
Background
Homologous recombination deficiency (HRD) has emerged as a functional biomarker reflecting genome-wide DNA repair defects and genomic instability. While the Cancer Genome Atlas (TCGA) molecular classification provides valuable prognostic guidance in endometrial cancer (EC), it lacks resolution for DNA repair competency and therapeutic responsiveness. This study aimed to investigate whether HRD subtyping could complement TCGA classification for improved prognostic stratification and therapeutic decision-making.
Methods
A total of 142 EC patients were analysed using a next-generation sequencing panel and genomic scar-based HRD scoring (loss of heterozygosity, telomeric allelic imbalance, large-scale state transitions). Unsupervised clustering stratified patients into HRD-High, -Middle, and -Low groups. Maximally selected rank statistics were used to identify prognostic thresholds for HRD scores; the tumour-immune microenvironment was characterised by RNA-based immune gene expression profiling and multiplex immunohistochemistry. A support vector machine (SVM) model was developed for recurrence prediction.
Results
HRD subtyping identified distinct genomic, pathological, and immunological features. HRD-High tumours were associated with advanced FIGO stages, TP53 mutations, higher chromosomal instability, and elevated CD8⁺PD-1⁺ T-cell infiltration. HRD subtyping independently predicted disease-free survival and showed superior prognostic accuracy (C-index = 0.857) compared to TCGA subtyping (C-index = 0.751). Integrating HRD and TCGA classifiers further improved predictive performance (C-index = 0.903). An SVM model incorporating HRD score and immune features achieved an AUC of 0.733 for recurrence prediction.
Conclusions
HRD subtyping refines risk stratification beyond traditional TCGA classification and identifies patients potentially responsive to immune checkpoint or DNA damage-targeted therapies. Integrating HRD-based genomic instability metrics with molecular and immune profiling supports precision oncology in endometrial cancer.
Similar content being viewed by others


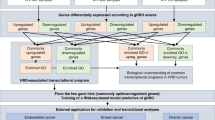
Introduction
Endometrial cancer (EC) is a common gynaecologic malignancy with a rising global burden. In 2022, more than 420,000 new cases and nearly 98,000 deaths were reported worldwide, with China accounting for the highest number of new diagnoses [1]. Over the past two decades, research on EC has undergone a significant shift, evolving from a primary focus on early detection to addressing the more urgent task of identifying high-risk patients with aggressive disease [2]. The molecular classification proposed by The Cancer Genome Atlas (TCGA) has demonstrated considerable prognostic value and has been clinically implemented through frameworks such as TransPORTEC and ProMisE, enhancing the feasibility of molecular subtyping in routine clinical practice [3, 4]. Despite the lack of standardised testing protocols and challenges related to implementation costs, molecular biomarkers have significantly advanced risk assessment, therapeutic decision-making, recurrence prediction, and target discovery. In 2023, the International Federation of Gynecology and Obstetrics (FIGO) incorporated TCGA subtypes into its staging system, integrating molecular features with conventional staging to improve the precision of risk stratification [5, 6]. Nonetheless, accurately forecasting patient outcomes and optimising adjuvant therapy selection remain among the most pressing challenges in EC management. Current TCGA-based molecular subtyping, although impactful, has inherent limitations: it does not assess the functional integrity of DNA repair pathways such as homologous recombination, lacks sensitivity to genome-wide chromosomal instability, and fails to account for intragroup heterogeneity—particularly within the MSI-H and TP53-wild-type subtypes. Furthermore, TCGA subtyping provides limited predictive insight into therapeutic response, especially in the context of DNA repair competence [7, 8]. These gaps underscore the need for a more integrative genomic profiling approach—one that not only captures established molecular subtypes but also delivers deeper DNA-level insights. Such a strategy could facilitate more nuanced patient stratification, elucidate subtype-specific biological variation, improve prognostic accuracy, and inform precision treatment strategies [9,10,11,12].
Homologous recombination deficiency (HRD) is a critical mechanism of genomic instability arising from defects in the homologous recombination repair (HRR) pathway, which plays a central role in the maintenance of DNA integrity [13, 14]. Loss of HRR function results in the accumulation of DNA double-strand breaks, chromosomal aberrations, and large-scale structural alterations that contribute to tumour progression and impact response to DNA-damaging agents such as platinum-based chemotherapy and PARP inhibitors [13, 15, 16]. HRD may also contribute to elevated tumour mutational burden and immune microenvironment variability, which have implications for therapeutic vulnerability [17, 18].
In EC, HRD is increasingly recognised as a biologically distinct and clinically relevant phenomenon. EC tumours with HRD features tend to display extensive chromosomal instability and are more likely to harbour mutations in DNA damage response genes such as TP53 and ARID1A, even in the absence of BRCA1/2 mutations [19]. These observations suggest that HRD in EC represents a convergent phenotype of genomic stress rather than a mutation-restricted event. Importantly, HRD-high represents a functional deficiency in HRR, distinct from TP53 mutations or generalised copy-number gains, which may reflect alternative pathways of genomic instability rather than specific DNA repair defects [20, 21]. Given these biological attributes, HRD serves as a promising complementary biomarker to existing classification frameworks. Unlike TCGA subtyping, which is primarily mutation- or pathway-based, HRD scoring quantifies structural consequences of impaired DNA repair across the genome. This functional perspective may enable improved resolution of molecular heterogeneity within TCGA categories and provide additional insight into therapeutic sensitivity.
Currently, several methods are available for assessing HRD status. These include: (1) identification of deleterious mutations in HRR genes such as BRCA1/2, (2) methylation analysis of the BRCA1 promoter, (3) mutational signature profiling, and (4) composite genomic scar assays based on next-generation sequencing (NGS) data [22]. Among these, SNP-based HRD scoring has gained widespread clinical acceptance. It quantifies HRD as the sum of three structural features of genomic instability—loss of heterozygosity (LOH), large-scale state transitions (LST), and telomeric allelic imbalance (TAI)—derived from high-density SNP array or NGS data [15]. This approach underpins the Myriad myChoice CDx test, which has been approved by the U.S. FDA as a companion diagnostic for PARP inhibitors in ovarian cancer.
However, applying uniform HRD score thresholds across different tumour types remains problematic. In ovarian cancer, the commonly used cutoff of ≥42 was established using BRCA-mutated cohorts and validated against treatment response [15]. In EC, where BRCA mutations are rare and HRD arises through more diverse and less-defined mechanisms, such a threshold may not accurately reflect clinically meaningful genomic instability [23]. More importantly, the lack of a validated surrogate or gold-standard comparator prevents the use of outcome-guided receiver operating characteristic (ROC) methods to define optimal cutoffs.
To address this limitation, unsupervised clustering based on the genomic distribution of LOH, LST, and TAI provides a data-driven approach to stratify patients. Unlike a fixed numerical threshold, clustering captures not only the total burden but also the chromosomal patterns of genomic scars, which may reflect distinct biological subtypes. This approach avoids arbitrary cutoffs and can be validated through associations with clinical, molecular, and immunologic features—offering a more flexible and biologically coherent classification framework, particularly in cancers like EC where HRD aetiology is heterogeneous and treatment paradigms remain under development [24, 25]. In parallel, we explored ROC curve analysis to identify optimal HRD score thresholds associated with survival outcomes; however, due to limited event rates, this approach did not yield a robust cutoff. Instead, we applied maximally selected rank statistics (maxstat) to define a prognostically relevant threshold. While clustering captures intrinsic genomic patterns and reflects underlying biology, maxstat-based cutoffs incorporate clinical outcome data to identify survival-associated inflection points. Together, these strategies provide complementary means of HRD stratification in EC. Standardisation of HRD assessment and prospective validation in clinical trials will be essential before widespread clinical adoption.
In this study, we developed a comprehensive DNA-based testing strategy to integrate genomic scar-based HRD scoring with broader NGS profiling. Our approach simultaneously supports TCGA subtyping, mutation analysis of 520 cancer-related genes, and assessment of genomic instability through HRD score components. This multi-dimensional platform is designed to facilitate biologically informed molecular stratification and to provide actionable insights for prognosis and treatment guidance in EC. Specifically, we aimed to characterise the clinicopathologic, molecular, and immune features associated with HRD subtypes, assess their distribution across TCGA groups, and evaluate their potential utility in refining prognostic prediction and adjuvant treatment decision-making.
Materials and methods
Patients and study design
This study included 142 patients who underwent surgical treatment for EC at Peking University Cancer Hospital between June 2011 and July 2020. All patients received no preoperative chemoradiotherapy or hormonotherapy and underwent at a minimum a total hysterectomy with bilateral salpingo-oophorectomy. Patients were eligible for inclusion if they had a histopathological diagnosis of EC based on primary tumour samples. FIGO stages ranged from I to IV. For inclusion, each patient was also required to have available tumour tissue providing at least 15 serial sections suitable for NGS and multiplex immunofluorescence staining. All cases were assigned a unique study identifier, and pathological diagnoses were independently reviewed and confirmed by two experienced gynaecologic pathologists. The study was approved by the Ethics Review Board of Peking University Cancer Hospital (Approval No. 2021YJZ21) and conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants.
Tissue DNA sequencing and data analyses
Tissue DNA was extracted from FFPE tumour samples using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). Capture-based targeted sequencing was conducted using a 520-gene panel (OncoScreen Plus™, Burning Rock Biotech, Guangzhou, China) [26]. Variant calling and annotation followed standardised pipelines. Tumour mutation burden per patient was calculated as the ratio of non-synonymous mutations to the total coding region size, including non-synonymous single-nucleotide variants and insertions/deletions (Indels) within the coding region and adjacent ±2 bp flanking regions. Copy-number variations (CNVs) were assessed using depth of coverage data for capture intervals. Bias corrections were applied for GC content and probe design. Normalisation used average coverage across all captured regions, with references from over 50 samples devoid of CNVs. CNVs were identified when the depth of coverage in tumour samples significantly deviated from reference values, with detection limits set at 1.5 for deletions and 2.64 for amplifications. Structural aberrations were counted as one per region of intrachromosomal gain or loss.
TCGA classifier
(1) POLE mutations were defined following NCCN guidelines, focusing on hotspot variants within the exonuclease domain, identified as causative for ultra-mutations [27]. (2) MSI status was determined using a read-count-distribution-based method as previously described [28, 29]. (3) TP53 mutations were identified through sequencing and annotated as missense, loss-of-function or gain-of-function mutations, reflecting their impact on tumour behaviour [30].
HRD score
The HRD score was calculated as the arithmetic sum of three genomic scar metrics: LOH, TAI, and LST. All three metrics were derived from high-depth targeted sequencing data using SNP-based copy-number segmentation algorithms, consistent with principles used in clinical HRD assays. LOH was defined as the number of contiguous regions of allelic loss greater than 15 Mb but not encompassing an entire chromosome. TAI was defined as the number of subchromosomal regions greater than 11 Mb in length that exhibited allelic imbalance extending to a subtelomeric region without crossing the centromere. LST was defined as the number of breakpoints between adjacent chromosomal segments greater than 10 Mb in length, excluding regions smaller than 3 Mb. The final HRD score was computed as HRD = LOH count + TAI count + LST count, as initially proposed by Telli et al. [15].
HRD subtype classification and the optimal threshold
HRD subtypes were derived using unsupervised hierarchical clustering based on genome-wide distributions of LOH, TAI, and LST scores. Tumours were grouped into HRD-High, HRD-Middle, and HRD-Low subtypes based on the relative burden and chromosomal distribution of genomic scars. To determine the optimal threshold for stratifying patients based on HRD score, we applied maximally selected rank statistics (maxstat) using the exact Gauss approximation. This method identifies the cutpoint that best separates survival curves according to the log-rank test. The analysis was performed separately for disease-free survival (DFS) and overall survival (OS).
Aneuploidy score, SCIN and WGD
Aneuploidy score was defined as the total number of chromosome arms that were gained or lost [31]. The structural complexity score (SCIN) was defined as the sum of all structurally aberrant regions [32]. Whole-genome doubling (WGD) status was inferred from allele-specific DNA copy-number analysis. Tumours were considered to have undergone WGD if more than 50% of their autosomal genome had a major copy number (the most frequent allele in a given segment, MCN) greater than or equal to two [33].
RNA sequencing and gene expression profiling
RNA was extracted from FFPE samples using the AllPrep DNA/RNA FFPE Kit (Qiagen, Germany). RNA quantity and quality were assessed with the Qubit RNA HS assay (Thermo Fisher Scientific, USA) and LabChip GXII Touch 24 system (PerkinElmer, USA), respectively. Fragmented RNA underwent strand-specific cDNA synthesis, dA-tailing, UMI adaptor ligation, PCR amplification, and hybridisation with capture probes from the gene expression profiling (GEP) panel (Burning Rock Biotech, China). The panel consists of 83 immunomodulatory genes (Table S1). The prepared libraries were sequenced, and the sequencing data were processed as previously described. Gene-level expression values were computed as transcripts per million and normalised to Z-scores before clustering. The score of 83 T-cell inflamed-related genes was calculated based on GEP data.
Multiplex immunohistochemistry assay and image analysis
Multispectral imaging, following a multiplex immunohistochemistry protocol, was utilised to analyse tumour-infiltrating lymphocytes in 5-μm-thick sections of FFPE tumour tissues. Using the PANO 7-plex IHC kit (Panovue, Beijing, China), these sections underwent epitope retrieval, blocking of endogenous peroxidase and proteins, antigen labelling, and signal amplification via tyramide (TSA). Two antibody panels were employed: one targeting PD-L1, PD-1, CD3, CD8, and Pan-CK, and another targeting CD68, CD163, CD56, and Pan-CK. After antigen labelling, sections were incubated with HRP-labelled secondary antibodies at 37 °C for 10 min, followed by microwave-based heat treatment during each TSA development step. Nuclear counterstaining was performed using DAPI. Multispectral scans were acquired with the Mantra Quantitative Pathology Imaging System (PerkinElmer, Waltham, Massachusetts, USA), collecting fluorescent spectra at 20-nm intervals across 420–720 nm. Composite images were generated by integrating these spectra, and a spectral library was constructed using images of unstained and singly stained sections to distinguish autofluorescence from specific fluorophore signals. Quantitative image analysis was conducted using InForm software (PerkinElmer, Waltham, Massachusetts, USA).
Immune cell populations were evaluated separately within the tumour parenchyma and stroma. The analysis included CD3+ T cells, CD8+ T cells, M1 macrophages, M2 macrophages, and natural killer (NK) cells, with associated markers such as PD-1, PD-L1, CD3, CD8, CD56, CD163, and CD68. Immune cell metrics were expressed as the positive ratio (proportion of positively stained cells among total cells). For analyses requiring dichotomisation, including CD8+PD-1+ T cells, patients were stratified into high and low groups based on an optimal cutoff determined by ROC curve analysis.
Statistical analyses
Statistical analyses were conducted using R (version 4.0.0). Chi-square or Fisher’s exact test compared categorical variables, while the Wilcoxon rank-sum or Kruskal–Wallis test compared continuous variables. Survival differences were examined using Kaplan–Meier (KM) curves with log-rank tests. Cox proportional hazards regression analysed relationships between variables and DFS/OS, including variables with p < 0.05 in univariate analysis. A two-sided p < 0.05 was considered significant. Harrell’s concordance index (C-index) was used to measure the discriminative ability of the molecular classifier model to predict the outcomes (DFS and OS) [29]. A C-index of 0.5 indicates that the model has no discriminative ability, and a C-index of 1 indicates that the model perfectly distinguishes between those who experienced an event and those who did not. For predicting recurrence, a support vector machine (SVM) model was developed using immune cell densities and HRD scores as input features. Model training and validation were performed using a stratified 5-fold cross-validation approach to ensure robustness. Model performance was evaluated using the area under the ROC area under curve (AUC) as the primary metric, with an AUC of 1 indicating perfect predictive accuracy and 0.5 indicating no predictive ability. Hyperparameters were optimised to minimise bias and overfitting, and feature importance was assessed through recursive feature elimination to determine the relative contributions of individual predictors.
Results
Patient characteristics
As illustrated in Table 1, patients’ age at diagnosis ranged from 30 to 78 (median 54) years old, with a median body mass index (BMI) of 25.66. The majority of cases (84.5%) were endometrioid, while 6 (4.2%) were serous, 4 (2.8%) were clear cell, and the remaining cases were mixed and other histological types. Grade distribution included 37 (26.1%) grade 1, 57 (40.1%) grade 2, and 48 (33.8%) grade 3. There were 98 (69.0%) patients diagnosed with FIGO stage I disease, while 14 (9.9%) were classified as stage II, 23 (16.2%) as stage III, and 4 (2.8%) as stage IV. Lymphovascular space invasion (LVSI) was evaluated in 138 cases, with 42 (29.6%) testing positive. Deep myometrial invasions were found in 62 (43.7%) cases. In total, 79 (55.7%) patients received adjuvant therapy such as chemotherapy, radiotherapy, and chemoradiotherapy. The median follow-up time of the cohort was 38.8 months (range, 1.9‒106.2 months), and 12.7% (18/142) of patients relapsed. The clinicopathological, molecular, and tumour-immune microenvironment data used in this study are summarised in Table S2.
Clinical and molecular characterisation of different HRD subtypes
We performed unsupervised clustering based on the distribution of LOH, TAI, and LST events across chromosomes, categorising 142 patients into three HRD subtypes: HRD-high, HRD-middle, and HRD-low, representing different levels of HRD (Fig. S1A). In the HRD-high group, genomic scars were present on nearly all chromosomes. In contrast, the HRD-middle group exhibited high-frequency genomic scars on specific chromosomes, such as chr1, chr2, and chr10, while the HRD-low group had almost no genomic scars. To further explore the prognostic relevance of HRD scores, we examined their distribution across the cohort and identified an optimal cutoff of 12 using maximally selected log-rank statistics (Fig. S1B). Based on this threshold, patients were dichotomised into HRD-high (≥12) and HRD-low (<12) groups. KM survival analysis revealed that the HRD-high group was associated with significantly improved outcomes in both DFS (HR = 0.08, 95% CI: 0.03–0.21, p = 1.73 × 10−7) and OS (HR = 0.08, 95% CI: 0.02–0.28, p = 7.73 × 10−5). These findings remained robust in multivariate Cox models adjusting for age, FIGO stage, and LVSI, where HRD-high status independently predicted longer DFS and OS (Table S3). The boxplot reveals significant differences among the three subgroups, with the HRD-High group showing a distinct cutoff compared to the other two groups (p < 0.05).
Notably, nearly 60% of HRD-high patients were in FIGO stages III and IV, significantly higher than the other two groups, with diverse histological subtypes observed (p = 0.006, p = 0.005; Fig. S1C). The incidence of deep myometrial invasion and LVSI also increased significantly with higher HRD levels (p = 0.031, p = 0.001; Fig. S1C).
We further analysed the molecular features of the three HRD subtypes. Figure 1a, b shows the mutation profiles of the top 20 most frequent mutated genes in our cohort of EC patients, highlighting the co-occurrence and mutually exclusive relationships between these mutations. Compared to the other two groups, the HRD-high group exhibited higher mutation frequencies in TP53, RAD51, and SPOP, but fewer mutations in PTEN, ARID1A, PIK3R1, CTNNB1, and CTCF (all P < 0.05; Fig. 1c). Notably, mutations in PTEN and TP53 were significantly mutually exclusive (p < 0.05; Fig. 1b).

a Oncoprint plot illustrating the distribution of genomic alterations across HRD subtypes (High, Middle, Low) and their associations with TCGA molecular classifiers, FIGO stages, histological types, and myometrial invasion. Key genomic events such as LOH, LST, TAI, and HRD scores are annotated. b Co-occurrence and mutual exclusivity analysis of frequently altered genes across the cohort. Heatmap displays −log10(p value) for co-occurrence, with significant relationships marked (*P < 0.05, **P < 0.01). c Bar plot showing the frequency of gene alterations across HRD subtypes. Light blue represents P < 0.05, and dark blue represents P > 0.05. d Correlation analysis of HRD scores with aneuploidy score (D-1) and SCIN (D-2). Boxplot compares HRD scores between whole-genome doubling (WGD-positive) and WGD-negative groups (D-3). e Stacked bar plot illustrating the distribution of TCGA molecular subtypes (MSI-H, POLE, TP53_mutant, TP53_wildtype) within HRD subtypes (High, Middle, Low).
The HRD score was strongly positively correlated with genomic instability markers, such as SCIN (R = 0.93, p < 0.01) and aneuploidy score (R = 0.83, p < 0.001). Moreover, HRD scores were significantly higher in WGD-positive samples compared to WGD-negative samples (p < 0.001; Fig. 1d).
To explore the relationship between TCGA molecular subtyping and HRD subtypes, we categorised the samples based on the TCGA classifier into POLE, MSI-H, TP53-mut, and TP53-wt groups. In the HRD-high group (n = 12), no patients carried POLE mutations, with 58% being TP53-mut and the remaining 42% being either MSI-H or TP53-mut. The HRD-middle group (n = 53) had the highest proportion of MSI-H and TP53-wt patients (30% each), while the HRD-low group (n = 75) consisted of over 50% TP53-wt patients (p < 0.001; Fig. 1e).
Prognostic value of HRD subtype stratification
We conducted survival analyses by grouping the study cohort according to both the TCGA molecular classifier and the HRD subtypes to assess OS among different patient subgroups. KM curves indicated that among the TCGA subtypes, POLE-mutant patients had the best prognosis, while TP53-mutant patients had the worst (Fig. 2a). This finding aligns with previous studies on TCGA molecular classification and its prognostic implications. Within the HRD subtype stratification, the HRD-High group demonstrated the worst prognosis, while the HRD-Low group exhibited the best (p < 0.001; Fig. 2b). Notably, the HRD-Low group (n = 75) identified a substantially larger subset of patients with a favourable prognosis compared to the POLE-mutant group (n = 10). Conversely, although the HRD-High group included only 12 patients, their median OS was just 30 months.

a Kaplan–Meier curve showing overall survival (OS) among TCGA molecular subtypes (MSI-H, POLE, TP53_mutant, TP53_wildtype). b Kaplan–Meier curve comparing OS among HRD subtypes (High, Middle, Low). c Bar plot of c-index values from Cox models evaluating the predictive accuracy of TCGA molecular classification, HRD subtype, and the combined model. Light green, dark green, and teal bars represent c-index values for the TCGA classifier, HRD subtype, and the combined model, respectively. d Kaplan–Meier curve showing OS stratified by HRD subtypes within the MSI-H group. e Kaplan–Meier curve showing OS stratified by HRD subtypes within the TP53 wild-type group. f Kaplan–Meier curve showing OS stratified by HRD subtypes within the TP53-mutant group.
We further evaluated the predictive performance of the TCGA molecular classifier and the HRD subtypes using c-index values derived from Cox models. The c-index scores for the TCGA classifier, HRD subtype, and the combined model were 0.751, 0.857, and 0.903, respectively (Fig. 2c). These results suggest that HRD subtype stratification provides superior prognostic accuracy compared to the TCGA molecular classification, and the interaction between the two classifiers enhances their predictive capability further.
Building on these findings, we incorporated HRD subtypes into the TCGA molecular classification for additional stratification. Despite the limited sample size, results showed significant prognostic differences among HRD levels within the MSI-H and TP53 wild-type subgroups (p = 0.01, p < 0.001; Fig. 2d, e). Similar trends were observed in the TP53-mutant subgroup (Fig. 2f).
Tumour-immune microenvironment characteristics across HRD subtypes
Clustering analysis revealed significant immune cell composition heterogeneity within each HRD subtype, with no significant differences in the positive ratio of eight immune cell types across the three groups, except for NK cells in the tumour parenchyma (Fig. S2A). Previous studies have highlighted that MSI-H and POLE-mutant tumours exhibit higher immunogenicity and distinct immune microenvironment features. Therefore, we grouped MSI-H and POLE-mutant patients as independent categories and compared them with the remaining samples stratified by HRD subtype.
Notably, MSI-H and POLE-mutant patients displayed significantly higher densities of CD3+ and CD8+ T cells both in the tumour and stroma compared to other subgroups (all p < 0.05; Fig. S2B). In addition, HRD-High patients exhibited significantly elevated numbers of CD3+PD-1+ T cells and CD8+PD-1+ T cells in their tumour-immune microenvironment compared to HRD-Middle and HRD-Low groups (all p < 0.05; Fig. 3a, b). The GEP score of T-cell inflamed-related genes, which reflects the intensity of the immune inflammatory response, exhibited considerable variability in the HRD-high group, but overall showed a strong inflammatory response (Fig. 3c).

a Violin plots showing the positive ratios of tumour-infiltrating CD3+PD-1+ T cells, CD8+PD-1+ T cells, and stromal CD3+PD-1+ T cells across HRD subtypes (High, Middle, Low) and MSI-H/POLE groups. b Violin plots showing the positive ratios of tumour-infiltrating CD3+PD-1+ T cells and CD8+PD-1+ T cells in the tumour and stroma across HRD subtypes (High, Middle, Low) and MSI-H/POLE groups. c Violin plot showing T-cell inflamed GEP scores across HRD subtypes and MSI-H/POLE groups. d Kaplan–Meier curve showing DFS stratified by high and low ratios of stromal CD8+PD-1+ T cells. e Kaplan–Meier curves showing DFS stratified by the positive ratios of tumour CD3+ T cells, stromal CD3+ T cells, tumour CD8+ T cells, stromal CD163+CD68+ macrophages, and tumour CD163-CD68+ macrophages.
Subsequently, we analysed the relationship between the positive ratio of these immune cell types and both OS and DFS. While immune cell positive ratio did not significantly affect OS in the context of the treatment regimen used in this cohort, we found that higher densities of CD3+ T cells (in both parenchyma and stroma), CD8+ T cells (in parenchyma), M1 macrophages (in parenchyma), and M2 macrophages (in stroma) were associated with a lower risk of recurrence or progression (Fig. 3e). Additionally, the DFS of patients with low-positive ratio CD8+PD-1+ T cells was significantly better than those with high-positive ratio CD8+PD-1+ T cells (Fig. 3d).
HRD subtypes as predictive markers for treatment efficacy and recurrence risk
Compared to the TCGA molecular classification, the HRD subtype provided superior prediction of DFS (Fig. 4a). To address potential confounding, we performed multivariate Cox proportional hazards regression, adjusting for age, FIGO stage, BMI, LVSI, and depth of myometrial invasion. HRD subtype remained an independent prognostic factor for survival, with both HRD-Middle and HRD-Low subtypes showing significantly reduced hazard ratios compared to the HRD-High group (HR = 0.04 and 0.01, respectively; both p < 0.001; Fig. S3A). The prediction performance of the HRD subtype model surpassed that of the TCGA molecular classification. The interactive model demonstrated the highest c-index, indicating the best predictive accuracy (TCGA classifier vs. HRD subtype vs. TCGA classifier: HRD subtype: 0.669: 0.787: 0.848; Fig. 4b). In clinical practice, patients classified as high or intermediate risk typically undergo adjuvant therapy. Among patients receiving such treatments, the HRD-High group exhibited the poorest prognosis, with DFS significantly lower than both the Middle (n = 35) and Low (n = 37) groups (Fig. 4c). Regardless of the treatment type—CR, R, or C—DFS demonstrated statistically significant differences among the HRD subgroups (all p < 0.05; Fig. 4d–f). In the CR treatment group, both DFS and OS exhibited significant differences across the HRD subgroups (Fig. S3B). Notably, 67% (8/12) of HRD-High patients experienced recurrence. In contrast, no significant DFS difference was observed between the HRD-Middle and Low groups in the low-risk clinical cohort (Fig. S3C). The treatment efficacy of adjuvant therapies according to the TCGA classification is presented in Fig. S4. Finally, we established a recurrence prediction model using SVM, which demonstrated that combining the immune cell positive ratio with HRD score yielded the best predictive performance (AUC = 0.733; Fig. 4g).

a Kaplan–Meier curves showing disease-free survival (DFS) among TCGA molecular subtypes (MSI-H, POLE, TP53_mutant, TP53_wildtype) and HRD subtypes (High, Middle, Low). b Bar plot of c-index values from Cox models evaluating the predictive performance of TCGA molecular classification, HRD subtype, and the combined model. Light blue, medium blue, and dark blue bars represent c-index values for the TCGA classifier, HRD subtype, and the combined model, respectively. c Kaplan–Meier curve showing DFS stratified by HRD subtypes (High, Middle, Low) in patients receiving adjuvant therapy. d Kaplan–Meier curve showing DFS stratified by HRD subtypes (High, Middle, Low) in the chemoradiotherapy (CR) treatment group. e Kaplan–Meier curve showing DFS stratified by HRD subtypes (High, Middle, Low) in the radiotherapy (R) treatment group. f Kaplan–Meier curve showing DFS stratified by HRD subtypes (High, Middle, Low) in the chemotherapy (C) treatment group. g ROC curve comparing the predictive performance of HRD subtypes, immune cell positive ratios, and the combined model (AUC = 0.733 for the combined model).
Discussion
HRD subtypes in EC were closely associated with adverse clinical features and aggressive tumour biology. HRD-High tumours were more frequently diagnosed at advanced stages and exhibited higher rates of deep myometrial invasion and LVSI, aligning with their elevated recurrence risk and poor survival outcomes [34]. Genomically, HRD-High tumours showed distinct mutational patterns—characterised by enrichment of TP53, RAD51, and SPOP mutations—and extensive chromosomal instability, including high SCIN and aneuploidy scores [35, 36]. These tumours also demonstrated widespread genomic scars and a high prevalence of WGD, reflecting their elevated intratumoral heterogeneity and capacity to tolerate DNA damage [37, 38]. In contrast, HRD-Middle and HRD-Low tumours were more often associated with PI3K-AKT or Wnt pathway alterations, suggesting alternative oncogenic drivers and more stable genomic architectures [39]. These findings support HRD-High as a biologically distinct and clinically high-risk subgroup with implications for treatment resistance and recurrence.
Beyond their association with aggressive biology, HRD subtypes also demonstrate meaningful divergence from existing molecular classification systems. When stratified by HRD subtypes, the HRD-high group demonstrated the worst prognosis, and the HRD-low group exhibited the best, indicating a strong association between HRD defects and tumour prognosis. Notably, the c-index evaluation suggested that HRD subtypes offer superior prognostic accuracy compared to the TCGA molecular classification. The data revealed that the HRD-low group, which includes a much larger sample size than the POLE group (75 vs. 10), contains 6 patients with TP53 mutations—patients who are typically associated with worse prognosis according to TCGA classification. Among the two groups with the poorest prognosis, the median OS for HRD-high patients was shorter than that of TP53-mutant patients. The c-index results also indicated that combining both classifiers could enhance the explanatory and predictive power regarding patient survival. HRD subtyping revealed additional prognostic information not captured by TCGA mutation-based groups. While HRD-High tumours overlapped with TP53-mutant cases and were absent among POLE-mutant tumours, HRD stratification further differentiated outcomes within TCGA-defined subtypes. For instance, multiple patients with serous carcinoma, FIGO stage III disease, and TP53 mutations received standard chemoradiotherapy. Among them, those with HRD-Low status (Patients 13 and 122) did not experience recurrence, whereas those classified as HRD-Middle or HRD-High (Patients 50 and 33) did. Similar patterns were observed among TP53-wildtype (Patients 132 and 66) and MSI-H tumours (Patients 16, 136, 49), where recurrence was more frequent in HRD-High cases despite otherwise comparable clinical features and treatment (Table S4). These real-world cases exemplify the ability of HRD subtyping to further stratify risk within clinically and molecularly homogeneous groups, reinforcing its potential as a complementary biomarker.
In addition to prognostic stratification, HRD status may influence tumour–immune interactions, which we further explored in this study. MSI-H and POLE-mutant patients exhibited significantly higher densities of CD3+ and CD8+ T cells in both tumour and stromal regions compared to other subtypes, suggesting a higher immunogenicity and more active immune microenvironment in these groups, consistent with prior studies [40,41,42]. High densities of CD3+, CD8+ T cells, and macrophages were associated with better DFS. Interestingly, the HRD-High group showed significantly higher levels of CD3+PD-1+ and CD8+PD-1+ T cells compared to the HRD-Middle and HRD-Low groups, indicating a dual immune phenotype characterised by T-cell recruitment and PD-1-mediated exhaustion [43]. HRD-driven genomic instability is known to increase tumour mutational burden and neoantigen generation, which may promote immune cell infiltration. However, this immunogenicity may be counterbalanced by immune evasion mechanisms such as PD-1 pathway upregulation, as reported in HR-deficient breast and ovarian cancers [18, 44, 45]. These findings suggest that HRD-High ECs may represent a subset of tumours with potential responsiveness to immune checkpoint inhibitors, particularly when used in combination with DNA damage-targeted therapies. Supporting this hypothesis, early clinical trials have demonstrated the promising efficacy of combined PARP inhibitor and anti-PD-1/PD-L1 regimens in HRD-associated tumours [46]. Although patients in our cohort did not receive immunotherapy, the presence of immune cell infiltration in HRD-High tumours highlights a tumour-intrinsic immune contexture that may be leveraged for future therapeutic strategies. CD8+PD-1+ T-cell enrichment, while associated with poor DFS in our study, may also serve as a biomarker for response to ICIs if immune evasion is pharmacologically reversed [47]. These observations underscore the translational potential of integrating HRD scoring with immune profiling to inform personalised treatment in EC.
Despite receiving standard adjuvant therapy, patients with HRD-High tumours experienced disproportionately high recurrence rates, suggesting a potential link between underlying genomic features and intrinsic resistance (Table S5). Mechanistically, HRD-High tumours exhibited extensive chromosomal instability, complex structural rearrangements, and frequent WGD—genomic hallmarks that may drive tumour plasticity and therapy evasion [48, 49]. These alterations have been associated with impaired apoptotic signalling, replication stress tolerance, and subclonal diversification, enabling tumour cells to adapt under therapeutic pressure [50]. Moreover, while HRD scoring reflects homologous recombination dysfunction, not all HRD-High tumours harbour functional BRCA1/2 inactivation [51]. This distinction may explain the heterogeneous therapeutic responses observed in HRD-High ECs and points to the need for more precise functional assays to identify repair-deficient, treatment-responsive subsets [52]. These findings underscore the importance of incorporating HRD subtype analysis not only for prognostic stratification, but also to unravel resistance mechanisms and guide future trials evaluating DNA damage-targeted and combination strategies in EC.
We developed a recurrence prediction model incorporating immune cell densities and HRD scores using an SVM framework. The model achieved an AUC of 0.733, indicating good predictive performance. These results suggest that integrating immune microenvironment features with HRD metrics may enhance recurrence risk stratification in EC.
This study has several limitations. The sample size was relatively small, which may limit the generalisability of our findings. Maximally selected rank statistics were used to identify prognostic thresholds for HRD scores; In addition, the absence of patients receiving immunotherapy limits the applicability of our immune microenvironment findings. Prospective studies with larger cohorts and therapeutic intervention arms are needed to validate the predictive and therapeutic relevance of HRD subtyping in EC.
Conclusions
This study demonstrates that HRD subtyping based on genomic scar patterns provides prognostic and predictive insights beyond the conventional TCGA molecular framework in EC. HRD-High tumours exhibited aggressive clinical behaviour, genomic instability, and immunologically active but potentially exhausted microenvironments, suggesting both poor prognosis and candidate sensitivity to combined immunotherapy and PARP inhibition. Integrating HRD subtyping with TCGA classification significantly improved prognostic stratification and recurrence prediction. These findings support the clinical utility of incorporating HRD-based assessment into molecular diagnostics to optimise adjuvant treatment decisions and guide biomarker-driven therapeutic strategies in EC.
Data availability
Data and results used in this study are available in the Supplementary Tables. Raw sequencing data are available at Peking University Cancer Hospital & Institute and can be made available upon reasonable request.
Code availability
Custom scripts used for statistical and survival analyses are available from the corresponding author upon reasonable request.
References
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63.
Urick ME, Bell DW. Clinical actionability of molecular targets in endometrial cancer. Nat Rev Cancer. 2019;19:510–21.
Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod Pathol. 2015;28:836–44.
Talhouk A, McConechy MK, Leung S, Yang W, Lum A, Senz J, et al. Confirmation of ProMisE: a simple, genomics-based clinical classifier for endometrial cancer. Cancer. 2017;123:802–13.
McAlpine J, Leon-Castillo A, Bosse T. The rise of a novel classification system for endometrial carcinoma; integration of molecular subclasses. J Pathol. 2018;244:538–49.
Stelloo E, Nout RA, Osse EM, Jürgenliemk-Schulz IJ, Jobsen JJ, Lutgens LC, et al. Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer-combined analysis of the PORTEC cohorts. Clin Cancer Res. 2016;22:4215–24.
de Jonge MM, Auguste A, van Wijk LM, Schouten PC, Meijers M, Ter Haar NT, et al. Frequent homologous recombination deficiency in high-grade endometrial carcinomas. Clin Cancer Res. 2019;25:1087–97.
Jiang F, Jiang S, Cao D, Mao M, Xiang Y. Immunologic signatures across molecular subtypes and potential biomarkers for sub-stratification in endometrial cancer. Int J Mol Sci. 2023;24:1791.
Raffone A, Travaglino A, Massimo M, Carbone L, Guida M, Insabato L, et al. TCGA molecular groups of endometrial cancer: pooled data about prognosis. Gynecol Oncol. 2019;155:374–83.
Makker V, MacKay H, Ray-Coquard I, Levine DA, Westin SN, Aoki D, et al. Endometrial cancer. Nat Rev Dis Prim. 2021;7:88.
Kommoss S, McConechy MK, Kommoss F, Leung S, Bunz A, Magrill J, et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann Oncol. 2018;29:1180–8.
Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer. 2015;113:299–310.
Groelly FJ, Fawkes M, Dagg RA, Blackford AN, Tarsounas M. Targeting DNA damage response pathways in cancer. Nat Rev Cancer. 2023;23:78–94.
Li W, Gao L, Yi X, Shi S, Huang J, Shi L, et al. Patient assessment and therapy planning based on homologous recombination repair deficiency. Genom Proteom Bioinform. 2023;21:962–75.
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22:3764–73.
O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–60.
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–25.
Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–98.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 2018;23:239–54.
Shi Z, Chen B, Han X, Gu W, Liang S, Wu L. Genomic and molecular landscape of homologous recombination deficiency across multiple cancer types. Sci Rep. 2023;13:8899.
Beinse G, Just PA, Le Frere Belda MA, Laurent-Puig P, Jacques S, Koual M, et al. Discovery and validation of a transcriptional signature identifying homologous recombination-deficient breast, endometrial and ovarian cancers. Br J Cancer. 2022;127:1123–32.
Witz A, Dardare J, Betz M, Michel C, Husson M, Gilson P, et al. Homologous recombination deficiency (HRD) testing landscape: clinical applications and technical validation for routine diagnostics. Biomark Res. 2025;13:31.
Shen M, Lin Y, Yao Z, Lin L, Sun Y. Homologous recombination deficiency in endometrial cancer: association with clinical and molecular characteristics. ESMO Open. 2024;9:16P.
Jamieson A, McAlpine JN. Molecular profiling of endometrial cancer from TCGA to clinical practice. J Natl Compr Cancer Netw. 2023;21:210–6.
Corr BR, Erickson BK, Barber EL, Fisher CM, Slomovitz B. Advances in the management of endometrial cancer. BMJ. 2025;388:e080978.
Wang M, Chen X, Dai Y, Wu D, Liu F, Yang Z, et al. Concordance study of a 520-gene next-generation sequencing-based genomic profiling assay of tissue and plasma samples. Mol Diagn Ther. 2022;26:309–22.
León-Castillo A, Britton Heidi, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol. 2020;250:323–35.
Zhu L, Huang Y, Fang X, Liu C, Deng W, Zhong C, et al. A novel and reliable method to detect microsatellite instability in colorectal cancer by next-generation sequencing. J Mol Diagn. 2018;20:225–31.
Cai Z, Wang Z, Liu C, Shi D, Li D, Zheng M, et al. Detection of microsatellite instability from circulating tumor DNA by targeted deep sequencing. J Mol Diagn. 2020;22:860–70.
Jamieson A, de Barros JS, Cochrane DR, Douglas JM, Shankar S, Lynch BJ, et al. Targeted and shallow whole-genome sequencing identifies therapeutic opportunities in p53abn endometrial cancers. Clin Cancer Res. 2024;30:2461–74.
Cohen-Sharir Y, McFarland JM, Abdusamad M, Marquis C, Bernhard SV, Kazachkova M, et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature. 2021;590:486–91.
Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–6.
Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet. 2018;50:1189–95.
Lin DI, Fine A, Danziger NA, Huang RSP, Mata DA, Decker B, et al. Molecular analysis of endometrial serous carcinoma reveals distinct clinicopathologic and genomic subgroups. Gynecol Oncol. 2022;164:558–65.
Thacker J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005;219:125–35.
Richardson C. RAD51, genomic stability, and tumorigenesis. Cancer Lett. 2005;218:127–39.
Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40.
Chang E, An JY. Whole-genome doubling is a double-edged sword: the heterogeneous role of whole-genome doubling in various cancer types. BMB Rep. 2024;57:125–34.
McConechy MK, Ding J, Senz J, Yang W, Melnyk N, Tone AA, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 2014;27:128–34.
Gargiulo P, Pepa CD, Berardi S, Califano D, Scala S, Buonaguro L, et al. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated endometrial cancers: new candidates for checkpoint blockade immunotherapy? Cancer Treat Rev. 2016;48:61–8.
Maiorano BA, Pio Maiorano MF, Cormio G, Maglione A, Lorusso D, Maiello E. How immunotherapy modified the therapeutic scenario of endometrial cancer: a systematic review. Front Oncol. 2022;12:844801.
Longoria TC, Eskander RN. Immunotherapy in endometrial cancer—an evolving therapeutic paradigm. Gynecol Oncol Res Pract. 2015;2:11.
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. 2018;48:812–30.
Wen WX, Leong CO. Association of BRCA1- and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS ONE. 2019;14:e0215381.
Wieser V, Gaugg I, Fleischer M, Shivalingaiah G, Wenze S, Sprung S, et al. BRCA1/2 and TP53 mutation status associates with PD-1 and PD-L1 expression in ovarian cancer. Oncotarget. 2018;9:17501–11.
Cao W, Ma X, Fischer JV, Sun C, Kong B, Zhang Q, et al. Immunotherapy in endometrial cancer: rationale, practice and perspectives. Biomark Biomark Res. 2021;9:49.
Nie Y, Yang L, Zhang Y, Guo H. CD8+CD28+PD1− T cells as a prognostic biomarker in endometrial cancer. Curr Oncol. 2025;32:121.
Comaills V, Castellano-Pozo M. Chromosomal instability in genome evolution: from cancer to macroevolution. Biology. 2023;12:671.
Guo X, Bian X, Li Y, Zhu X, Zhou X. The intricate dance of tumor evolution: exploring immune escape, tumor migration, drug resistance, and treatment strategies. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167098.
Zhou J, Zhou XA, Zhang N, Wang J. Evolving insights: How DNA repair pathways impact cancer evolution. Cancer Biol Med. 2020;17:805–27.
Ladan MM, van Gent DC, Jager A. Homologous recombination deficiency testing for BRCA-like tumors: the road to clinical validation. Cancers. 2021;13:1004.
Meijer TG, Martens JWM, Prager-van der Smissen WJC, Verkaik NS, Beaufort CM, van Herk S, et al. Functional homologous recombination (HR) screening shows the majority of BRCA1/2-mutant breast and ovarian cancer cell lines are HR-proficient. Cancers. 2024;16:741.
Acknowledgements
DNA large panel sequencing, RNA immune-related panel sequencing, and multiplex immunofluorescence assay were performed at Burning Rock Biotech (Guangzhou, China). The authors wish to thank the patients and their families. The authors would also like to acknowledge Ms. Lan Su, Dr. Fei Zhao, Dr. Qiang Lu, Dr Guoqiang Wang from Burning Rock Biotech for their assistance and suggestions in data interpretation and manuscript writing.
Funding
This work was supported by the National Natural Science Foundation of China (No. 62172047) and Bethune · “Research Wings Boost Growth”—Supporting Scientific Research in the Field of Therapy (No. 2024-YJ-226-J-005). Funds were used for data collection, statistical analysis, manuscript writing and publication.
Author information
Authors and Affiliations
Contributions
Wei Wang and Min Gao designed and supervised the study. Yunong Gao, Hong Zheng, and Nan Song participated in the idea of the article. Naiyi Zhang, Tong Shu, Weijiao Gao, Nan Zhang, and Hongguo Wang acquired samples and collected clinical data. Wei Wang, Min Gao, Nan Song, and Hong Zheng performed the bioinformatic analysis and statistical analysis. Wei Wang and Min Gao wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was approved by the Ethics Review Board at Peking University Cancer Hospital & Institute with the reference number 2021YJZ21 and was performed under the Declaration of Helsinki. At the time of sample collection, all participants provided written informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, W., Gao, M., Gao, Y. et al. Integrating homologous recombination deficiency subtyping with TCGA molecular classification for enhanced prognostic stratification and personalised therapy in endometrial cancer. Br J Cancer (2025). https://doi.org/10.1038/s41416-025-03179-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41416-025-03179-y
