
Abstract
Juvenile myelomonocytic leukemia (JMML) is caused by constitutively activated RAS signaling and characterized by increased proliferation and predominant myelomonocytic differentiation of hematopoietic cells. Using MxCre;Ptpn11D61Y/+ mice, which model human JMML, we show that RAS pathway activation affects apoptosis signaling through cell type-dependent regulation of BCL-2 family members. Apoptosis resistance observed in monocytes and granulocytes was mediated by overexpression of the anti-apoptotic and down-regulation of the pro-apoptotic members of the BCL-2 family. Two anti-apoptotic proteins, BCL-XL and MCL-1, were directly regulated by the oncogenic RAS signaling but, in addition, were influenced by microenvironmental signals. While BCL-XL and BCL-2 were required for the survival of monocytes, MCL-1 was essential for neutrophils. Interestingly, stem and progenitor cells expressing the oncogenic PTPN11 mutant showed no increased apoptosis resistance. BCL-XL inhibition was the most effective in killing myeloid cells in vitro but was insufficient to completely resolve myeloproliferation in vivo.
Similar content being viewed by others



Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare and highly aggressive myeloid neoplasia of early childhood, characterized by constitutive active RAS signaling. Mutations in KRAS, NRAS, RRAS, or RRAS2 or in their regulators PTPN11 (encoding for SHP2), CBL, or NF1 [1,2,3,4,5,6,7] in JMML cells, enhance proliferation and myelomonocytic differentiation upon cytokine stimulation (i.e., GM-CSF) [8]. Monocytes and granulocytes infiltrate bone marrow (BM), spleen, liver and other organs, causing fever, thrombocytopenia, hepatosplenomegaly, respiratory distress, and diarrhea [1, 9,10,11,12]. Most patients require allogeneic stem cell transplantation (HSCT).
Resistance to apoptosis is a key cancer hallmark contributing to tumor emergence and persistence [13,14,15]. The intrinsic apoptosis pathway is controlled by BCL-2 family proteins [16,17,18,19,20,21], divided into anti-apoptotic (BCL-2, BCL-XL, MCL-1), pro-apoptotic BH3-only (BIM, PUMA, BID, NOXA, BMF, BAD, BIK, HRK), and pro-apoptotic effector proteins (BAX, BAK, BOK) [22,23,24,25,26,27,28,29]. Once activated, effector proteins multimerize and lead to mitochondrial outer membrane permeabilization, cytochrome c release, and cell death [28, 30]. Pro- and anti-apoptotic proteins interact to regulate cell fate. Every cell requires one or more BCL-2 protein/s for survival. In cancer cells, this “BCL-2 protein addiction” is modulated by the driver mutations and microenvironmental signals [31,32,33]. The fact that cancer cells, at the same time, accumulate pro-apoptotic proteins (known as “mitochondrial priming”) is used during cancer therapy since cytotoxic drugs induce apoptosis more readily in cancer than in healthy cells.
RAS activation influences cell death decisions by regulating BCL-2 proteins, by inhibiting pro-apoptotic and activating anti-apoptotic proteins, as observed mostly in cell lines [34, 35]. Here, we investigated BCL-2-regulated apoptosis in a genetically engineered mouse model for JMML. Using mice expressing the PTPN11/SHP2D61Y mutation in the hematopoietic system [1, 36], we found survival benefits in leukemic monocytes and neutrophils, mediated by higher BCL-XL and/or MCL-1 expression. Adjacent non-leukemic cells acquired similar but transient apoptosis resistance. Surprisingly, SHP2D61Y expressing hematopoietic stem and progenitor cells (HSPCs) showed no apoptosis resistance. Inhibition of BCL-2 proteins by selective BH3-mimetics revealed differential “addiction” in monocytes and neutrophils. In vivo treatment with the BCL-XL inhibitor A1155463 reduced myeloproliferation but did not cure the mice. In conclusion, apoptosis resistance in JMML is restricted to few cell types and influenced by microenvironmental signals.
Methods
Genetically engineered JMML mouse model
Animal procedures were carried out in accordance with the regulatory requirements of German law. MxCre and Ptpn11D61Y/+ were provided by Benjamin Neel and bred to obtain MxCre;Ptpn11D61Y/+. All mice received three doses of poly I: C (300 μg/dose/mouse) every other day. Leukemic mice were euthanized and analyzed at different disease stages (Supplementary Fig. 6). All mice were euthanized when showing distress.
Mice genotyping
The DNA from ear punches was PCR assayed for Neo-Stop cassette and MxCre using the primers shown in Supplementary Table 4 (Supplementary Fig. 6).
In vivo treatment
The BCL-XL inhibitor A1155463 (Selleck Chemicals) was dissolved in 2% DMSO, 30% PEG300, 2%Tween80 and administered intraperitoneally (ip), daily for 28 days (5 mg/kg/day). Mice were euthanized one day after treatment, and hematopoietic cell compartments were analyzed.
Transplantation JMML mouse model
CD45.1+ (WT) mice were sub-lethally irradiated (3 Gy). Leukemic splenocytes from terminally ill S3-MxCre;Ptpn11D61Y/+ mice were isolated and transplanted intravenously (1 × 106 CD45.2 in PBS) into the irradiated mice. Recipients received riketron in drinking water for four weeks after irradiation. Engraftment and myeloproliferation were assessed in peripheral blood via flow cytometry.
Flow cytometry
For surface staining, single-cell suspensions from BM, spleen, blood, and liver and lung MNCs were stained with antibodies as in Supplementary Table 3, and the gating strategy was performed as shown on Supplementary Fig. 7. For intracellular staining, cells were stained for surface markers, then fixed and permeabilized using a buffer set (eBioscience concentrate and diluent), followed by intracellular protein staining per the manufacturer’s instructions. For phospho-flow, cells were stained for surface markers, incubated with 3.7% formaldehyde, then 90% cold methanol, and stained with pMAPK and pmTOR phosphoantibodies. For apoptosis staining, after surface staining, cells were resuspended in binding buffer (BB) containing Annexin V (AV) and 7AAD, incubated for 15 min, and measured on a BD Fortessa. Freshly isolated CD11b+ or LSK cells were cultured for the specified time points before apoptosis staining.
Cell isolation and culture
LSK cells
Lineage-negative (Lin-) cells were isolated from BM using the lineage depletion Kit (Miltenyi Biotec). Followed by sorting of Sca+cKit+ cells and cultured in IMDM medium (Gibco), with 10% FCS (Thermo Fisher), 1% penicillin/streptomycin (ThermoFisher), stem cell factor (SCF), thrombopoietin (TPO) and Fms-related receptor tyrosine kinase 3 ligand (FLT3L) at 100 ng/ml each (ImmunoTools).
CD11b+ cells
CD11b+ myeloid cells were isolated using a MACS kit (Miltenyi Biotec) with two column separation rounds and cultured in IMDM medium supplemented with 30% FCS, 1% P/S, 25 mM HEPES (Sigma-Aldrich), 0,1 mM MEM non-essential amino acids (Gibco) and 0,1 mM sodium pyruvate (Gibco), 2-B-mercaptoethanol (Gibco). Where indicated, CD11b+ cells were stained for CD45.1 and CD45.2 and sorted. CD11b+ CD45.1 and CD45.2 were cultured individually or in co-cultures. Neutralizing antibodies for anti-CD74 (R&D systems), anti-ITGAM (Invitrogen) or TNFα (Immunotools) were added as indicated.
Apoptosis induction
Apoptosis of LSK and CD11b+ cells was assessed after treatment with BH3-mimetics: ABT737 (Selleck Chemicals), ABT199 (Selleck Chemicals), A115546, S63845 (Synthesis) and Runx inhibitor (Tocris). After BH3 mimetic treatment, cells were stained for myeloid markers, stained with AV and 7AAD, and measured on a flow cytometer. Specific apoptosis was calculated as: 100 × (% live cells without treatment − % live cells with treatment)/% live cells without treatment.
Cytokine bead array
Cytokines found in BM and spleen secretomes were quantified using a cytokine bead array (CBA) kit, according to the manufacturer’s instructions (BD Bioscience).
RT-MLPA
mRNA was isolated from BM-LSK cells and spleen CD11b+ of MxCre and S3 MxCre;Ptpn11D61Y/+ mice using Fast Spin columns (Zymo Research) and reverse transcribed into cDNA for RT-MLPA (MRC-Holland). cDNA was ligated to two oligonucleotides, and the amplicons were separated by capillary electrophoresis (ABI-3130xl Genetic Analyzer). Data was analyzed using Sequence Pilot software (JSI Medical Systems) and normalized taking the sum of all peaks as 100%.
Western blot
CD11b+ myeloid cells were lysed with 1x Laemmeli buffer and sonicated. Lysates were resolved in 12% SDS-PAGE and transferred to PVDF membrane, which were incubated overnight with primary antibodies diluted in 5% BSA in TBST: Bim (C.Signaling Technology), Bid (BD), Noxa (Enzo), Bmf (NBPI), Bak and Bax (C.Signaling Technology). Mouse or rabbit HRP-conjugated secondary antibodies were incubated with the respective membranes. β-actin (Sigma-Aldrich) was used as a loading control. Membranes were developed using Fusion equipment. For protein determination, equal volumes extracted from CD11b+ cells were run per Western blot, and β-actin band intensities were quantified using ImageJ. Supplementary Table 2 lists the antibodies.
RNA isolation and sequencing
RNA isolation
RNA was isolated from CD11b+ cells (BM and spleen of S3 MxCre;Ptpn11D61Y/+ and MxCre mice; 3 mice/genotype) with the RNeasy Micro kit (Qiagen) following the kit instructions. RNA sequencing was performed at the Genomic and Proteomic Core Facility of the German Cancer Research Center.
RNA Sequencing and data analysis
RNA sequencing libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina) according to the manufacturer’s protocol. The 100 bp paired-end reads were trimmed using TrimGalore (v0.6.5) and mapped to the mouse reference genome (GRCm38) using STAR (v2.5.3a). Read counts were normalized, and differential expression was calculated using the R packages DESeq2 (v1.22.2) and limma (v3.38.3). Gene set enrichment analysis was performed using GAGE tools (v2.44.0). The next-generation sequencing data is deposited at NCBI GEO (GSE277667).
Whole exome sequencing (WES) and data analysis
Genomic DNA was extracted from splenocytes of control MxCre and MxCre;Ptpn11D61Y/+ S3 mice by using the DNeasy Blood &Tissue Kit (Qiagen). Skin DNA was used as a germline control. WES libraries were prepared using the Agilent SureSelectXT Mouse All Exon kit. Adapter and quality trimming were performed using Trimmomatic (v0.39), and the trimmed reads were mapped to the mouse reference genome (GRCm38) using the BWA-MEM aligner. The aligned reads were processed using Samtools and the GATK toolkit (v3.8.1). Variants were identified using Samtools mpileup and VarScan (v2.4.3) and annotated with Annovar and SnpEff (v4.3).
BM and spleen secretome
Two femurs per mouse were flushed with 2 ml PBS to collect secretome content. Spleens were smashed and resuspended in 2 ml PBS. The supernatants were used for TNFα cytokine assay or further processed for mass spectrometry.
Mass spectrometry-based proteomics
Tryptic peptides from secretome lysate were labeled with TMT10plex reagent, pooled and fractioned as described previously [74] [75]. Nanoflow LC-MS/MS was performed on a Dionex 3000 (Thermo) coupled online to an Orbitrap Fusion LUMOS (Thermo). Samples were measured in DDA mode with a MS3 method and 50 min linear gradient. Database search against the mouse reference proteome (UP000000589) and common contaminants was performed with MaxQuant (v. 1.6.3.3) with standard settings for reporter ion MS3 [78]. The spleen and bone marrow TMT set corrected TMT reporter intensities were total sum and row-wise normalized for statistical analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (PMID 34723319) partner repository with the dataset identifier PXD055845.
Histology
BM/sternum, spleen, liver, and lung sections from indicated mice were fixed in 4% paraformaldehyde. The samples were decalcified in EDTA for 5–10 days and embedded in paraffin. The sections were stained with haematoxylin and eosin (H&E). Images were acquired using a Zeiss microscope.
Statistics
The unpaired/non-parametric Mann-Whitney test was used to analyze all experiments. P values: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Analysis was performed on GraphPad Prism.
Results
Monocytes and granulocytes isolated from MxCre;Ptpn11 D61Y/+ mice show increased apoptosis resistance
Mice expressing SHP2D61Y in the hematopoietic system and known to develop JMML-like myeloproliferation were generated by treating MxCre;Ptpn11D61Y/+ mice with PolyI:C [1]. We defined terminally ill mice as “stage 3” (S3) and earlier disease stages as “stage 1 and 2” (S1, S2), respectively (Fig. 1A). MxCre;Ptpn11D61Y/+ mice developed massive splenomegaly and succumbed 130–400 days post polyI:C injection (median 274 days, Fig. 1B, C). Flow cytometry revealed increasing infiltration of monocytic, granulocytic, and erythroid progenitors in the spleen, lung, and liver (Supplementary Fig. 1A–F). The number of HSPCs defined as lineage marker negative, Sca1 and cKit positive (LSK) cells, remained stable in the BM but increased in the S2–3 spleens (Supplementary Fig. 1G). Activated SHP2 signaling was confirmed by increased MEK and mTOR phosphorylation in SHP2D61Y expressing myeloid cells (Supplementary Fig. 1H). Histopathology confirmed myelomonocytic organ infiltration (Supplementary Fig. 2). Flow cytometry immediately after organ isolation revealed higher viability of monocytes and neutrophils from spleen, especially in S2-3 mice (Fig. 1D). BM myeloid cells were generally more viable, with no significant differences between the genotypes (Supplementary Fig. 3A). SHP2D61Y expressing LSK cells, lymphocytes and erythroid progenitors from the spleen (Fig. 1E) and BM (Supplementary Fig. 3B) showed no survival benefit.

A Schematic representation of the experimental procedure. All experimental mice (controls -WT, MxCre, Ptpn11D61Y/+ and JMML mice - MxCre;Ptpn11D61Y/+) were injected with 300 μg poly I:C/mouse (in 3 doses every other day) to activate the oncogenic mutation. The experimental mice were analyzed at different stages of the disease: preleukemic - defined as stage 1 - S1 (5 weeks after polyI:C injection); leukemic - defined as stage 2 - S2 (5 months after polyI:C injection) and full-blown myeloproliferative disease - defined as stage 3 - S3 (7+ months after poly I:C injection - terminally ill mice). B Representative spleens are shown for: control mice (MxCre, Ptpn11D61Y/+) and JMML mice MxCre;Ptpn11D61Y/+ (S1, S2, S3). C Survival curves are shown for control mice (WT) and JMML MxCre;Ptpn11D61Y/+ mice. The percentage of live cells (AV− 7AAD−) was determined in the following splenic hematopoietic cell types: D CD11b+ myeloid cells; CD11b+Ly6Cmedium - circulatory monocytes; CD11b+Ly6Chigh - inflammatory monocytes and CD11b+Ly6G+ - neutrophils; E LSK - stem and progenitor hematopoietic cells, B220+ - B cells, TCR-β+ - T cells and Ter119+ - erythrocytes obtained from the spleen. All the above cell populations were determined ex vivo and assayed by flow cytometry for the indicated genotypes. The gating strategy was performed as described in Supplementary Fig. 7. All data are presented as mean ± SEM (n = 3–10; ≥5 independent experiments).
To confirm their reduced susceptibility to apoptosis, myeloid cells from S2 and S3 spleens were cultured for 24 and 48 h without pro-survival cytokines. Increased survival of S2-3 bulk myeloid cells (CD11b+), monocytes and granulocytes were observed compared to controls (Fig. 2A–C), with stronger effects in S3 cells. SHP2 mutant LSK cells showed no viability differences when cultured with or without cytokines compared to controls (Fig. 2D). Clonal evolution as the reason for the increased apoptosis resistance in late disease stages was excluded by whole exome sequencing (WES). In splenocytes of three S3 mice, no JMML-associated mutations (i.e., secondary RAS pathway lesions, mutations in JAK2, JAK3, or SETBP1) [37] were identified. Mutation numbers and variant allele frequencies (VAF) were similar in leukemic and control mice (Supplementary Fig. 3C, D, Supplementary Table 1).

The primary murine LSK and CD11b+ cells were cultured for 24 h and 48 h. The percentage of living cells (AV-7AAD-) was determined by flow cytometry for the following cell populations: A myeloid cells - CD11b+ cells; B monocytes - CD11b+Ly6C+; C neutrophils - CD11b+Ly6G+ after for 24 h (left panel) and 48 h (right panel) of culture. D LSK-stem and progenitor cells were cultured for 48 h in the presence and absence of cytokines (FLT3L, TPO and SCF), and living cells were determined as AV-7AAD- by flow cytometry. The gating strategy was performed as described in Supplementary Fig. 7. All data are presented as mean ±SEM (n = 1–10; ≥3 independent experiments).
BCL-XL and MCL1 are upregulated in SHP2D61Y expressing myeloid cells
To understand SHP2D61Y mediated apoptosis resistance at the molecular level, we analyzed levels of pro- and anti-apoptotic BCL-2 proteins. RT-MLPA revealed an approximately twofold increase of Bcl-xl and Mcl-1 mRNA in spleen-isolated CD11b+ cells. While proapoptotic antagonists Bim, Bad and Bok were mildly but not significantly upregulated (Fig. 3A). Increased mRNA levels of Bcl-2, Bcl-xl, Mcl-1 and A1, but also Bid and Bax were observed in the non-resistant LSK cells (Supplementary Fig. 3E). BCL-XL and MCL-1 protein levels were slightly but not significantly higher in CD11b+ and LSK cells from S1-S3 mice, when compared to controls (Fig. 3B, C). In cultured SHP2D61Y monocytes, BCL-XL and MCL-1 levels were higher than in controls. In neutrophils, only MCL-1 was upregulated (Fig. 3D). Western blot analysis revealed upregulation of several pro-apoptotic proteins in S1 myeloid cells but downregulation during disease progression (Fig. 3E, F).
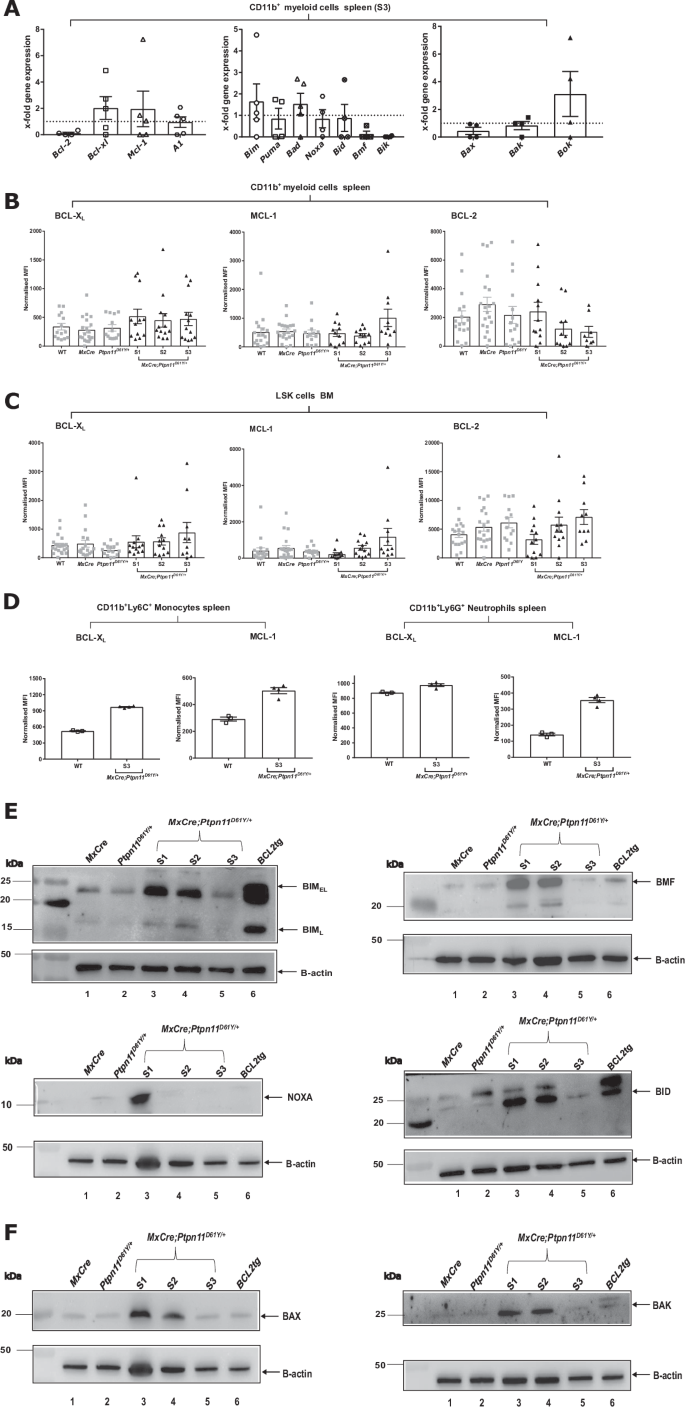
A The mRNA levels of BCL-2 family members (anti-apoptotic, pro-apoptotic and pore-forming) were determined by RT-MLPA in primary CD11b+ myeloid murine cells. Myeloid cells were isolated from S3 MxCre;Ptpn11D61Y/+ and control MxCre mice. Protein levels of pro-survival BCL-2 family proteins (BCL-XL, MCL-1 and BCL-2) were determined ex vivo by intracellular flow cytometry for the following cell types: B CD11b+ myeloid cells and C LSK–stem and progenitor cells. D The freshly isolated murine CD11b+ cells from disease stage 3 were cultured for 24 h and the pro-survival BCL-2 family proteins (BCL-XL, MCL-1) were determined in CD11b+Ly6C+ - monocytes and CD11b+Ly6G+ - neutrophils. Representative western blots showing the expression levels of pro-apoptotic BCL-2 proteins for the following BCL-2 family members: E BIMEL and BIML, BMF, NOXA, BID and pore-former proteins F BAX and BAK. BCL2 tg cells are the positive control for western blot. All data are presented as Mean ±SEM (n = 1–10; ≥3 independent experiments). The western blot analysis were performed on freshly isolated primary murine CD11b+ cells for an n = 2 with 5–7 mice/lysates. Antibodies used for WB are listed in Supplementary Table 2.
Apoptosis resistance is driven by the oncogenic signaling
RNAseq with gene set enrichment analysis (GSEA) revealed strong enrichment of cell cycle-related gene sets and downregulation of the apoptosis gene set in S3 leukemic spleen myeloid cells when compared to MxCre-tg controls (Fig. 4A). Unexpectedly, the levels of PTPN11 mRNA were higher in BM LSK than in splenic myeloid cells, this not correlating with the increased apoptosis resistance (Fig. 4B). Upregulated KRAS signaling (HALLMARK_KRAS_SIGNALING_UP) in myeloid cells versus immature LSK cells suggests that RAS/MAPK activity downstream of SHP2 explains their differing apoptosis susceptibility (Fig. 4C, Supplementary Fig. 4). To confirm that SHP2 and the RAS/MAPK signaling directly regulate BCL-XL and MCL-1 expression, we inhibited MEK and, additionally the transcription factor RUNX1, which has been shown to drive myeloproliferation downstream of SHP2 [38]. BCL-XL expression was reduced in bulk CD11b+ cells and specifically in monocytes, when both MEK and RUNX1 were inhibited (Fig. 4D). In contrast, MCL-1 expression was most profoundly reduced by MEK inhibition alone (Fig. 4E). Only combined MEK and RUNX1 inhibition induced apoptosis in both cell types (Fig. 4F).

A The bubble plot representing the top up-(left) and downregulated (top) hallmark gene sets in the comparison of S3 CD11b+ leukemic cells with the control (Mxcre or WT) cells. The gene ratio is represented on the y-axis, the bubble size corresponds to the gene count and the color map represents the adjusted p value. B The Ptpn11 mRNA expression level in the bone marrow LSK and spleen CD11b+ cells in the S3 leukemic (MxCre;Ptpn11D61Y/+) and control (MxCre or WT) cells. The y-axis represents the logCPM (CPM- counts per million reads). C The gene set enrichment analysis of hallmark KRAS signaling up-regulated gene sets in the comparison of S3 spleen CD11b+ leukemic cells and LSK leukemic cells. The genes are ranked based on the fold change and the y-axis represents the running enrichment score (top) and the ranked list metric (bottom). The normalized enrichment score (NES), p value and adjusted p-value are shown in the table. Myeloid CD11b+ cells isolated from leukemic S2 mice or MxCre controls were treated with trametinib and/or Runx inhibitor for 24 h and then analyzed for expression of pro-survival proteins: D BCL-XL and E MCL-1 and F % of specific apoptosis for myeloid cells (CD11b+ cells), monocytes (CD11b+Ly6C+) and neutrophils (CD11b+Ly6G+). All data are presented as Mean ±SEM (n = 1–10; ≥3 independent experiments).
Microenvironmental signals contribute to apoptosis resistance in vivo
The generally higher viability of myeloid cells in BM than in the spleen prompted us to study the microenvironmental contribution to cell death decisions in leukemia. First, we transplanted leukemic mice into CD45.1 + WT recipients to generate chimeras (Fig. 5A, left). Suppl Fig. 5A shows the proportion of leukemic cells. Again, BM cells were more viable than spleen cells (Supplementary Fig. 5B). In chimeric spleens, increased survival rates were noted for both leukemic and recipient WT cells (Fig. 5B). Next, we performed co-culture experiments with WT and leukemic cells (Fig. 5A, right). We observed survival advantages (Fig. 5C) and elevated BCL-XL and MCL-1 levels in leukemic myeloid cells, both in the absence and presence of WT cells (Fig. 5D, E). Interestingly, also WT cells and, particularly, neutrophils showed elevated BCL-XL levels comparable to the ones of leukemic cells (Fig. 5D). Yet, they were not apoptosis-resistant (Fig. 5C), indicating that SHP2D61Y expressing myeloid cells influence the survival of adjacent WT cells only in vivo. To identify pro-survival molecules in the leukemic microenvironment, we performed secretome mass spectrometry. Spleen supernatants contained more proteins than the BM supernatant (Fig. 5F). We identified proteins with known anti-apoptotic function at high abundance in leukemic spleens (i.e.ITGAM/CD11b, TNFα and CD74) and confirmed these findings by flow cytometry and cytokine assay (Fig. 5G, Supplementary Fig. 5C–F). Blocking either ITGAM or CD74 or adding TNFα in vitro did not induce apoptosis in WT or S3 myeloid cells (Supplementary Fig. 5G–I), indicating that other not yet identified microenvironmental signals affect cell survival within the leukemic spleen.

A Schematic representation of the experimental procedure. Sub-lethally irradiated (3 Gy) WT (CD45.1) mice were transplanted with splenocytes (1 × 106 cells) from S3 - terminally ill MxCre;Ptpn11D61Y/+ mice (CD45.2) (left panel). Representative spleens are shown for the control and the engrafted mice (middle panel). Schematic representation of the 24 h single cultures and co-cultures of leukemic - CD45.2 and WT - CD45.1 myeloid CD11b+ cells (right panel). The single cultures consisted of control CD45.1 - designated as c-CD45.1; single culture CD45.1- designated as s-CD45.1; single culture CD45.2 - designated as s-CD45.2. The co-culture conditions consisted of co-culture of CD45.1 and CD45.2 - designated as co-CD45.1 or co-CD45.2. The cells used for the condition: single cultures CD45.1 and CD45.2 were sorted from successfully engrafted recipient mice, while the control CD45.1 was obtained from not transplanted mice. Leukemic CD45.2 and WT CD45.1 myeloid cells were determined via flow cytometry. B Percentages of live cells (7AAD-) within the spleen residing CD45.1 and CD45.2 determined in engrafted and control mice for CD11b+ - myeloid cells, CD11b+Ly6Cmedium - circulatory monocytes, CD11b+Ly6Chigh - inflammatory monocytes, CD11b+Ly6G+ - neutrophils, was assessed ex vivo. C The CD11b+ cells were cultured for 24 h in single cultures or co-cultures. The percentage of living cells (AV−7AAD−) was determined by flow cytometry for: CD11b+ - myeloid cells; CD11b+Ly6C+ - monocytes and CD11b+Ly6G+ - neutrophils from BM and spleen. In vitro expression of pro-survival proteins: D BCL-XL and E MCL-1 was determined in the myeloid compartment isolated from spleen, CD11b+ - myeloid cells, CD11b+Ly6C+ - monocytes and CD11b+Ly6G+ - neutrophils via intracellular staining after 24 h culture. F Significantly differently expressed proteins identified by mass spectrometry were selected and plotted based on log2 fold change obtained for BM or spleen supernatant from S3 leukemic vs MxCre. Significant changes in BM (turquoise), spleen (orange), or both (gray). Significant proteins only identified in either BM or spleen are marked by dashed boxes. The total number of identified proteins for BM and spleen are depicted as box insert. The gating strategy was performed as described in Supplementary Fig. 7. All data are presented as Mean ± SEM (n = 7–9; ≥3 independent experiments).
BCL-XL inhibition has potent anti-leukemic activity in vivo
To identify the anti-apoptotic BCL-2 protein(s) required for leukemic cell survival, we used selective BH3-mimetics in vitro [39,40,41,42,43,44]. Splenic myeloid cells were most sensitive to BCL-XL inhibition (Fig. 6A). Leukemic monocytes were sensitive to both combined and selective inhibition of BCL-XL and BCL-2 (Fig. 6B), while neutrophils were most sensitive to MCL-1 inhibition (Fig. 6C). Greatest sensitivity to BH-3 mimetics was found in the terminal S3 stage. BM-derived LSK cells did not show increased sensitivity to BH3-mimetics when cultured in the presence of cytokines (Fig. 6D). Under cytokine deprivation, they became slightly sensitive to BCL-XL and MCL-1 inhibition (Fig. 6E). Based on the promising in vitro effects, we used the BCL-XL inhibitor A1155463 in vivo (Fig. 7A). Treatment for 28 days resulted in significant reduction of the spleen size in S2 mice (Fig. 7B). Analysis of all hematopoietic cells revealed significant decrease in monocytes and neutrophils but also HSPCs and erythroid progenitors in the spleen of treated mice (Fig. 7C–G), along with reduced infiltration in the blood and lungs (Fig. 7E). Interestingly, no effects were observed in BM or liver (Fig. 7D, E).
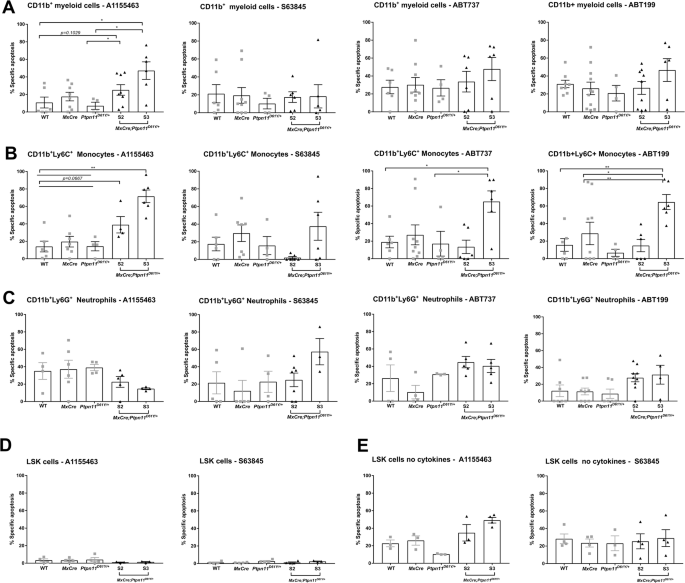
CD11b+ myeloid cells were treated with BH-3 mimetics, A1155463 (10 μM), S63845 (1 μM), ABT737 (3 μM), ABT199 (5 μM) for 48 h. Then, cells were stained for the surface markers CD11b+, Ly6G+, and Ly6C+ followed by staining for AV and 7AAD, and the viable cells were determined as AV-7AAD- on flow cytometry. The percentage of specific apoptosis induced by BH3 mimetics was calculated for the specific cell types: A CD11b+ - myeloid cells, B CD11b+Ly6C+ - monocytes and C CD11b+Ly6G+ - neutrophils. Freshly isolated LSK stem and progenitor cells (cultured with or without cytokines) were treated with the BCL-XL (A1155463) or MCL-1 (S63845) inhibitor for 48 h. The percentage of specific apoptosis was calculated for LSK cells: D in the presence (of TPO, FLT3, and SCF) and E in the absence of cytokines. The gating strategy was performed as described in Supplementary Fig. 7. All data are presented as Mean ±SEM (n = 1–3; ≥3 independent experiments).

A Schematic representation of experimental procedure. Mice were treated with the BCL-XL inhibitor A1155463 (5 mg/kg/day) for 28 days (daily); on day 29 the mice were sacrificed and analyzed, and the leukemic burden was assessed. B Representative spleens (figure and weight) after in vivo treatment with the A1155463 or vehicle mice. Absolute cell counts are shown for the following cell populations of the BCL-XL inhibitor A1155463 or vehicle-treated mice: myeloid cells - CD11b+ - myeloid cells; CD11b+Ly6Cmedium – circulatory monocytes; CD11b+Ly6Chigh – inflammatory monocytes; CD11b+Ly6G+ – neutrophils residing in the C spleen and D BM. E Number of CD11b+ myeloid cells residing in the blood, liver and lung. F LSK stem and progenitor hematopoietic cells. G Erythrocytes: - CD71+Ter119high - Early erythroblasts (EB); (EB) - CD71+Ter119intermediate - Pro-erythroblasts; CD71-Ter119+ - late EB. All data are presented as Mean ±SEM (n = 3–7; =3 independent experiments).
Discussion
Our study aimed to characterize and target BCL-2-regulated apoptosis signaling, with the ultimate goal of identifying novel therapeutic approaches for high-risk JMML. Constitutively active RAS signaling, downstream of SHP2, was shown to exert anti-apoptotic effects [34, 35]. In hematopoietic cells, SHP2D61Y or SHP2E76K mutants were shown to have increased viability by upregulation of BCL-2 and BCL-XL and downregulation of BIM. Similarly, Ptpn11E76K/+ transformed erythroleukemic TF-1 cells showed BCL-XL upregulation [45]. Here, we compare different primary cell types at the same time, using a mouse model reminiscent of JMML and different culture conditions. We found that oncogenic SHP2D61Y conferred survival advantages specifically to myeloid cells but not to HSPCs, despite high Ptpn11 mRNA levels in LSK cells. Survival advantages were accompanied by BCL-XL and/or MCL-1 upregulation in Ptpn11 mutant myeloid cells and reversed by inhibition of the SHP2 downstream signaling, establishing a direct link between the oncogene and these anti-apoptotic proteins. This suggests that apoptosis resistance, alongside increased proliferation and differentiation, drives SHP2 myeloproliferation [13, 15]. The increased activation of RAS signaling in mature but not immature cells may explain why PTPN11 mutations, when acquired as a first hit, lead to differentiating myelomonocytic leukemia rather than acute myeloid leukemia. It raises, however, the question, of why PTPN11 and/or KRAS mutations in HSCs allow relapse after transplantation. Most likely, PTPN11 and/or KRAS mutated HSCs acquire features providing selective fitness not linked to cell death signaling (i.e. immune escape mechanisms, metabolic fitness or others). Understanding the specific reasons for higher KRAS signaling in mature cells and the selective fitness of HSCs requires further detailed studies of the differentiation process, the regulatory networks involved, and the functional demands placed on both immature and mature cells.
In monocytes, both BCL-XL and MCL-1 expression were directly linked to SHP2D61Y but regulated differently: BCL-XL expression was regulated by both MEK and RUNX1, whereas MCL-1 was regulated by MEK alone. In neutrophils, no clear link between SHP2D61Y and the anti-apoptotic proteins was established. In addition, cell death decisions were affected by extracellular signals, which increased survival even in WT bystander cells. It was shown earlier that an abnormal microenvironment contributes to myeloproliferation in JMML mouse models, with CCL3/MIP-1 α playing an important role [46,47,48]. Here, we identified BCL-XL as a novel player in this setting.
Our model revealed a cell type-specific “BCL-2 protein addiction”, where BCL-XL and BCL-2 expression were essential for monocyte survival, whereas MCL-1 was crucial for neutrophils. Further analysis is needed to elucidate the regulation and engagement of these proteins. Differences in SHP2 signaling itself (i.e. activation levels and abundance of downstream signaling components), and other cell type-specific pathways, such as lineage-specific transcription factors, may influence the expression of BCL-2 family members. Potential candidates include NFkB and ETS, which were shown to influence BCL-2 protein expression, or the master transcription factors for myelomonocytic differentiation GATA2, PU.1 and GFI1 [49,50,51]. Similarly, the abundance of pro-apoptotic BH3-only proteins activated by developmental cues may differ between monocytes and granulocytes. Transcriptomic analyses and co-immunoprecipitation assays using highly purified cell populations will be necessary to fully delineate the apoptosis signaling pathways for each cell type.
Interestingly, apoptosis resistance increased during disease progression, with the most resistant granulocytes and monocytes at the lethal leukemia stage. Since there was no evidence of clonal evolution, we hypothesize that this is due to a gradual expansion of Ptpn11-mutant cells following polyI:C induction, combined with their selective advantages. However, we observed a strong downregulation of pro-apoptotic BH3-only and effector BCL-2 proteins in the late disease stage. At the same time, the response to BH3-mimetics changed, with a newly acquired BCL-2 dependency at the late disease stage. Most likely, niche-derived signals influence not only the abundance of BCL-XL but also of pro-apoptotic proteins. Along this line, apoptosis resistance could be transferred to bystander WT cells in vivo but was not maintained in culture, suggesting a complex interplay of cell types and soluble factors contributing to apoptosis resistance. Indeed, several proteins with known anti-apoptotic function in granulocytes (i.e., CD74, TNFα, and ITGAM/CD11b) [52,53,54,55] were enriched in the microenvironment of leukemic mice. While these signals may cooperate to increase the viability of bystander cells in vivo, oncogene-driven cell-intrinsic signals are sufficient to ensure the survival of leukemic SHP2D61Y cells.
Finally, we asked whether JMML could be cured by the BCL-XL inhibitor, based on our in vitro data. Treatment significantly reduced leukemic burden and interestingly affected cells not sensitive in vitro (i.e HSPcs and erythroid progenitors). Again, this supports the hypothesis that niche-derived signals change apoptosis signaling in vivo [46,47,48] and highlights the need for disease-relevant in vivo models. Not unexpectedly, BCL-XL inhibition used as monotherapy did not cure the mice. In patient-derived JMML cells, we recently identified synergistic effects of BCL-XL and MCL-1 inhibitors. Along this line, azacitidine reduced MCL-1 levels, explaining its synergy with BCL-XL inhibition in PDX mice [56]. Alternatively, therapeutic modulation of the microenvironment might increase the susceptibility of JMML cells towards BCL-xL inhibition. Future strategies could focus on disrupting LSK-niche interactions using small molecule inhibitors, integrin blockers to modulate ECM dynamics, or interventions targeting niche-derived survival cytokines. Anti-CXCR4 therapies, such as plerixafor, used in AML [57] to disrupt interactions with the bone marrow microenvironment, could be explored as a potential treatment in JMML.
The JMML PDX and genetically modified mouse models provide complementary insights into RAS dysregulation on apoptotic signaling. While the PDX model excels in translational relevance by mimicking key JMML features in patients (including methylation signatures), its reliance on immunodeficient mice limits the study of RAS effects on the complete hematopoietic and immune system. The genetically modified mouse model used in this study, in contrast, preserves immune interactions and enables detailed investigation of leukemogenesis in an immunocompetent setting, offering a more comprehensive view of disease progression on the full hematopoietic compartment. A more comprehensive understanding of apoptosis and other cell death forms, such as necroptosis and pyroptosis [58] in both models are crucial for harnessing cell death in therapeutic applications and to develop effective combination therapies.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (PMID 34723319) partner repository with the dataset identifier PXD055845. The next-generation sequencing data is deposited at NCBI GEO (GSE277667). All other data generated and analyzed during this study are included in this published article and supplementary information.
References
Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113:4414–24.
Calvo KR, Price S, Braylan RC, Oliveira JB, Lenardo M, Fleisher TA, et al. JMML and RALD (Ras-associated autoimmune leukoproliferative disorder): common genetic etiology yet clinically distinct entities. Blood. 2015;125:2753–8.
Niemeyer CM, Flotho C. Juvenile myelomonocytic leukemia: who’s the driver at the wheel? Blood. 2019;133:1060–70.
Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet. 2014;51:689–97.
Louka E, Povinelli B, Rodriguez-Meira A, Buck G, Wen WX, Wang G, et al. Heterogeneous disease-propagating stem cells in juvenile myelomonocytic leukemia. J Exp Med. 2021;218:e20180853.
Niemeyer CM. JMML genomics and decisions. Hematology Am Soc Hematol Educ Program. 2018;2018:307–12.
Yang Z, Li Y, Yin F, Chan RJ. Activating PTPN11 mutants promote hematopoietic progenitor cell-cycle progression and survival. Exp Hematol. 2008;36:1285–96.
Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77:925–9.
Liu W, Yu WM, Zhang J, Chan RJ, Loh ML, Zhang Z, et al. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia. 2017;31:1415–22.
Krombholz CF, Aumann K, Kollek M, Bertele D, Fluhr S, Kunze M, et al. Long-term serial xenotransplantation of juvenile myelomonocytic leukemia recapitulates human disease in Rag2-/-gammac-/- mice. Haematologica. 2016;101:597–606.
Tarnawsky SP, Yu WM, Qu CK, Chan RJ, Yoder MC. Hematopoietic-restricted Ptpn11E76K reveals indolent MPN progression in mice. Oncotarget. 2018;9:21831–43.
Pandey R, Saxena M, Kapur R. Role of SHP2 in hematopoiesis and leukemogenesis. Curr Opin Hematol. 2017;24:307–13.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Weinberg RA. Oncogenes and the molecular biology of cancer. J Cell Biol. 1983;97:1661–2.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Davids MS, Deng J, Wiestner A, Lannutti BJ, Wang L, Wu CJ, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012;120:3501–9.
Potter DS, Du R, Bhola P, Bueno R, Letai A. Dynamic BH3 profiling identifies active BH3 mimetic combinations in non-small cell lung cancer. Cell Death Dis. 2021;12:741.
Sanchez-Rivera FJ, Ryan J, Soto-Feliciano YM, Clare Beytagh M, Xuan L, Feldser DM, et al. Mitochondrial apoptotic priming is a key determinant of cell fate upon p53 restoration. Proc Natl Acad Sci USA 2021;118:e2019740118.
Bhatt S, Pioso MS, Olesinski EA, Yilma B, Ryan JA, Mashaka T, et al. Reduced mitochondrial apoptotic priming drives resistance to BH3 mimetics in acute myeloid leukemia. Cancer Cell. 2020;38:872–90.e6.
Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–55.
Yin W, Xia X, Wu M, Yang H, Zhu X, Sun W, et al. The impact of BCL-2/MYC protein expression and gene abnormality on primary central nervous system diffuse large B-cell lymphoma. Int J Clin Exp Pathol. 2019;12:2215–23.
Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–93.
Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer. 2016;16:539–48.
Labi V, Erlacher M. How cell death shapes cancer. Cell Death Dis. 2015;6:e1675.
Kollek M, Muller A, Egle A, Erlacher M. Bcl-2 proteins in development, health, and disease of the hematopoietic system. FEBS J. 2016;283:2779–810.
Sochalska M, Tuzlak S, Egle A, Villunger A. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: implications for targeted therapy. FEBS J. 2015;282:834–49.
Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–42.
Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80.
Erlacher M, Labi V. MCL-1 and BCL-XL: blood brothers. Blood. 2021;137:1850–1.
Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714.
Munk Pedersen I, Reed J. Microenvironmental interactions and survival of CLL B-cells. Leuk Lymphoma. 2004;45:2365–72.
Packham G, Stevenson FK. Bodyguards and assassins: Bcl-2 family proteins and apoptosis control in chronic lymphocytic leukaemia. Immunology. 2005;114:441–9.
Audrito V, Vaisitti T, Serra S, Bologna C, Brusa D, Malavasi F, et al. Targeting the microenvironment in chronic lymphocytic leukemia offers novel therapeutic options. Cancer Lett. 2013;328:27–35.
Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284:4177–95.
Vitagliano O, Addeo R, D’Angelo V, Indolfi C, Indolfi P, Casale F. The Bcl-2/Bax and Ras/Raf/MEK/ERK signaling pathways: implications in pediatric leukemia pathogenesis and new prospects for therapeutic approaches. Expert Rev Hematol. 2013;6:587–97.
Usenko T, Chan G, Torlakovic E, Klingmuller U, Neel BG. Leukemogenic Ptpn11 allele causes defective erythropoiesis in mice. PLoS ONE. 2014;9:e109682.
Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45:937–41.
Acosta ED, Huang H, Garcia SP, Stieglitz E, Loh ML, Yuan GC, et al. RUNX1 is a candidate transcriptional effector in juvenile myelomonocytic leukemia. Blood. 2016;128:2699.
Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015;22:1071–80.
Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018;25:56–64.
Adams JM. Therapeutic potential of a peptide targeting BCL-2 cell guardians in cancer. J Clin Invest. 2012;122:1965–7.
Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med Chem Lett. 2014;5:1088–93.
Wang H, Guo M, Wei H, Chen Y. Targeting MCL-1 in cancer: current status and perspectives. J Hematol Oncol. 2021;14:67.
Juarez-Salcedo LM, Desai V, Dalia S. Venetoclax: evidence to date and clinical potential. Drugs Context. 2019;8:212574.
Ren Y, Chen Z, Chen L, Woods NT, Reuther GW, Cheng JQ, et al. Shp2E76K mutant confers cytokine-independent survival of TF-1 myeloid cells by up-regulating Bcl-XL. J Biol Chem. 2007;282:36463–73.
Dong L, Zheng H, Qu CK. CCL3 is a key mediator for the leukemogenic effect of Ptpn11-activating mutations in the stem-cell microenvironment. Blood. 2017;130:1471–4.
Baba T, Naka K, Morishita S, Komatsu N, Hirao A, Mukaida N. MIP-1alpha/CCL3-mediated maintenance of leukemia-initiating cells in the initiation process of chronic myeloid leukemia. J Exp Med. 2013;210:2661–73.
Deng L, Chan RJ. Cleaning up the environment in juvenile myelomonocytic leukemia. Transl Cancer Res. 2017;6:S36–S8.
Grad JM, Zeng XR, Boise LH. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin Oncol. 2000;12:543–9.
Stavast CJ, Leenen PJM, Erkeland SJ. The interplay between critical transcription factors and microRNAs in the control of normal and malignant myelopoiesis. Cancer Lett. 2018;427:28–37.
Rosmarin AG, Yang Z, Resendes KK. Transcriptional regulation in myelopoiesis: Hematopoietic fate choice, myeloid differentiation, and leukemogenesis. Exp Hematol. 2005;33:131–43.
Maharshak N, Cohen S, Lantner F, Hart G, Leng L, Bucala R, et al. CD74 is a survival receptor on colon epithelial cells. World J Gastroenterol. 2010;16:3258–66.
Wirtz TH, Saal A, Bergmann I, Fischer P, Heinrichs D, Brandt EF, et al. Macrophage migration inhibitory factor exerts pro-proliferative and anti-apoptotic effects via CD74 in murine hepatocellular carcinoma. Br J Pharmacol. 2021;178:4452–67.
Wolf Y, Shemer A, Polonsky M, Gross M, Mildner A, Yona S, et al. Autonomous TNF is critical for in vivo monocyte survival in steady state and inflammation. J Exp Med. 2017;214:905–17.
Yamashita M, Passegue E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25:357–72.e7.
Wu Y, Zehnle PMA, Rajak J, Koleci N, Andrieux G, Gallego-Villar L, et al. BH3 mimetics and azacitidine show synergistic effects on juvenile myelomonocytic leukemia. Leukemia. 2024;38:136–48.
Uy GL, Rettig MP, Stone RM, Konopleva MY, Andreeff M, McFarland K, et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 2017;7:e542.
Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun. 2020;11. 1659.
Acknowledgements
We thank Ursula Kern, Charlotte M. Niemeyer and the members of DFG FOR2036 and CRC353 for insightful discussion. We thank N. Kaltenbach, C. Ambs and J. Viere for excellent technical assistance, and N. Krause and her team of the Center for Experimental Models and Transgenic Services (CEMT) for animal care. We are grateful to M. Follo and her team at the Lighthouse Fluorescence Technologies Core Facility, Freiburg for cell sorting and maintenance of flow cytometers. The work was supported by grants from the European Research Council (ERC Starting Grant no. 638145 “ApoptoMDS” to ME), the German Federal Ministry of Education and Research (BMBF), Berlin (“MyPred - Network for young individuals with syndromes predisposing to myeloid malignancies” to ME, BS, GG and CMN; no 01GM1911A and no 01GM2207A), DFG FOR2036 (”New Insights into Bcl-2 family interactions: from biophysics to function”; to ME and GH) and Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - TRR 353/1 - 471011418 (“Regulation of cell death decisions”; to ME, GH and MB). The Orbitrap Fusion Lumos mass spectrometer was partly funded by the German Research Foundation (INST 95/1436-1 FUGG).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
NK, NAW, JM and ME designed, performed, and analyzed the experiments. NK performed all experiements, except those specified. NAW performed and analyzed the CD74 data. JM performed and analyzed the mass spectrometry secretome data. SR and MB analyzed the WES and RNA sequencing data. KA conducted the histology. SK and GH assisted with the WB protocols and antibodies. VRM assisted with the mice genotyping protocol by sequencing. YW, JR, HX, MW, JW and SB assisted with mice sacrificing and organ processing. HX also contributed to the TNFα blocking experiments. NK and ME interpreted the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All methods were performed in accordance with the good scientific conduct guidelines and regulations. All animal procedures have complied with the ethics committee of the University of Freiburg as well as the local authorities - Regierungspräsidium Freiburg with registration number G18/143.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Gerry Melino
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koleci, N., Wu, Y., Wehner, N.A. et al. Oncogenic and microenvironmental signals drive cell type specific apoptosis resistance in juvenile myelomonocytic leukemia. Cell Death Dis 16, 165 (2025). https://doi.org/10.1038/s41419-025-07479-2
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-025-07479-2
This article is cited by
-
Oleuropein regulates ubiquitination-mediated Mcl-1 turnover and exhibits antitumor activity
Cancer Gene Therapy (2025)
