplants in adult primary non-seminoma mediastinal germ-cell tumors. A report from the Cellular Therapy and Immunobiology working party of the EBMT. ESMO Open. 2024;9:103692.
Ladenstein R, Pötschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28:3284–91.
Heilig CE, Badoglio M, Labopin M, Fröhling S, Secondino S, Heinz J, et al. Hae- matopoietic stem cell transplantation in adult soft-tissue sarcoma: an analysis from the European Society for Blood and Marrow Transplantation. ESMO Open. 2020;5:e000860.
Spreafico F, Massimino M, Gandola L, Cefalo G, Mazza E, Landonio G, et al. Survival of adults treated for medulloblastoma using paediatric protocols. Eur J Cancer. 2005;41:1304–10.
Alexander T, Greco R. Hematopoietic stem cell transplantation and cellular therapies for autoimmune diseases: overview and future considerations from the Autoimmune Diseases Working Party (ADWP) of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transpl. 2022;57:1055–62.
Muraro PA, Pasquini M, Atkins HL, Bowen JD, Farge D, Fassas A, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017;74:459–69.
Burt RK, Balabanov R, Burman J, Sharrack B, Snowden JA, Oliveira MC, et al. Effect of nonmyeloablative hematopoietic stem cell transplantation vs continued disease-modifying therapy on disease progression in patients with relapsing-remitting multiple sclerosis: a randomized clinical trial. JAMA. 2019;321:165–74.
Tappenden P, Wang Y, Sharrack B, Burman J, Kazmi M, Saccardi R, et al. Evaluating the clinical effectiveness of autologous haematopoietic stem cell transplantation versus disease-modifying therapy in multiple sclerosis using a matching-adjusted indirect comparison: an exploratory study from the Autoimmune Diseases Working Party (ADWP) of the European Society of Bone and Marrow Transplantation (EBMT). Bone Marrow Transpl. 2020;55:1473–5.
Boffa G, Massacesi L, Inglese M, Mariottini A, Capobianco M, Moiola L, et al. Long-term clinical outcomes of hematopoietic stem cell transplantation in multiple sclerosis. Neurology. 2021;96:e1215–e26.
Das J, Snowden JA, Burman J, Freedman MS, Atkins H, Bowman M, et al. Autologous haematopoietic stem cell transplantation as a first-line disease-modifying therapy in patients with «aggressive» multiple sclerosis. Mult Scler. 2021;27:1198–204.
Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011;378:498–506.
van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311:2490–8.
Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. 2018;378:35–47.
Henes J, Oliveira MC, Labopin M, Badoglio M, Scherer HU, Del Papa N, et al. Autologous stem cell transplantation for progressive systemic sclerosis: a prospective non-interventional study from the European Society for Blood and Marrow Transplantation Autoimmune Disease Working Party. Haematologica. 2021;106:375–83.
Burt RK, Han X, Quigley K, Arnautovic I, Shah SJ, Lee DC, et al. Cardiac safe hematopoietic stem cell transplantation for systemic sclerosis with poor cardiac function: a pilot safety study that decreases neutropenic interval to 5 days. Bone Marrow Transpl. 2021;56:50–9.
Muraro PA, Mariottini A, Greco R, Burman J, Iacobaeus E, Inglese M, et al. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis and neuromyelitis optica spectrum disorder—recommendations from ECTRIMS and the EBMT. Nat Rev Neurol. 2025.
Hawkey CJ, Allez M, Clark MM, Labopin M, Lindsay JO, Ricart E, et al. Autologous hematopoetic stem cell transplantation for refractory crohn disease: a randomized clinical trial. Jama. 2015;314:2524–34.
Lindsay JO, Hind D, Swaby L, Berntsson H, Bradburn M, Bannur CU, et al. Safety and efficacy of autologous haematopoietic stem-cell transplantation with low-dose cyclophosphamide mobilisation and reduced intensity conditioning versus standard of care in refractory Crohn’s disease (ASTIClite): an open-label, multicentre, randomised controlled trial. Lancet Gastroenterol Hepatol. 2024;9:333–45.
Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. 2009;113:214–23.
Burt RK, Han X, Gozdziak P, Yaung K, Morgan A, Clendenan AM, et al. Five year follow-up after autologous peripheral blood hematopoietic stem cell transplantation for refractory, chronic, corticosteroid-dependent systemic lupus erythematosus: effect of conditioning regimen on outcome. Bone Marrow Transpl. 2018;53:692–700.
Alchi B, Jayne D, Labopin M, Demin A, Sergeevicheva V, Alexander T, et al. Autologous haematopoietic stem cell transplantation for systemic lupus erythematosus: data from the European Group for Blood and Marrow Transplantation registry. Lupus. 2013;22:245–53.
Greco R, Bondanza A, Oliveira MC, Badoglio M, Burman J, Piehl F, et al. Autologous hematopoietic stem cell transplantation in neuromyelitis optica: a registry study of the EBMT Autoimmune Diseases Working Party. Mult Scler. 2015;21:189–97.
Burt RK, Balabanov R, Han X, Burns C, Gastala J, Jovanovic B, et al. Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology. 2019;93:e1732–e41.
Burt RK, Balabanov R, Tavee J, Han X, Sufit R, Ajroud-Driss S, et al. Hematopoietic stem cell transplantation for chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol. 2020;267:3378–91.
Bryant A, Atkins H, Pringle CE, Allan D, Anstee G, Bence-Bruckler I, et al. Myasthenia gravis treated with autologous hematopoietic stem cell transplantation. JAMA Neurol. 2016;73:652–8.
Kass-Iliyya L, Snowden JA, Thorpe A, Jessop H, Chantry AD, Sarrigiannis PG, et al. Autologous haematopoietic stem cell transplantation for refractory stiff-person syndrome: the UK experience. J Neurol. 2021;268:265–75.
Burt RK, Balabanov R, Han X, Quigley K, Arnautovic I, Helenowski I, et al. Autologous hematopoietic stem cell transplantation for stiff-person spectrum disorder: a clinical trial. Neurology. 2021;96:e817–e30.
Alexander T, Samuelson C, Daikeler T, Henes J, Akil M, Skagerlind L, et al. Autologous haematopoietic stem cell transplantation (HSCT) for anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis: a retrospective survey of patients reported to European Society for Blood and Marrow Transplantation (EBMT) registry. Bone Marrow Transpl. 2020;55:1512–5.
Laurent C, Marjanovic Z, Ricard L, Henes J, Dulery R, Badoglio M, et al. Autologous hematopoietic stem cell transplantation with reduced-intensity conditioning regimens in refractory Takayasu arteritis: a retrospective multicenter case-series from the Autoimmune Diseases Working Party (ADWP) of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transpl. 2020;55:2109–13.
Puyade M, Patel A, Lim YJ, Blank N, Badoglio M, Gualandi F, et al. Autologous hematopoietic stem cell transplantation for Behçet’s Disease: a retrospective survey of patients treated in Europe, on Behalf of the Autoimmune Diseases Working Party of the European Society for Blood and Marrow Transplantation. Front Immunol. 2021;12:638709.
Nijeboer P, van Wanrooij R, van Gils T, Wierdsma NJ, Tack GJ, Witte BI, et al. Lymphoma development and survival in refractory coeliac disease type II: histological response as prognostic factor. U Eur Gastroenterol J. 2017;5:208–17.
Snowden JA, Badoglio M, Labopin M, Giebel S, McGrath E, Marjanovic Z, et al. Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv. 2017;1:2742–55.
Burt RK, Tappenden P, Han X, Quigley K, Arnautovic I, Sharrack B, et al. Health economics and patient outcomes of hematopoietic stem cell transplantation virsus disease-modifying therapies for relapsing remitting multiple sclerosis in the United States of America. Mult Scler Relat Disord. 2020;45:102404.
Burt RK, Tappenden P, Balabanov R, Han X, Quigley K, Snowden JA, et al. The cost-effectiveness of immunoglobulin vs. hematopoietic stem cell transplantation for CIDP. Front Neurol. 2021;12:645263.
Greco R, Labopin M, Badoglio M, Veys P, Furtado Silva JM, Abinun M, et al. Allogeneic HSCT for autoimmune diseases: a retrospective study from the EBMT ADWP, IEWP, and PDWP working parties. Front Immunol. 2019;10:1570.
Sharrack B, Saccardi R, Alexander T, Badoglio M, Burman J, Farge D, et al. Autologous haematopoietic stem cell transplantation and other cellular therapy in multiple sclerosis and immune-mediated neurological diseases: updated guidelines and recommendations from the EBMT Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of EBMT and ISCT (JACIE). Bone Marrow Transpl. 2020;55:283–306.
Farge D, Burt RK, Oliveira MC, Mousseaux E, Rovira M, Marjanovic Z, et al. Cardiopulmonary assessment of patients with systemic sclerosis for hematopoietic stem cell transplantation: recommendations from the European Society for Blood and Marrow Transplantation Autoimmune Diseases Working Party and collaborating partners. Bone Marrow Transpl. 2017;52:1495–503.
Del Galdo F, Lescoat A, Conaghan PG, Bertoldo E, Čolić J, Santiago T, et al. EULAR recommendations for the treatment of systemic sclerosis: 2023 update. Ann Rheum Dis. 2024.
Snowden JA, Panés J, Alexander T, Allez M, Ardizzone S, Dierickx D, et al. Autologous Haematopoietic Stem Cell Transplantation (AHSCT) in Severe Crohn’s Disease: A Review on Behalf of ECCO and EBMT. J Crohns Colitis. 2018;12:476–88.
Jessop H, Farge D, Saccardi R, Alexander T, Rovira M, Sharrack B, et al. General information for patients and carers considering haematopoietic stem cell transplantation (HSCT) for severe autoimmune diseases (ADs): A position statement from the EBMT Autoimmune Diseases Working Party (ADWP), the EBMT Nurses Group, the EBMT Patient, Family and Donor Committee and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Bone Marrow Transpl. 2019;54:933–42.
Roberts F, Hobbs H, Jessop H, Bozzolini C, Burman J, Greco R, et al. Rehabilitation before and after Autologous Haematopoietic Stem Cell Transplantation (AHSCT) for patients with Multiple Sclerosis (MS): consensus guidelines and recommendations for best clinical practice on Behalf of the Autoimmune Diseases Working Party, Nurses Group, and Patient Advocacy Committee of the European Society for Blood and Marrow Transplantation (EBMT). Front Neurol. 2020;11:556141.
Alexander T, Roldan E, Del Papa N, Farge D, Henes J, Marjanovic Z, et al. Autologous haematopoietic stem cell transplantation for rheumatic diseases: best practice recommendations from the EBMT Practice Harmonization and Guidelines committee. Bone Marrow Transpl. 2025; https://doi.org/10.1038/s41409-025-02695-y (Online ahead of print).
Greco R, Alexander T, Burman J, Del Papa N, de Vries-Bouwstra J, Farge D, et al. Hematopoietic stem cell transplantation for autoimmune diseases in the time of COVID-19: EBMT guidelines and recommendations. Bone Marrow Transpl. 2021;56:1493–508.
Brittain G, Roldan E, Alexander T, Saccardi R, Snowden JA, Sharrack B, et al. The role of chimeric antigen receptor T-cell therapy in immune-mediated neurological diseases. Ann Neurol. 2024;96:441–52.
Schett G, Mackensen A, Mougiakakos D. CAR T-cell therapy in autoimmune diseases. Lancet. 2023.
Mougiakakos D, Kronke G, Volkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. 2021;385:567–9.
Mackensen A, Muller F, Mougiakakos D, Boltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022;28:2124–32.
Li M, Zhang Y, Jiang N, Ning C, Wang Q, Xu D, et al. Anti-CD19 CAR T cells in refractory immune thrombocytopenia of SLE. N Engl J Med. 2024;391:376–8.
Auth J, Müller F, Völkl S, Bayerl N, Distler JHW, Tur C, et al. CD19-targeting CAR T-cell therapy in patients with diffuse systemic sclerosis: a case series. Lancet Rheumatol. 2024.
Muller F, Boeltz S, Knitza J, Aigner M, Volkl S, Kharboutli S, et al. CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet. 2023;401:815–8.
Pecher AC, Hensen L, Klein R, Schairer R, Lutz K, Atar D, et al. CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase syndrome. JAMA. 2023;329:2154–62.
Wang X, Wu X, Tan B, Zhu L, Zhang Y, Lin L, et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell. 2024;187:4890–904.e9.
Wang L, Luo YC, Wang H, Zou YB, Yao HQ, Ullah S, et al. Azure-winged magpies fail to understand the principle of mirror imaging. Behav Process. 2020;177:8.
Haghikia A, Hegelmaier T, Wolleschak D, Böttcher M, Pappa V, Motte J, et al. Clinical efficacy and autoantibody seroconversion with CD19-CAR T cell therapy in a patient with rheumatoid arthritis and coexisting myasthenia gravis. Ann Rheum Dis. 2024;83:1597–8.
Lidar M, Rimar D, David P, Jacoby E, Shapira-Frommer R, Itzhaki O, et al. CD-19 CAR-T cells for polyrefractory rheumatoid arthritis. Ann Rheum Dis. 2024.
Haghikia A, Hegelmaier T, Wolleschak D, Bottcher M, Desel C, Borie D, et al. Anti-CD19 CAR T cells for refractory myasthenia gravis. Lancet Neurol. 2023;22:1104–5.
Fischbach F, Richter J, Pfeffer LK, Fehse B, Berger SC, Reinhardt S, et al. CD19-targeted chimeric antigen receptor T cell therapy in two patients with multiple sclerosis. Med. 2024;5:550–8 e2.
Faissner S, Motte J, Sgodzai M, Geis C, Haghikia A, Mougiakakos D, et al. Successful use of anti-CD19 CAR T cells in severe treatment-refractory stiff-person syndrome. Proc Natl Acad Sci USA. 2024;121:e2403227121.
Qin C, Tian DS, Zhou LQ, Shang K, Huang L, Dong MH, et al. Anti-BCMA CAR T-cell therapy CT103A in relapsed or refractory AQP4-IgG seropositive neuromyelitis optica spectrum disorders: phase 1 trial interim results. Signal Transduct Target Ther. 2023;8:5.
Granit V, Benatar M, Kurtoglu M, Miljkovic MD, Chahin N, Sahagian G, et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. 2023;22:578–90.
Burns SO, Morris EC. How I use allogeneic HSCT for adults with inborn errors of immunity. Blood. 2021;138:1666–76. de noviembre de
Baronciani D. Age is a crucial determinant of GFRS with incidence of severe chronic GVHD reducing over time in haemopoietic cell transplantation for transfusion dependent thalassaemia: real world data from 2010–2021. an analysis of the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Working Party. ASH 2024 Abstract. En.
Angelucci E, Pilo F, Coates TD. Transplantation in thalassemia: revisiting the Pesaro risk factors 25 years later. Am J Hematol 2017;92:411–3. mayo de
Angelucci E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica 2014;99:811–20. de mayo de
De La Fuente J. Significant growth in HCT use for haemoglobinopathies in the last decade driven by sickle cell disease across all ages with excellent outcomes: an analysis of EBMT Haemoglobinopathies Working Party. ASH 2024 Abstract. En.
Niewerth D, Creutzig U, Bierings MB, Kaspers GJ. A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia. Blood. 2010;116:2205–14.
Burke MJ, Wagner JE, Cao Q, Ustun C, Verneris MR. Allogeneic hematopoietic cell transplantation in first remission abrogates poor outcomes associated with high-risk pediatric acute myeloid leukemia. Biol Blood Marrow Transpl. 2013;19:1021–5.
Klusmann JH, Reinhardt D, Zimmermann M, Kremens B, Vormoor J, Dworzak M, et al. The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica. 2012;97:21–29.
Creutzig U, Zimmermann M, Bourquin JP, Dworzak MN, Kremens B, Lehrnbecher T, et al. Favorable outcome in infants with AML after intensive first- and second-line treatment: an AML-BFM study group report. Leukemia. 2012;26:654–61.
Locatelli F, Pende D, Maccario R, Mingari MC, Moretta A, Moretta L. Haploidentical hemopoietic stem cell transplantation for the treatment of high-risk leukemias: how NK cells make the difference. Clin Immunol. 2009;133:171–8.
Marks D, Khattry N, Cummins M, Goulden N, Green A, Harvey J, et al. Haploi- dentical stem cell transplantation for children with acute leukaemia. Br J Haematol. 2006;134:196–201.
Hasle H. A critical review of which children with acute myeloid leukaemia need stem cell procedures. Br J Haematol. 2014;166:23–33.
Mann G, Attarbaschi A, Schrappe M, De Lorenzo P, Peters C, Hann I, et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood. 2010;116:2644–50.
von Stackelberg A, Volzke E, Kuhl JS, Seeger K, Schrauder A, Escherich G, et al. Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer. 2011;47:90–97.
Peters C, Cornish JM, Parikh SH, Kurtzberg J. Stem cell source and outcome after hematopoietic stem cell transplantation (HSCT) in children and adolescents with acute leukemia. Pedia Clin North Am. 2010;57:27–46.
Schrauder A, von Stackelberg A, Schrappe M, Cornish J, Peters C. Allogeneic hematopoietic SCT in children with ALL: current concepts of ongoing prospective SCT trials. Bone Marrow Transpl. 2008;41:S71–74.
Peters C, Schrappe M, von Stackelberg A, Schrauder A, Bader P, Ebell W, et al. Stem-cell transplantation in children with acute lymphoblastic leukemia: a prospective international multicenter trial comparing sibling donors with matched unrelated donors—The ALL-SCT-BFM-2003 trial. J Clin Oncol. 2015;33:1265–74.
Pulsipher MA, Peters C, Pui CH. High-risk pediatric acute lymphoblastic leuke- mia: to transplant or not to transplant? Biol Blood Marrow Transpl. 2011;17:S137–148.
Bader P, Kreyenberg H, Henze GH, Eckert C, Reising M, Willasch A, et al. Prog- nostic value of minimal residual disease quantification before allogeneic stem- cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol. 2009;27:377–84.
Eckert C, Henze G, Seeger K, Hagedorn N, Mann G, Panzer-Grumayer R, et al. Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol. 2013;31:2736–42.
Bader P, Kreyenberg H, von Stackelberg A, Eckert C, Salzmann-Manrique E, Meisel R, et al. Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: results of the ALL-BFM-SCT 2003 trial. J Clin Oncol. 2015;33:1275–84.
Beck JC, Cao Q, Trotz B, Smith AR, Weigel BJ, Verneris MR, et al. Allogeneic hematopoietic cell transplantation outcomes for children with B-precursor acute lymphoblastic leukemia and early or late BM relapse. Bone Marrow Transpl. 2011;46:950–5.
Meisel R, Klingebiel T, Dilloo D. German/Austrian Pediatric Registry for Stem Cell Transplantation Peripheral blood stem cells versus bone marrow in pediatric unrelated donor stem cell transplantation. Blood. 2013;121:863–5.
Peters C, Dalle JH, Locatelli F, Poetschger U, Sedlacek P, Buechner J, et al. Total body irradiation or chemotherapy conditioning in childhood ALL: a multinational, randomized, noninferiority Phase III study. J Clin Oncol. 2021;39:295–307.
Kalwak K, Moser L, Poetschger U, et al. Comparable outcomes after busulfan- or treosulfan-based conditioning for allo-HSCT in children with ALL. Results of FORUM. Blood Adv. 2025;9:741–51. https://doi.org/10.1182/bloodadvances.2024014548. Feb
Suttorp M, Eckardt L, Tauer JT, Millot F. Management of chronic myeloid leukemia in childhood. Curr Hematol Malign Rep. 2012;7:116–24.
Suttorp M, Yaniv I, Schultz KR. Controversies in the treatment of CML in children and adolescents: TKIs versus BMT? Biol Blood Marrow Transpl. 2011;17:S115–122.
de la Fuente J, Baruchel A, Biondi A, de Bont E, Dresse MF, Suttorp M, et al. Managing children with chronic myeloid leukaemia (CML): recommendations for the management of CML in children and young people up to the age of 18 years. Br J Haematol. 2014;167:33–47.
Jaeger BA, Tauer JT, Ulmer A, Kuhlisch E, Roth HJ, Suttorp M. Changes in bone metabolic parameters in children with chronic myeloid leukemia on imatinib treatment. Med Sci Monit. 2012;18:721–8.
Ulmer A, Tabea Tauer J, Glauche I, Jung R, Suttorp M. TK inhibitor treatment disrupts growth hormone axis: clinical observations in children with CML and experimental data from a juvenile animal model. Klin Padiatr. 2013;225:120–6.
Suttorp M, Claviez A, Bader P, Peters C, Gadner H, Ebell W, et al. Allogeneic stem cell transplantation for pediatric and adolescent patients with CML: results from the prospective trial CML-paed I. Klin Padiatr. 2009;221:351–7.
Millot F, Claviez A, Leverger G, Corbaciglu S, Groll AH, Suttorp M. Imatinib cessation in children and adolescents with chronic myeloid leukemia in chronic phase. Pediatr Blood Cancer. 2014;61:355–7.
Pichler H, Sedlacek P, Meisel R, et al. Haematopoietic stem cell transplantation after reduced intensity conditioning in children and adolescents with chronic myeloid leukaemia: a prospective multicentre trial of the I-BFM Study Group. Br J Haematol. 2024;205:268–79.
Locatelli F, Crotta A, Ruggeri A, Eapen M, Wagner JE, Macmillan ML, et al. Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood. 2013;122:2135–41.
Madureira AB, Eapen M, Locatelli F, Teira P, Zhang MJ, Davies SM, et al. Analysis of risk factors influencing outcome in children with myelodysplastic syndrome after unrelated cord blood transplantation. Leukemia. 2011;25:449–54.
Strahm B, Nollke P, Zecca M, Korthof ET, Bierings M, Furlan I, et al. Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia. 2011;25:455–62.
Burkhardt B, Reiter A, Landmann E, Lang P, Lassay L, Dickerhoff R, et al. Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the Berlin-Frankfurt-Muenster group. J Clin Oncol. 2009;27:3363–9.
Gross TG, Hale GA, He W, Camitta BM, Sanders JE, Cairo MS, et al. Hematopoietic stem cell transplantation for refractory or recurrent non-Hodgkin lymphoma in children and adolescents. Biol Blood Marrow Transpl. 2010;16:223–30.
Woessmann W, Peters C, Lenhard M, Burkhardt B, Sykora KW, Dilloo D, et al. Allogeneic haematopoietic stem cell transplantation in relapsed or refractory anaplastic large cell lymphoma of children and adolescents-a Berlin-Frankfurt-Munster group report. Br J Haematol. 2006;133:176–82.
Kelly KM. Hodgkin lymphoma in children and adolescents: improving the therapeutic index. Blood. 2015;126:2452–8.
Kahn JM, Kelly KM. Adolescent and young adult Hodgkin lymphoma: raising the bar through collaborative science and multidisciplinary care. Pediatr Blood Cancer. 2018;65:e27033.
Ladenstein R, Potschger U, Hartman O, Pearson AD, Klingebiel T, Castel V, et al. 28 years of high-dose therapy and SCT for neuroblastoma in Europe: lessons from more than 4000 procedures. Bone Marrow Transpl. 2008;41:S118–127.
Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a Children’s Oncology Group study. J Clin Oncol. 2009;27:1007–13.
Del Bufalo F, De Angelis B, Caruana I, Del Baldo G, De Ioris MA, Serra A, et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N Engl J Med. 2023;388:1284–95. de abril de
Samarasinghe S, Marsh J, Dufour C. Immune suppression for childhood acquired aplastic anemia and myelodysplastic syndrome: where next? Haematologica. 2014;99:597–9.
Dufour C, Veys P, Carraro E, Bhatnagar N, Pillon M, Wynn R, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of EBMT. Br J Haematol. 2015;171:585–94.
Burman J, Kirgizov K, Carlson K, Badoglio M, Mancardi GL, De Luca G, et al. Autologous hematopoietic stem cell transplantation for pediatric multiple sclerosis: a registry-based study of the Autoimmune Diseases Working Party (ADWP) and Pediatric Diseases Working Party (PDWP) of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transpl. 2017;52:1133–7.
Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol. 2018;141:1036–49.e5.
Devillier R, Dalle JH, Kulasekararaj A, D’aveni M, Clément L, Chybicka A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the EBMT Severe Aplastic Anemia Working Party. Haematologica. 2016;101:884–90.
Medeiros C, Zaris-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anaemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transpl. 1999;24:849–52.
De Latour RP, Porcher R, Dalle J-H, Aljurf M, Korthof ET, Svahn J, et al. Allogeneic haemopoietic stem cell transplantation in Fanconi anaemia: the European Group for Blood and Bone Marrow Transplantation experience. Blood. 2013;122:4279–86.
Mehta PA, Davies SM, Leemhuis T, Myers K, Kernan NA, Prockop SE, et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood. 2017;129:2308–15.
Dufour C, Pierri F. Modern management of Fanconi anemia. Hematology. 2022;2022:649–57. de diciembre de
Fioredda F, Iacobelli S, Korthof ET, Knol C, van Biezen A, Bresters D, et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br J Haematol. 2018;183:110–8.
Gadalla SM, Sales-Bonfim C, Carreras J, Alter BP, Antin JH, Ayas M, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. l Biol Blood Marrow Transpl. 2013;19:1238–43.
Ayas M, Nassar A, Hamidieh AA, Kharfan-Dabaja M, Othman TB, Elhaddad A, et al. Reduced intensity conditioning is effective for hematopoietic SCT in dyskeratosis congenita-related BM failure. Bone Marrow Transpl. 2013;48:1168–72.
Fagioli F, Quarello P, Zecca M, Lanino E, Corti P, Favre C, et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol. 2014;165:673–81.
Achini-Gutzwiller FR, Snowden JA, Corbacioglu S, Greco R. Haematopoietic stem cell transplantation for severe autoimmune diseases in children: a review of current literature, registry activity and future directions on behalf of the autoimmune diseases and paediatric diseases working parties of the European Society for Blood and Marrow Transplantation. Br J Haematol. 2022;198:24–45.
Rabusin M, Snowden JA, Veys P, Quartier P, Dalle JH, Dhooge C, et al. Long-term outcomes of hematopoietic stem cell transplantation for severe treatment-resistant autoimmune cytopenia in children. Biol Blood Marrow Transpl. 2013;19:666–9.
Silva JMF, Ladomenou F, Carpenter B, Chandra S, Sedlacek P, Formankova R, et al. Allogeneic hematopoietic stem cell transplantation for severe, refractory juvenile idiopathic arthritis. Blood Adv. 2018;2:777–86.
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42:1473–507. octubre de
Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, et al. EBMT/ ESID inborn errors working party guidelines for hematopoietic stem cell trans- plantation for inborn errors of immunity. Bone Marrow Transpl. 2021;56:2052–62.
Schuetz C, Gerke J, Ege M, Walter J, Kusters M, Worth A, et al. Hypomorphic RAG deficiency: impact of disease burden on survival and thymic recovery argues for early diagnosis and HSCT. Blood. 2023;141:713–24. de febrero de
Albert MH, Sirait T, Eikema DJ, Bakunina K, Wehr C, Suarez F, et al. Hematopoietic stem cell transplantation for adolescents and adults with inborn errors of immunity: an EBMT IEWP study. Blood. 2022;140:1635–49. de octubre de
Lum SH, Albert MH, Gilbert P, Sirait T, Algeri M, Muratori R, et al. Outcomes of HLA-mismatched HSCT with TCRαβ/CD19 depletion or post-HSCT cyclophosphamide for inborn errors of immunity. Blood. 2024;144:565–80. de agosto de
Lum SH, Neven B, Slatter MA, Gennery AR. Hematopoietic cell transplantation for MHC Class II deficiency. Front Pediatr. 2019;7:516.
Aydin SE, Freeman AF, Al-Herz W, Al-Mousa HA, Arnaout RK, Aydin RC, et al. Hematopoietic stem cell transplantation as treatment for patients with DOCK8 deficiency. J Allergy Clin Immunol Pr. 2019;7:848–55.
Ferrua F, Galimberti S, Courteille V, Slatter MA, Booth C, Moshous D, et al. Hematopoietic stem cell transplantation for CD40 ligand deficiency: results from an EBMT/ESID-IEWP-SCETIDE-PIDTC study. J Allergy Clin Immunol. 2019;143:2238–53.
Shah RM, Elfeky R, Nademi Z, Qasim W, Amrolia P, Chiesa T, et al. T-cell receptor αβ + and CD19 + cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J Allergy Clin Immunol. 2018;141:1417–26.e1.
Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, et al. HLA- haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. 2014;124:822–6.
Laberko A, Sultanova E, Gutovskaya E, Shipitsina I, Shelikhova L, Kurnikova E, et al. Mismatched related vs matched unrelated donors in TCRαβ/CD19-depleted HSCT for primary immunodeficiencies. Blood. 2019;134:1755–63.
Neven B, Diana JS, Castelle M, Magnani A, Rosain J, Touzot F, et al. Haploi- dentical hematopoietic stem cell transplantation with post-transplant cyclophosphamide for primary immunodeficiencies and inherited disorders in children. Biol Blood Marrow Transpl. 2019;25:1363–73.
Folloni Fernandes J, Nichele S, Arcuri LA, Ribeiro L, Zamperlini-Netto G, et al. Outcomes after haploidentical stem cell transplantation with post-transplantation cyclophosphamide in patients with primary immunodeficiency diseases. Biol Blood Marrow Transpl. 2020;26:1923–9.
Merli P, Jodele S, Cook E, Myers KC, Lane A, Ryhänen S, et al. Emapalumab for the treatment of immune-mediated graft failure after HSCT. Bone Marrow Transplant. 2024;
Tsilifis C, Speckmann C, Lum SH, Fox TA, Soler AM, Mozo Y, et al. Hematopoietic stem cell transplantation for CTLA-4 insufficiency across Europe: a European Society for Blood and Marrow Transplantation Inborn Errors Working Party study. J Allergy Clin Immunol. 2024;154:1534–44. diciembre de
Fischer M, Olbrich P, Hadjadj J, Aumann V, Bakhtiar S, Barlogis V, et al. JAK inhibitor treatment for inborn errors of JAK/STAT signaling: An ESID/EBMT-IEWP retrospective study. J Allergy Clin Immunol. 2024;153:275–286.e18. enero de
Lehmberg K, Moshous D, Booth C. Haematopoietic stem cell transplantation for primary haemophagocytic lymphohistiocytosis. Front Pediatr. 2019;7:435.
Lucchini G, Marsh R, Gilmour K, Worth A, Nademi Z, Rao A, et al. Treatment dilemmas in asymptomatic children with primary hemophagocytic lymphohis-tiocytosis. Blood. 2018;132:2088–96.
Bakhtiar S, Salzmann-Manrique E, Blok HJ, Eikema DJ, Hazelaar S, Ayas M, et al. Allogeneic hematopoietic stem cell transplantation in leukocyte adhesion deficiency type I and III. Blood Adv. 2021;5:262–73.
Chiesa R, Wang J, Blok HJ, Hazelaar S, Neven B, et al. Despina Moshous hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood. 2020;136:1201–11.
Migliavacca M, Barzaghi F, Fossati C, Rancoita PMV, Gabaldo M, Dionisio F, et al. Long-term and real-world safety and efficacy of retroviral gene therapy for adenosine deaminase deficiency. Nat Med 2024;30:488–97. febrero de
Kohn DB, Booth C, Shaw KL, Xu-Bayford J, Garabedian E, Trevisan V, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med. 2021;384:2002–13.
Cowan MJ, Yu J, Facchino J, Fraser-Browne C, Sanford U, Kawahara M, et al. Lentiviral gene therapy for artemis-deficient SCID. N Engl J Med. 2022;387:2344–55. de diciembre de
Kohn DB, Booth C, Kang EM, Pai SY, Shaw KL, Santilli G, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med. 2020;26:200–6.
Ferrua F, Cicalese MP, Galimberti S, Giannelli S, Dionisio F, Barzaghi F, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6:e239–e253.
Fox T, Bueren J, Candotti F, Fischer A, Aiuti A, Lankester A, et al. Access to gene therapy for rare diseases when commercialization is not fit for purpose. Nat Med 2023;29:518–9. marzo de
Booth C, Aiuti A. Realizing the potential of gene therapies for rare and ultra-rare inherited diseases. Hum Gene Ther. 2023;34:776–81. septiembre de
Tan EY, Boelens JJ, Jones SA, Wynn RF. Hematopoietic stem cell transplantation in inborn errors of metabolism. Front Pediatr. 2019;7:433.
Tucci F, Consiglieri G, Cossutta M, Bernardo ME. Current and future perspective in hematopoietic stem progenitor cell-gene therapy for inborn errors of metabolism. Hemasphere. 2023;7:e953. octubre de
Fumagalli F, Calbi V, Natali Sora MG, Sessa M, Baldoli C, Rancoita PMV, et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399:372–83. 22 de enero de
Gentner B, Tucci F, Galimberti S, Fumagalli F, De Pellegrin M, Silvani P, et al. Hematopoietic stem- and progenitor-cell gene therapy for Hurler syndrome. N Engl J Med. 2021;385:1929–40.
Consiglieri G, Tucci F, De Pellegrin M, Guerrini B, Cattoni A, Risca G, et al. Early skeletal outcomes after hematopoietic stem and progenitor cell gene therapy for Hurler syndrome. Sci Transl Med. 2024;16:eadi8214. mayo de
Federico A, de Visser M. New disease-modifying therapies for two genetic childhood-onset neurometabolic disorders (metachromatic leucodystrophy and adrenoleucodystrophy). Neurol Sci. 2021;42:2603–6.
Duncan CN, Bledsoe JR, Grzywacz B, Beckman A, Bonner M, Eichler FS, et al. Hematologic cancer after gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2024;391:1287–301. de octubre de
Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9:522–36.
De La Fuente J, Dhedin N, Koyama T, Bernaudin F, Kuentz M, Karnik L, et al. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international learning collaborative. Biol Blood Marrow Transplant. 2019;25:1197–209. junio de
Forni GL, Gianesin B, Musallam KM, Longo F, Rosso R, Lisi R, et al. Overall and complication‐free survival in a large cohort of patients with β‐thalassemia major followed over 50 years. Am J Hematol 2023;98:381–7. marzo de
Baronciani D, Angelucci E, Potschger U, Gaziev J, Yesilipek A, Zecca M, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000–2010. Bone Marrow Transpl. 2016;51:536–41. abril de
Galambrun C, Pondarré C, Bertrand Y, Loundou A, Bordigoni P, Frange P, et al. French multicenter 22-year experience in stem cell transplantation for beta-thalassemia major: lessons and future directions. Biol Blood Marrow Transplant. 2013;19:62–8. enero de
Oostenbrink LVE, Pool ES, Jol-van Der Zijde CM, Jansen-Hoogendijk AM, Vervat C, Van Halteren AGS, et al. Successful mismatched hematopoietic stem cell transplantation for pediatric hemoglobinopathy by using ATG and post-transplant cyclophosphamide. Bone Marrow Transpl. 2021;56:2203–11. septiembre de
Vellaichamy Swaminathan V, Ravichandran N, Ramanan KM, Meena SK, Varla H, Ramakrishnan B, et al. Augmented immunosuppression and PTCY-based haploidentical hematopoietic stem cell transplantation for thalassemia major. Pediatr Transpl. 2021;25:e13893.
Anurathapan U, Hongeng S, Pakakasama S, Songdej D, Sirachainan N, Pongphitcha P, et al. Hematopoietic stem cell transplantation for severe thalassemia patients from haploidentical donors using a novel conditioning regimen. Biol Blood Marrow Transplant. 2020;26:1106–12. junio de
Matthes‐Martin S, Lawitschka A, Fritsch G, Lion T, Grimm B, Breuer S, et al. Stem cell transplantation after reduced‐intensity conditioning for sickle cell disease. Eur J Haematol. 2013;90:308–12. abril de
Eapen M, Brazauskas R, Walters MC, Bernaudin F, Bo-Subait K, Fitzhugh CD, et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. 2019;6:e585–96. noviembre de
Foell J, Schulte JH, Pfirstinger B, Troeger A, Wolff D, Edinger M, et al. Haploidentical CD3 or α/β T-cell-depleted HSCT in advanced-stage sickle cell disease. Bone Marrow Transpl. 2019;54:1859–67. noviembre de
Locatelli F, Lang P, Wall D, Meisel R, Corbacioglu S, Li AM, et al. Exagamglogene autotemcel for transfusion-dependent β-thalassemia. N Engl J Med. 2024;390:1663–76. de mayo de
Frangoul H, Locatelli F, Sharma A, Bhatia M, Mapara M, Molinari L, et al. Exagamglogene autotemcel for severe sickle cell disease. N Engl J Med. 2024;390:1649–62. 9 de mayo de
Kwiatkowski JL, Walters MC, Hongeng S, Yannaki E, Kulozik AE, Kunz JB, et al. Betibeglogene autotemcel gene therapy in patients with transfusion-dependent, severe genotype β-thalassaemia (HGB-212): a non-randomised, multicentre, single-arm, open-label, single-dose, phase 3 trial. Lancet. 2024;404:2175–86. noviembre de
Kanter J, Walters MC, Krishnamurti L, Mapara MY, Kwiatkowski JL, Rifkin-Zenenberg S, et al. Biologic and clinical efficacy of lentiglobin for sickle cell disease. N Engl J Med. 2022;386:617–28. de febrero de
Thuret I, Ruggeri A, Angelucci E, Chabannon C. Hurdles to the adoption of gene therapy as a curative option for transfusion-dependent thalassemia. Stem Cells Transl Med. 2022;11:407–14. de abril de
Schoemans H, Burns LJ, Liptrott SJ, Murray J, Kenyon M, Barata A, et al. Patient engagement in hematopoietic stem cell transplantation and cell therapy: a survey by the EBMT patient engagement task force & transplantation complications working party. Bone Marrow Transpl. 2024;59:1286–94. septiembre de
Acknowledgements
The authors are grateful for the advice and helpful comments received from a number of EBMT fellow specialists in HCT, including Michael Albert, Luca Castagna, Yves Chalandon, Ghandi Damaj, Régis Peffault de Latour, Peter Dreger, Carlo Dufour, Rémy Dulery, Bertram Glass, Patrick Hayden, Olivier Hermine, Charalimpia Kyriakou, Lynn Manson, Silvia Montoto, Nour Moukalled, Guillermo Ortí, Anna Ossami Saidi, Marie Robin, Yasmina Serroukh, Norbert Schmitz, Alina Tanase, Catherine Thieblemont.
Author information
Authors and Affiliations
Contributions
RG, AR, IS-O and JAS coordinated the writing of the manuscript following approval of the structure and authorship at the EBMT Board and Scientific Council on 19 October 2024. Relevant sections were drafted by authors designated according to roles within EBMT Scientific Council, Working Parties (WPs) and other roles: namely, AR (Cellular Therapy and Immunobiology WP), TA with RG (Autoimmune Diseases WP); DPM (Chronic Malignancies WP), FC (Acute Leukaemia WP), ZP (Transplant Complications WP), AB (Lymphoma WP); PP (for Solid Tumours, within Cellular Therapy and Immunobiology WP chaired by AR); KK (Paediatric Diseases WP), AMR (Severe Aplastic Anaemia WP); EA (Haemoglobinopathies WP), BN (Inborn Errors WP), with input from DA (Infectious Diseases WP), JAS (for Quality, JACIE and EBMT Benchmarking), MK and HM (Nurses Group). RG, AR, IS-O, JAS, MDH and AS provided cross-cutting advisory expertise throughout. RG, AR and IS-O edited the manuscript and all authors provided input across all sections in the final drafts of the manuscript according to expertise. Other specialists from EBMT are acknowledged. The manuscript was agreed and finalised at the EBMT Board and Scientific Council Meeting on 29 March 2025. All authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
RG discloses speaking honoraria from Biotest, Pfizer, Medac, BMS, MSD, Neovii, Kyverna and Magenta; advisory board from Pfizer. AR: declares honoraria from Gilead and Kyte. JAS declares advisory board membership for the last 36 months with Medac, Jazz, Vertex and BMS. TA received study support from Johnson & Johnson. DA delcares MSD – local PI in a clinical trial, advisory board. AB: declares Speaker bureau or advisory board: Novartis, Roche, Sanofi, Jazz, Takeda, Hikma, Biologix, Jansen, MSD, Abbvie, Pfizer and Amgen and Research support: Novartis, Roche, Takeda, Biologix, Hikma, Pfizer and Jansen. MDH: none MK advisory board Jazz, Alexion. KK: declares speaker’s bureau for Novartis, medac, Pierre Fabre. ZP declares advisory board for Amgen, Novartis and Pfizer. AMR: declares payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing, or educational events from Alexion (AstraZeneca Rare Disease), Apellis, Kira, Novartis, Omeros, Pfizer, Roche, and Sobi; and support for attending meetings and/or travel from Novartis and Sobi. AS declares TakedaBMS/Celgene, MSD. DPM, EA, MK, HM, BN, PP, ISO and FC declare no conflict of interests.
Ethics approval and consent to participate
This manuscript did not require ethical approval as it did not involve human participants, human tissue, animals, or identifiable personal data. It is based on EBMT recommendations, providing guidance on the indications for HCT and CAR-T in haematological diseases, solid tumours, and immune disorders, with reference to previously published literature, guidelines, and supporting evidence.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Greco, R., Ruggeri, A., McLornan, D.P. et al. Indications for haematopoietic cell transplantation and CAR-T for haematological diseases, solid tumours and immune disorders: 2025 EBMT practice recommendations. Bone Marrow Transplant (2025). https://doi.org/10.1038/s41409-025-02701-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41409-025-02701-3
