
Abstract
Diabetes mellitus (DM), a metabolic disease of globally health concern, is pathologically attributed to mitochondrial dysfunction, an essential component in disease progression. Mitochondrial quality control (MQC) acts as a critical defense mechanism for metabolic homeostasis, yet its implications in DM and its complications remain incompletely understood. This study thoroughly summarizes emerging evidence that delineates the molecular processes of MQC, with an emphasis on effector protein post-translational regulation, upstream signaling hubs, and interactions with other metabolic processes including ferroptosis and lipid metabolism. We highlight newly discovered processes involving mitochondrial-derived vesicles, licensed mitophagy, and mitocytosis that broaden the regulatory landscape of MQC, going beyond the traditionally recognized process including biogenesis, dynamics and mitophagy. MQC imbalance exacerbates insulin resistance, while impaired insulin signaling reciprocally compromises mitochondrial function, creating a vicious cycle of metabolic deterioration. Despite tissue-specific pathophysiology, diabetic complications exhibit identical MQC impairment including suppressed biogenesis, fission-fusion imbalance, and deficient mitophagy. Emerging therapies including clinical hypoglycemic agents and bioactive phytochemicals demonstrate therapeutic potential by restoring MQC. However, current strategies remain anchored to classical pathways, neglecting novel MQC mechanisms such as mitocytosis. Addressing this gap demands integration of cutting-edge MQC insights into drug discovery, particularly for compounds modulating upstream regulators. Future studies must prioritize mechanistic dissection of MQC novel targets and their translational relevance in halting metabolic collapse of diabetes progression. Since mitochondrial function is a cornerstone of metabolic restoration, synergizing precision MQC modulation with multi-target interventions, holds transformative potential for refine diabetic complications therapeutics.

Similar content being viewed by others


Facts
-
MQC governs insulin signaling and cellular maturation in metabolic tissues, maintaining metabolic homeostasis and function.
-
Despite the tissue-specific manifestations, MQC impairment in diabetes accelerates systemic metabolic collapse with conserved pathogenic features including suppressed biogenesis and fusion, excessive fission, and deficient mitophagy.
-
Emerging therapeutic strategies including clinical hypoglycemic agents and natural products exhibit great potential in the treatment of diabetes and complications by targeting MQC.
Introduction
Diabetes mellitus (DM), a systemic metabolic disease characterized by chronic hyperglycemia and insulin resistance, poses a mounting global health burden, with a prevalence estimated to reach 783.2 million adults by 2045 [1]. Mitochondria are dynamic organelles essential for cellular energy metabolism [2, 3]. Metabolic stress including persistent hyperglycemia and dysregulated glucolipid metabolism could induce pancreatic mitochondrial dysfunction, especially in β cells, which is key to the onset and progression of diabetes, aggravating oxidative stress, inflammation, and glucolipotoxicity, driving diabetic multi-organ damage [4,5,6,7,8].
Mitochondrial quality control (MQC), a network encompassing biogenesis, dynamics (fission and fusion), and mitophagy, ensures mitochondrial integrity and metabolic flexibility [9]. Emerging evidence positions MQC as a linchpin in DM pathophysiology. MQC imbalance disrupts cellular ATP synthesis, amplifies oxidative damage, and perpetuates metabolic stress across insulin-sensitive tissues [10,11,12,13]. While the role of MQC in diabetes is well-documented previously, its tissue-specific regulation in diabetic complications, ranging from cardiac lipotoxicity to renal tubular injury, remains underexplored [14, 15].
This review synthesizes emerging insights into MQC mechanisms, elucidating their physiological and pathological interplay with DM. We posit that MQC imbalance serves as a co-pathogenic aspect of diabetic complications, and summarize emerging evidence of MQC molecular targets on diabetic complications. What is more, we discuss the therapeutic strategies targeting MQC to break the cycle of mitochondrial collapse and systemic metabolic decompensation. We searched PubMed for relevant English-language articles published up to April 2025 (final search date). The query terms related to MQC and diabetic complications: (“mitochondrial quality control” OR “mitochondrial biogenesis” OR “mitochondrial dynamics” OR “mitochondrial fission” OR “mitochondrial fusion” OR “mitophagy”) AND (“diabetes mellitus” OR “insulin resistance”) AND (“pancreas” OR “liver” OR “muscle” OR “adipose” OR “diabetic complications” OR “diabetic heart” OR “diabetic cardiomyopathy” OR “diabetic nephropathy” OR “diabetic kidney” OR “diabetic retinopathy” OR “diabetic retina” OR “diabetic neuropathy” OR “diabetic peripheral neuropathy” OR “diabetic brain”). Studies were included if they investigated mechanistic links between MQC and diabetes or its complications, provided molecular or therapeutic insights, and were peer-reviewed original research. Excluded articles included reviews, editorials, non-English publications, and studies lacking direct relevance to DM and MQC. Two independent reviewers screened titles and abstracts, followed by full-text assessment of eligible articles.
Mitochondrial quality control
To ensure optimal function, mitochondria undergo a finely tuned quality control mechanism involving mitochondrial motility, mitochondrial-derived vesicles (MDVs), the ubiquitin-proteasome system, and cellular environmental factors [16].
Mitogenesis
Mitogenesis replenishes mitochondrial mass via coordinated nuclear DNA (nDNA)- and mitochondrial DNA (mtDNA)-encoded programs. The peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1) family, including PGC-1α, PGC-1β, and PRC, orchestrate this process by activating transcription factors NRF1, NRF2, and ERRα, as well as respiratory complex assembly [17,18,19,20]. While PGC-1α drives biogenesis in energy-demanding tissues including heart and muscle, PGC-1β compensates during brown adipogenesis [21]. PRC regulates NRF1/CREB-dependent transcription but is dispensable for basal biogenesis [22, 23]. Upstream regulators and post-translational modifications like PGC-1α monomethylation [24] fine-tune biogenesis, while mitochondrial preprotein translocases TOM and TIM complexes ensure nuclear-encoded protein import [25, 26]. Under stress, mitochondrial also transfer from neighboring cells and can rescue bioenergetic deficits [27, 28].
Mitochondrial dynamics
Mitochondrial morphology adapts to metabolic demands via fission and fusion, governed by post-translational modifications (PTMs) of dynamin-related GTPases [29, 30]. Fusion merges outer (MFN1/MFN2) and inner (OPA1) membranes, enabling content exchange and cristae stabilization [31, 32]. Transient “kiss-and-run” fusion events allow selective matrix communication [33]. Fission, mediated by Drp1 recruitment to mitochondrial receptors (Mff, Mid49/51, Fis1), partitions damaged regions for degradation [34,35,36,37,38]. Imbalanced dynamics including excessive fission and defective fusion disrupt metabolic flexibility and amplify oxidative stress.
Mitophagy
Mitophagy, a specialized form of cellular autophagy, selectively degrades damaged mitochondria via the Ser/Thr kinase PINK1/ the E3 ubiquitin ligase Parkin-dependent and -independent pathways. Depolarized mitochondria stabilize PINK1, which phosphorylates ubiquitin to recruit Parkin for OMM protein ubiquitination [39,40,41]. Parkin-independent pathways utilize OMM receptors including BCL2 and adenovirus E1B 19-kDa-interacting protein3 (BNIP3), BNIP3-like (BNIP3L/NIX), Bcl-2-like protein 13 (BCL2L13), FUN14 domain containing 1 (FUNDC1) and tacrolimus (FK506)-binding protein8 (FKBP8), as well as IMM phospholipids cardiolipin to recruit LC3-labeled autophagosomes [42, 43]. Regulatory crosstalk involves prohibitin 2 (PHB2), autophagy-beclin1-regulator1(AMBRA1), and Myosin VI (MYO6), which promote autophagosome-lysosome fusion [44,45,46,47,48].
Novel horizons for MQC
Beyond canonical mitophagy, MQC encompasses adaptive mechanisms to preserve homeostasis under stress. MDVs act as a compensatory pathway when LC3-dependent autophagosome formation is impaired, selectively packaging misfolded proteins, oxidized lipids or damaged mtDNA into mitochondrial-derived compartments (MDCs) for lysosomal or peroxisomal degradation [49,50,51,52,53]. MDV biogenesis is regulated by microtubule-associated motor proteins MIRO1/2 and fission machinery components (Drp1, Mff, Mid49, Mid51), linking vesicle formation to mitochondrial dynamics [53].
Cells unable to recycle dysfunctional mitochondria intracellularly may transfer damaged components via extracellular vesicles for phagocytic clearance by neighboring cells (e.g., macrophages), a process termed “licensed mitophagy” [27, 28]. Recent work also identifies mitocytosis, a migrasome-mediated expulsion of mitochondria during cell migration. Under mild stress, motor proteins direct damaged mitochondria to migrasomes at the cell periphery, enabling extracellular disposal [54].
Furthermore, mitochondria-lysosome crosstalk enables localized repair through inner mitochondrial membrane-derived vesicles (VDIMs). Cristae damage triggers ROS-dependent activation of lysosomal TRPML1 channels, promoting VDAC1 oligomerization and herniation pore formation on the OMM. This facilitates extrusion of damaged IMM components, which are engulfed by lysosomes as VDIMs [55].
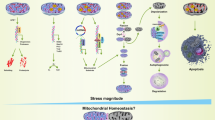
These emerging pathways underscore MQC’s plasticity, offering novel therapeutic entry points to disrupt the cycle of mitochondrial dysfunction and metabolic disease (Fig. 1).

Mitogenesis: Nuclear DNA and mitochondrial DNA are responsible for the synthesis of new mitochondria, a process regulated by coactivators and transcription factors through membrane protein transport mechanisms. Mitochondrial dynamics: The fusion is mediated by OMM proteins (MFN1 and MFN2), and the fission is mediated by the interaction between DRP1 and fission factors (Fis1. Mff, Mid49, Mid51). Mitophagy: The process is mainly mediated by the PINK1/Parkin pathway and mitochondrial membrane protein receptor-dependent signaling pathway. MDVs biogenesis: Mitochondria selectively release components and even damaged mtDNA through MDVs, which are degraded through specific transport pathways to lysosomes. Mitocytosis: Mitochondria are translocated to migrasomes, and migrating cells selectively bind damaged mitochondria to motor proteins, localize them to the cell’s periphery, and ultimately discharge them outside the cell to be removed. Licensed mitophagy: A cell transfers its own damaged mitochondrial components to extracellular vesicles to be released and eliminated with the help of mitophagy of another cell. Mitochondria transfer: When endogenous mitogenesis is deficient, the cell replenishes exogenous healthy mitochondria from outside other cells via mitochondrial transfer.
The role of mitochondria in pancreatic beta cells
Mitochondria regulate pancreatic β-cell insulin secretion and peripheral insulin sensitivity through redox balance, metabolic flux, and interorganellar communication [56, 57]. Maintaining mitochondria turnover ensures establishment of sufficient mitochondria OXPHOS for islet β cells differentiation and maturation [58]. The mitochondrial tRNA-derived fragment interacts with electron transfer system complexes and paly pivotal role in mitochondrial oxidative phosphorylation and its coupling to insulin secretion of pancreatic β-cells [59].
In pancreatic β cells from type 2 diabetic subjects, the impaired secretory response to glucose is associated with a marked alteration of mitochondrial function and morphology. In particular, uncoupling protein-2 expression is increased, which leads to lower ATP, decreased ATP/ADP ratio, with consequent reduction of insulin release [60]. Hyperglycemic environment induces metabolic changes in β cells that significantly reduce mitochondrial metabolism and ATP synthesis, which contribute to the progressive failure of β-cells to respond to glucose in diabetes [61]. Inactivation of transcriptional factor YY1 impairs mitochondrial oxidative phosphorylation and mitochondrial function, resulting in the reduction of insulin secretion, β-cell mass, and glucose tolerance [62]. Lipotoxicity including saturated free fatty acids (FFAs) accumulation alters β-cell signal transduction [63], induce mitochondrial fragmentation and production of ROS in β cells, thereby promoting β-cell dysfunction and pancreatic insulin sensitivity changes [64,65,66]. Effective mitochondrial adaptive responses including mitochondrial redox signaling and mild uncoupling reduce β-cell oxidative stress and dysfunction under metabolic stresses like hypertriglyceridemia [67].
MQC in the pathophysiology of insulin resistance and diabetes
Insulin resistance (IR), a hallmark of type 2 diabetes mellitus (T2DM), arises from impaired cellular responsiveness to insulin, disrupting glucose homeostasis and driving hyperglycemia. Both insulin insufficiency and IR converge on mitochondrial dysfunction, where defective MQC exacerbates metabolic dysregulation. Targeting MQC pathways offers a strategic avenue to uncouple mitochondrial failure from disease progression.
MQC-insulin signaling crosstalk
Enhanced mitochondrial biogenesis improves ATP synthesis, facilitating insulin-stimulated glucose uptake via GLUT4 translocation [68]. Mitochondrial dynamics proteins directly modulate insulin signaling: Mfn2 stabilizes insulin receptor substrate 1 (IRS1) and coordinates mitochondrial-ER crosstalk [69], while phosphorylation of mitochondrial fission factor (MFFS131) promotes hepatic insulin sensitivity by tuning mitochondrial fragmentation [30]. Conversely, insulin regulates MQC via Akt/mTOR/FOXO1 signaling, suppressing Drp1/Fis1-mediated fission and enhancing PGC-1α-driven biogenesis [70,71,72]. Disruption of this bidirectional crosstalk which is evidenced by mitophagy-deficient β-cells with impaired insulin secretion [73, 74] fuels a self-perpetuating cycle of IR and mitochondrial collapse [75].
MQC dysregulation in metabolic stress
Chronic hyperglycemia and lipid overload impair mitochondrial oxidative phosphorylation, elevating ROS production and depleting ATP synthesis [76]. ROS inactivate IRS1/2 via serine phosphorylation, exacerbating β-cell apoptosis and peripheral IR [77]. Downregulation of PGC-1α and NRF1 in T2DM reduces mitochondrial biogenesis, fatty acid oxidation, and respiratory capacity [78,79,80], while hyperglycemia-driven the upregulation of fission proteins fragments mitochondria, further amplifying oxidative stress [81,82,83]. Restoring deficient fusion or inhibiting excessive fission rescues IRS1/Akt signaling and glucose uptake, underscoring MQC’s role in metabolic flexibility [84, 85].
Defective mitophagy exacerbates IR by accumulating damaged mitochondria, triggering NLRP3 inflammasome activation and adipose inflammation [13, 86]. FUNDC1 knockout mice exhibit worsened obesity and IR under high-fat diets, linking impaired mitophagy to adipose metabolic dysfunction [87]. These findings position MQC as a nodal regulator of inflammation-metabolism crosstalk in diabetes.
Impaired MQC in the pathophysiology of metabolic tissues associated with IR
MQC dysregulation induces a retrograde signaling program that impairs identity and maturity in insulin-sensitive tissues including pancreas, liver and adipose, yielding metabolic disorders and driving systemic IR and diabetes progression [88]. Restoring MQC plasticity in these tissues offers a strategic avenue to mitigate metabolic dysfunction (Table 1).
Pancreas
The pancreatic endocrine β-cells, central to systemic glucose homeostasis [89], are critically vulnerable to mitochondrial dysfunction in diabetes. The disruption of mitochondrial turnover and function in islets contribute to chronic exposure of pancreatic β-cell to high concentrations of FFAs, aggravating lipotoxicity-mediated suppression of glucose-stimulated insulin secretion [90,91,92]. Emerging evidence highlights that MQC failure in islets drives β-cell dysfunction through impaired glucose tolerance and exacerbated insulin resistance, accelerating diabetes progression [93, 94]. Notably, NRF2 deficiency under metabolic stress disrupts mitogenesis, triggering a marked decline in functional β-cell mass and systemic glucose dysregulation [95]. Concurrently, O-GlcNAc transferase (OGT) sustains β-cell physiology by enhancing pancreatic and duodenal homeobox 1 (Pdx1)-dependent mitogenesis, thereby preserving insulin secretion [96]. Dysregulated mitochondrial dynamics directly underpins β-cell death. For instance, RNA binding protein HuD overexpression rescues diabetic β-cell dysfunction by restoring MFN2-mediated mitochondrial fusion, whereas palmitate (a C16-FFA)- induced activation of TRPM2 elevates mitochondrial Zn²⁺, potentiating DRP1-driven mitochondrial fission, β-cell apoptosis, and insulin secretory failure [97, 98]. The Activation of LRH-1/NR5A2 targeting mitochondrial dynamics attenuates the autoimmune attack coupled to beta cell regeneration and islet survival in type 1 diabetes mellitus (T1DM) [99]. Furthermore, human islet amyloid polypeptide (hIAPP) aggregation in diabetes exacerbates mitochondrial fragmentation and suppresses mitophagy, leading to ROS overproduction and irreversible β-cell damage [100]. Disruption of the PPP3/calcineurin/TFEB axis impairs mitophagy, culminating in β-cell dysfunction and glucose intolerance [101]. Under glucotoxic conditions, GRP78 upregulation cause depletion of the antioxidant pool and promote mitophagy in pancreatic cells in T1DM [102]. Collectively, MQC impairment contributes to β-cell demise in diabetes, offering potential targets for interventions to preserve pancreatic β-cell mass and function.
Liver
Hepatic MQC directly modulates glucose and lipid homeostasis. Activation of the AMPK/SIRT1/PGC-1α axis enhances mitogenesis, suppresses gluconeogenesis, and promotes fatty acid oxidation, reversing lipid accumulation and IR in T2DM [80, 103]. CerS6-derived sphingolipids, as a kind of hepatic lipid deposition, promote hepatic mitochondrial fragmentation and glucose disorders in a Mff-dependent manner, exacerbating obesity and IR [34]. Nutrient-sensing pathways further fine-tune mitochondrial dynamics. AKT-dependent phosphorylation of mitochondrial fission factor MFFS131 induces transient mitochondrial fragmentation, priming the liver for metabolic adaptation to feeding cycles [30]. Conversely, Drp1 and Fis1 upregulation disrupts oxidative phosphorylation, exacerbating hepatic steatosis and hyperglycemia [104]. As a catabolic process, mitophagy promotes mitochondrial fatty acid oxidation to inhibit hepatic fatty acid accumulation, thereby improving hepatic IR and metabolic syndrome [105].
Skeletal muscle
MQC governs insulin-stimulated glucose uptake in skeletal muscle. Upregulation of miR-30d-5p undermines glycolipid metabolism in skeletal muscle by targeting SIRT1/PGC-1α-dependent mitogenesis [106]. Lipid infusion increases the expression of DRP1 and PINK1, regulating MQC networks to suppress insulin sensitivity in human skeletal muscle [75]. Nutrient storage stress triggers a BNIP3L-mediated signaling cascade that promotes mitochondrial fission and mitophagy, impairing myocyte glucose uptake with the activation of MTOR-RPS6KB and IRS1 phosphorylation [107]. In diabetic sarcopenia, MFG-E8 (lactadherin) is upregulated and induces mitophagy deficiency via the HSPA1L-Parkin pathway, thereby aggravating diabetic muscle atrophy [108]. Deubiquitinating enzymes including USP15 and USP30 activates DRP1-dependent mitochondrial fission, thereby regulating skeletal muscle MQC and insulin sensitivity in T2DM patients [109].
Adipose tissue
Adipocyte MQC regulates systemic energy balance. JMJD1A, a histone demethylase, activates PGC-1-mediated mitogenesis and the formation of beige adipocytes in white adipose tissues (WAT), alleviating obesity and metabolic dysfunction [110]. In WAT, activation of small GTPase RalA promotes excessive mitochondrial fission, suppresses thermogenesis, and drives obesity-associated metabolic dysfunction [111]. TMEM135 overexpression promotes brown fat mitochondrial fission, counteracts obesity and insulin resistance, and rescues thermogenesis in adipose-specific peroxisome-deficiency [112]. FUNDC1-dependent mitophagy maintains adipose homeostasis, whereas FUNDC1 ablation in WAT disrupts mitochondrial integrity, exacerbates diet-induced obesity and IR [87]. Brown adipose tissue (BAT) relies on mitochondrial protein succinylation and malonylation to sustain thermogenic capacity, with defects in these post-translational modifications impairing mitophagy and glucose homeostasis in T2DM [86, 113].
Impaired MQC in diabetic complications
Individuals with diabetes face a significantly elevated risk of macrovascular events, such as stroke and myocardial infarction, as well as microvascular complications, including retinopathy and nephropathy, compared to non-diabetic populations. Emerging evidence underscores MQC dysfunction as a pivotal contributor to diabetic vascular endothelial dysfunction. Hyperglycemia induces mitochondrial fragmentation, impaired fusion, and excessive fission and autophagy across various tissues, including the cardiovascular system, liver, and pancreas [114, 115]. FOXO1 inhibition mitigates high glucose (HG)-induced DRP1 overexpression and mitochondrial fission in endothelial cells, reducing reactive oxygen species (mtROS) overproduction and ameliorating vascular dysfunction [116]. Given that mtROS-driven endothelial injury underlies both macro- and microvascular diabetic complications [117, 118], targeting MQC represents a promising therapeutic avenue to attenuate disease progression [119].
Diabetic cardiomyopathy
Diabetic cardiomyopathy (DCM), characterized by myocardial fibrosis and impaired cardiac function independent of coronary artery disease, is linked to MQC defects involving calcium dysregulation, oxidative stress, and lipid accumulation [120]. Cardiac mitochondrial damage and biogenesis are induced by oxidative stress in a chronic model of type 1 diabetes [121]. While compensatory mitophagy may protect against early lipid overload in high fat diet-induced DCM [122], persistent imbalances in mitochondrial dynamics which is marked by excessive fission and deficient fusion and mitophagy that drive DCM pathogenesis.
Recent research has identified novel targets for DCM that alleviate mitochondrial dysfunction and cellular apoptosis by modulating post-translational modifications (PTMs) of MQC proteins, including phosphorylation, acetylation, and methylation. PGAM5 dephosphorylates PHB2 at Ser91, disrupting MQC and exacerbating myocardial injury [123, 124]. Downregulated miR144 elevates RAC1, impairing AMPK/PGC-1α-mediated mitogenesis and promoting apoptosis [125]. CaSR enhances ubiquitination of MFN1, MFN2, and CX43 via GP78, disrupting mitochondrial fusion and energy metabolism [126]. PFKFB3 deficiency in diabetic hearts destabilizes OPA1, whereas NEDD4L-mediated OPA1 ubiquitination preserves mitochondrial integrity [127]. MAP4K4 disrupts GPX4-dependent redox balance, inducing Drp1 S-nitrosylation (SNO-Drp1), excessive fission, and ferroptosis [128]. MST1 inhibits SIRT3/PARKIN mitophagy and promotes DRP1 phosphorylation, accelerating mitochondrial fragmentation [129, 130]. By performing targeted metabolomics to characterize the metabolic phenotype of human myocardium of DCM, it is founded that RCAN1 upregulation activates DRP1 Ser616, driving lipid accumulation and metabolic remodeling in cardiomyocytes [131].
Epigenetic and epitranscriptomic mechanisms further modulate MQC in DCM. The NOTCH1/ALKBH5/YTHDF axis, regulated by m6A-mediated phase separation, suppresses NOTCH1 expression, promoting fission and fibrosis [132]. Similarly, BRD4 binds H3K27ac at the PINK1 promoter, repressing PINK1/Parkin mitophagy; BRD4 inhibition restores mitophagy and alleviates DCM [133].
MQC dysregulation intersects with lipid metabolism, calcium signaling, and ferroptosis in DCM. SFRP2, an adipokine, modulates mitochondrial dynamics via AMPK/PGC-1α and activates calcineurin/TFEB-dependent mitophagy [134, 135]. ORAI1-mediated Ca2+ influx activates DRP1, exacerbating fission and hypertrophy. Targeting ORAI1/p-ERK1/2/CnA normalizes calcium homeostasis [136]. DUSP26 rescues mitochondrial dynamics and lipid metabolism via FAK/ERK signaling [137]. LGR6 deficiency impairs STAT3/PGC-1α, aggravating ferroptosis and biogenesis defects [138].
Key transcription factors integrate MQC with metabolic stress. STAT3 activation by BNP sustains OPA1-mediated fusion, while FOXO1 competes with STAT3 for 14-3-3 binding, impairing mitochondrial homeostasis under hyperglycemia [139, 140]. PPARα downregulation reduces MFN2 expression, contributing to dynamics imbalance [141]. ALDH2 preserves mitochondrial function via Akt/GSK3β/Foxo3a signaling and PARKIN-dependent mitophagy [142].
DCM pathogenesis involves a complex interplay of MQC defects, PTMs, and transcriptional and epigenetic dysregulation. Targeting nodes such as DRP1 phosphorylation, mitophagy pathways, or redox balance offers therapeutic potential. Future studies should clarify the spatiotemporal dynamics of MQC disruption and optimize strategies to restore mitochondrial homeostasis in diabetes. (Table 2)
Diabetic nephropathy
Diabetic kidney disease (DKD), a chronic complication driven by persistent hyperglycemia and glomerular hyperfiltration, remains a leading cause of end-stage renal disease globally [143]. Pathologically, DKD is marked by altered glomerular filtration rates, proteinuria, and dysregulation of metabolites and ions. Podocytes, essential components of the glomerular filtration barrier, are particularly vulnerable to hyperglycemia-induced damage, contributing to mitochondrial dysfunction and disease progression. Emerging evidence underscores the centrality of mitochondrial homeostasis in maintaining renal health under diabetic conditions [144].
Mitogenesis is critically regulated in DKD through metabolic and signaling pathways. Hyperglycemia suppresses pyruvate kinase M2 (PKM2) tetramerization and activity via sulfenylation in podocytes, impairing glycolytic flux and amplifying toxic glucose metabolite accumulation. Restoring PKM2 activity enhances glucose metabolism, upregulates PGC-1α expression, and promotes mitochondrial biogenesis, thereby mitigating renal injury [145]. Conversely, overexpression of p66SHC, a redox-sensitive adaptor protein, suppresses mitogenesis by downregulating SIRT1, PGC-1α, NRF1, and TFAM, exacerbating mitochondrial ROS production and podocyte damage [146]. Similarly, mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH) deficiency in diabetic models disrupts PGC-1α-dependent mitogenesis, activating the RAGE-S100A10 axis to drive oxidative stress and renal injury [147]. Additional regulators include AKAP1, which phosphorylates Larp1 via PKC signaling to inhibit TFAM-mediated mtDNA replication, and adiponectin receptor 1 (AdipoR1), whose deficiency impairs the CREB/PGC-1α/TFAM axis. Exogenous adiponectin supplementation rescues mitogenesis and alleviates tubular dysfunction, highlighting therapeutic potential [148, 149]. Progranulin (PGRN) depletion further disrupts mitochondrial homeostasis via Sirt1/PGC-1α/FOXO1 signaling, while the lncRNA PVT1 exacerbates fission and bioenergetic deficits by destabilizing AMPKα through TRIM56 interaction [150, 151]. Telomerase reverse transcriptase (TERT) also safeguards renal function by sustaining AMPK/PGC-1α-mediated mitochondrial energy balance [152].
Dysregulated mitochondrial dynamics, intertwined with hypoxia, lipid abnormalities, and calcium dyshomeostasis, further propagate DKD. Hypoxia-inducible factor-1α (HIF-1α) upregulation in tubular cells induces heme oxygenase-1 (HO-1) to stabilize mitochondrial dynamics and attenuate hypoxia-driven damage [153]. ALCAT1 expression is increased in the glomeruli of DKD patients, accompanying with significant mitochondrial damage [154]. Lipidomic perturbations, such as ALCAT1-mediated cardiolipin oxidation, impair mitophagy via AMPK signaling, while the ChREBP/GNPAT/plasmalogen axis links lipid metabolism to mitochondrial fission [154, 155]. RIPK3 overexpression exacerbates podocyte injury by activating PGAM5/Drp1-mediated fission through necroptosis, illustrating crosstalk between cell death pathways and organelle dynamics [156].
Electron transport chain (ETC) dysfunction exacerbates bioenergetic failure in DKD. NDUFS4 deficiency disrupts complex I integrity, impairing cristae morphology via STOML2 interaction, whereas IGF2BP3 stabilizes CAMK1 to inhibit fission and apoptosis through m6A modification [157, 158]. Calcium signaling further modulates mitochondrial fitness. CaMKKβ sustains AMPK phosphorylation to support function, but NEDD4L ubiquitination under hyperglycemia destabilizes CaMKKβ, promoting fission [159]. TRIM22 and WTAP form an m6A-dependent regulatory axis targeting OPA1 ubiquitination, thereby disrupting fusion and amplifying injury [160]. HGF maintaining mitochondrial homeostasis in type 1 diabetic podocytes via decreasing ARF6-dependent DRP1 translocation [161].
Impaired mitophagy, coupled with MAM disruption, ferroptosis, and oxidative stress, is a hallmark of DKD. KCa3.1 channel deficiency impedes TGF-β1/BNIP3-dependent mitophagy, while DsbA-L depletion activates JNK/MFF/DRP1 signaling to drive fission. Conversely, DsbA-L preserves MAM integrity via HELZ2/MFN2 to promote mitophagy, illustrating context-dependent roles [162,163,164]. TNFAIP8L1/TIPE1 exacerbates tubular injury by degrading the mitophagy receptor PHB2, whereas UHRF1 enhances PINK1-dependent mitophagy and suppresses ferroptosis via TXNIP inhibition [165, 166]. MFN2-SERCA2 interactions and VDR activation restore MAMs and mitophagy through the FUNDC1 pathway, while PACS-2 coordinates fission inhibition and BECN1-dependent mitophagosome formation [167, 168]. YAP1 inactivation disrupts both mitogenesis and PINK1/Parkin-mediated mitophagy, skewing macrophage polarization via CXCL1, whereas TRPC6/calpain-1 signaling impairs mitophagy to fuel inflammation [169, 170]. DLAT, downregulated in diabetes, enhances AMPK-driven mitophagy, offering protection against renal dysfunction [171]. N-Acetyl-Cysteine combined with insulin reverses glomerular wrinkling and fibrosis by regulating the mitochondrial dynamics and FUNDC1-mediated mitophagy in type 1 diabetic nephropathy canine [172].
These findings underscore the multifaceted interplay between mitochondrial homeostasis and diabetic renal pathology, with emerging therapeutic targets summarized in Table 3.
Diabetic retinopathy
Diabetic retinopathy (DR), a neurovascular complication of chronic hyperglycemia, is characterized by retinal glial network dysfunction, microvascular damage, and pathological neovascularization, culminating in vision loss and blindness [173]. The retina, composed of neurovascular units integrating endothelial cells, Müller glia, pericytes, ganglion cells, and pigment epithelial cells, undergoes mitochondrial dysregulation under hyperglycemic stress, marked by impaired mitophagy and aberrant mitochondrial dynamics. Emerging evidence highlights MQC as a pivotal therapeutic target to counteract metabolic memory and mitigate DR progression [174, 175].
Retinal vascular endothelial cells (RECs), critical for nutrient exchange and barrier integrity, exhibit disrupted mitochondrial homeostasis in diabetes. Hypermethylation of the MFN2 promoter, driven by Dnmt1 binding and diminished SP1 recruitment, suppresses MFN2 expression, while SIRT1 overexpression reduces MFN2 acetylation and GTPase activity, impairing mitochondrial fusion [176, 177]. Hyperglycemia further upregulates SENP3, which deSUMOylates Drp1 to promote fission, exacerbating blood-retinal barrier dysfunction [178]. Loss of PON2, glycated by N-carboxymethyl lysine (CML), amplifies ER stress, inflammation, and mitochondrial fragmentation, whereas TGR5 activation inhibits Drp1 via Ca2+-PKCδ signaling and enhances PINK1/Parkin-mediated mitophagy by displacing mitochondrial hexokinase 2 (HK2) [179, 180]. Methylglyoxal (MGO)-mediated glycation suppresses glyoxalase 1 (GLO1) and downregulates OPA1/MFN1, aggravating oxidative injury, while VDAC1 sustains mitophagy and restrains NLRP3 inflammasome activation in RECs [181, 182].
Pericytes, essential for vascular stability, succumb to hyperglycemia-induced mitochondrial fission via EPAC1-DRP1 signaling, driving ROS overproduction and microvascular damage [183]. In retinal pigment epithelial cells (RPECs), copper transporter 1 (CTR1) overexpression disrupts copper homeostasis, inducing oxidative stress and mitochondrial dysfunction, while TIN2 accumulation impairs PINK1-dependent mitophagy via mTOR, exacerbating cellular senescence and tight junction disruption [184, 185]. SIRT3, however, counteracts hyperglycemia by activating AMPK/mTOR/ULK1 and FOXO3a/PINK1/Parkin pathways, restoring mitophagy and attenuating RPEC apoptosis [186, 187].
Retinal ganglion cells (RGCs), vulnerable to oxidative stress, are protected by DJ-1, which scavenges ROS and restores mitochondrial homeostasis under hyperglycemic conditions [188]. Müller glia, pivotal for retinal repair, exhibit TXNIP-driven mitochondrial fission and Parkin-dependent mitophagy in response to hyperglycemia, alongside reactive gliosis marked by GFAP upregulation [189].
Collectively, these findings delineate mitochondrial dysregulation as a central axis in DR pathogenesis, with therapeutic strategies targeting MQC components offering promise to preserve retinal integrity (Table 4).
Diabetic neuropathy
Diabetic neuropathy, encompassing central neurocognitive decline and diabetic peripheral neuropathy (DPN), represents a debilitating complication of chronic hyperglycemia, driven by glucotoxicity-induced neuronal and cerebrovascular dysfunction [190,191,192]. As mitochondria serve as the primary hubs of energy synthesis, MQC imbalances emerge as critical contributors to diabetic encephalopathy (DE) and peripheral nerve damage, disrupting cellular resilience and metabolic homeostasis [193].
Impaired mitogenesis underpins cognitive deficits in diabetes. Phosphorylation of WW domain-containing oxidoreductase 1 (WWOX) at tyrosine 33 under hyperglycemia exacerbates mitogenesis defects and ROS overproduction, triggering neuronal apoptosis in neuroblastoma models [194]. Conversely, activation of dopamine D1 receptors (DRD1) rescues AMPK/PGC-1α signaling, restoring mitochondrial biogenesis and alleviating cognitive dysfunction in diabetic mice [195]. Similarly, sodium butyrate (NaB), a gut-derived metabolite, enhances hippocampal synaptic plasticity by reviving AMPK/PGC-1α-driven mitogenesis, highlighting metabolic-epigenetic crosstalk in T2DM neuroprotection [196].
Dysregulated mitochondrial dynamics further propagate neuronal injury. Diabetic models reveal diminished OPA1 and MFN1/2 alongside elevated Drp1 and Fis1, reflecting fission-fusion imbalance in hippocampal and peripheral neurons [197]. Lipin1 deficiency, by altering mitochondrial membrane phospholipids, disrupts synaptic mitochondrial dynamics, exacerbating cognitive decline [197]. Caveolin-1 (Cav-1) counters these effects by suppressing GSK3β/Drp1-mediated fission and enhancing AMPK/PINK1/Parkin- and ULK1-dependent mitophagy, though excessive Drp1 inhibition may paradoxically worsen neurodegeneration [198, 199]. In peripheral nerves, mitochondrial calcium uniporter (MCU) deletion mitigates neuropathic pain by curbing calcium-dependent fission, preserving small-fiber integrity in diabetic mice [200].
Mitophagy dysfunction amplifies diabetic neuropathology. Hyperglycemia and advanced glycation end-products (AGEs) impair hippocampal mitophagy via Keap1/Nrf2/PHB2 suppression, accelerating cognitive decline [201]. Brain-derived neurotrophic factor (BDNF) counteracts oxidative stress by activating TrkB/HIF-1α/BNIP3 signaling, restoring mitophagy in cerebral microvasculature [202]. NaB further rescues neuronal mitophagy by blocking RELA-HDAC8 repression of Parkin, while the SIRT1 activator piceatannol (PCN) enhances both mitogenesis and mitophagy via SIRT1/PGC1α and SIRT1/PINK1/Parkin axes, alleviating peripheral neuropathy [203, 204]. SIRT3 overexpression attenuates painful DPN through FoxO3a/PINK1/Parkin-mediated mitophagy, whereas PARP1 hyperactivity drives peripheral nerve injury via mitophagy suppression [205, 206]. These mechanisms underscore mitochondrial dysregulation as a nexus of diabetic neuropathies, with therapeutic strategies targeting MQC offering promise to preserve neuronal integrity and function.
Overall, MQC imbalance plays an important role in the pathophysiology of diabetes and diabetic complications (Fig. 2).

Pancreas: MQC failure in islets impairs β-cell mass, insulin secretion and glucose tolerance by harming insulin signaling, which excerbates insulin resistance and elevates blood glucose. Liver: Hepatic MQC imbalance induces hepatic steatosis and fatty acid oxidation. Adipose: Adipocyte MQC imbalance decreases adipocytes thermogenic capacity and influences systemic energy balance. Muscle: MQC failure impairs insulin-stimulated myocyte glucose uptake and promotes muscle atrophy. Retina: MQC damage induces retinal glial network dysfunction and microvascular damage. Heart: The myocardial fibrosis and cardiac dysfunction in DCM are aggravated by an interplay of MQC defects, ferrotopsis and oxidation stress. Kidney: MQC dysfunction is contributed to glomerular injury in diabetic nephropathy through promoting mitochondrial ROS, MAM dysruption and ferrotopsis. Nerve: MQC imbalance induce both central neurocognitive decline and peripheral nerve damage by promoting apoptosis, ROS and calcium metabolism disorder.
Therapeutic potentials for diabetic complications by targeting MQC
Current therapeutic paradigms for diabetic vascular complications, often siloed by organ-specific pathology, inadequately address the systemic MQC dysfunction underpinning these conditions. MQC has emerged as a unifying therapeutic axis across diabetic angiopathies. Aligning with clinical guidelines, repurposing existing therapies and exploring natural compounds to modulate MQC pathways offers transformative potential for multisystem vascular protection.
Clinically approved hypoglycemic agents
Several glucose-lowering agents exert their pleiotropic effect on diabetic complications by targeting MQC (Table 5). Metformin, an AMPK activator, efficiently prevents high glucose induced mitochondrial ultrastructural and functional abnormalities in human islet cells [207]. It has beneficial clinical implications on promoting mitochondrial function and mitophagy in type 2 diabetic patients, which alleviates retinopathy and nephropathy by restoring AMPK-dependent MQC balance and inhibiting Drp1-mediated fission in atherosclerosis models. It further rescues cognitive deficits via PGC-1α-driven mitogenesis and enhances post-stroke recovery through mitophagy activation [208,209,210,211,212,213]. DPP-4 inhibitors demonstrate class-wide vascular benefits. Alogliptin and evogliptin enhance cardiac mitogenesis via PGC-1α/NRF1/TFAM signaling in DCM, while sitagliptin restores renal mitochondrial dynamics through SDF-1α/CXCR4/STAT3. Vildagliptin attenuates endothelial fission via AMPK/Drp1 inhibition and reduces arrhythmia risk by promoting mitogenesis [214,215,216,217,218,219]. SGLT2 inhibitors, including empagliflozin and canagliflozin, mitigate renal and cardiac injury by suppressing fission and enhancing mitophagy. Empagliflozin alleviates tubulopathy and myocardial microvascular damage, whereas canagliflozin activates PINK1/Parkin-mediated mitophagy in DCM [220,221,222,223,224]. GLP-1 receptor agonists exhibit tissue-specific modulation. Exendin-4 reverses cardiac mitochondrial abnormalities and curbs vascular calcification via AMPK-dependent mitophagy, while liraglutide paradoxically suppresses PINK1/Parkin signaling to protect endothelial function [225,226,227]. Pioglitazone and Exendin-4 exert neuroprotective effects by targeting altered brain mitogenesis in T1DM [228].
Natural products and herbal compounds
Natural products, rich in bioactive phytochemicals, offer diverse mechanisms to restore MQC (Table 6). Rosmarinic acid and resveratrol attenuate cardiac dysfunction through SIRT1-mediated PGC-1α deacetylation, while Astragalus-derived astragaloside IV enhances mitogenesis via SIRT1/PGC1α/NRF1 and stabilizes mitochondrial dynamics in nephropathy [229,230,231,232,233]. Salidroside and astragalin activate AMPK/SIRT3-PGC-1α axes, mitigating renal and myocardial fibrosis [234,235,236]. Rhein and Gynostemma pentaphyllum combat retinal and cardiac injury via AMPK/NRF2 signaling, whereas berberine restores podocyte homeostasis by balancing PGC-1α-mediated biogenesis and Drp1 inhibition [237,238,239,240,241,242,243]. Traditional formulas like Jinlida granules and Si-Miao-Yong-An decoction improve renal and cardiac outcomes through AMPK/PGC-1α activation and PPARα modulation [244, 245].
Mitochondrial dynamics are further regulated by compounds such as tanshinone IIa, which activates GLO1 to counteract retinal fission, and paeonol, which enhances OPA1 via CK2α/JAK2-STAT3 signaling in DCM [181, 246,247,248]. Modified Hu-lu-ba-wan and notoginsenoside Fc restore glomerular integrity by modulating PKM2/PGC-1α/OPA1 and HMGCS2-mediated dynamics [249, 250].
Natural products also alleviate diabetic complications by targeting mitophagy [251,252,253,254]. Notoginsenoside R1 and germacrone enhance PINK1-dependent pathways in retinopathy and nephropathy, while Huangqi-Danshen decoction and hyperforin activate STING1/PINK1 or DLAT-AMPK axes to bolster autophagic flux [255,256,257,258,259,260,261]. Ginsenoside Ro, a Panax ginseng-derived compound, preserves retinal endothelial integrity by restoring EPAC1/AMPK-mediated mitophagy, countering diabetic retinopathy [262]. Poricoic acid A, sourced from Poria cocos, mitigates podocyte injury via FUNDC1 activation, a receptor-dependent mitophagy pathway critical for renal protection [263]. Heyingwuzi formulation, a traditional Chinese medicine, alleviates retinal damage by suppressing ROS and apoptosis through HIF-1α/BNIP3/NIX-driven mitophagy, highlighting alternative pathways for vascular rescue [264].
Emerging therapeutic strategies
Beyond conventional and natural therapies, exogenous agents like melatonin restore cardiac MQC and myocardial ischemia/reperfusion injury in T1DM via AMPK/PGC-1αsignaling [265, 266]. Photobiomodulation modulates mitochondrial dynamics in the sciatic nerve and in the dorsal root ganglia neurons, alleviating peripheral nervous system in T1DM [267]. Immunotherapeutic approach anti-CD3 monoclonal antibodies targets exhausted CD8 + T cells’ energy metabolism and suppresses T cell signaling through regulation of Drp1-mediated mitochondrial dynamics in T1DM [268]. Finerenone rescues renal mitophagy through PI3K/Akt/eNOS pathways [269]. Placental mesenchymal stem cells rejuvenate podocyte mitophagy via SIRT1/PGC-1α/TFAM, highlighting regenerative medicine’s potential [270].
In a word, these findings underscore MQC as a nexus for therapeutic innovation in diabetic angiopathy. While repurposed drugs and natural compounds show promise, challenges remain in optimizing tissue-specific targeting, resolving mechanistic contradictions, and advancing combinatorial approaches. Translational validation and clinical trials are imperative to harness MQC modulation for multisystem vascular protection.
Conclusions
This review synthesizes emerging insights into MQC as a central axis in the pathophysiology of DM and its vascular complications, emphasizing the interplay between mitochondrial dysfunction and metabolic dysregulation. Mitochondrial anomalies drive oxidative stress, inflammation, and cellular demise, positioning MQC as a pivotal therapeutic frontier. Despite advances in understanding MQC mechanisms, translational gaps persist, particularly in targeting diabetic complications, which remain understudied compared to primary metabolic defects. Current therapies, including clinical hypoglycemic agents and natural compounds, demonstrate pleiotropic benefits by restoring mitochondrial homeostasis, yet lack specificity for vascular pathologies. Natural products, with their multi-target capacity to modulate MQC pathways, offer promising scaffolds for drug development but require rigorous clinical validation to address challenges such as bioavailability and tissue selectivity. Furthermore, although majority of the therapeutic drugs have proved to be clinical effective, the reported data related to MQC mostly come from experiments with rodent and cell models, while studies conducted in humans (ex vivo and in vivo) are still relatively few and lack higher quality evidence, which provides further direction for our future research. Future research must prioritize combinatorial strategies, precision targeting of MQC nodes, and mechanistic elucidation of organ-specific mitochondrial crosstalk. By bridging molecular insights with therapeutic innovation, this review underscores the potential of MQC-centric approaches to redefine the management of diabetic angiopathies, urging interdisciplinary efforts to transform preclinical promise into clinical reality.
References
Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diab Res Clin Pract. 2022;183:109119 Erratum in: Diabetes Res Clin Pract. 2023; 204:110945.
Talari NK, Mattam U, Meher NK, Paripati AK, Mahadev K, Krishnamoorthy T, et al. Lipid-droplet associated mitochondria promote fatty-acid oxidation through a distinct bioenergetic pattern in male Wistar rats. Nat Commun. 2023;14:766.
Greiser M, Karbowski M, Kaplan AD, Coleman AK, Verhoeven N, Mannella CA, et al. Calcium and bicarbonate signaling pathways have pivotal, resonating roles in matching ATP production to demand. Elife. 2023;12:e84204.
Hrabalova P, Bohuslavova R, Matejkova K, Papousek F, Sedmera D, Abaffy P, et al. Dysregulation of hypoxia-inducible factor 1α in the sympathetic nervous system accelerates diabetic cardiomyopathy. Cardiovasc Diabetol. 2023;22:88.
Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. 2020;126:1501–25.
Ueki K, Sasako T, Okazaki Y, Miyake K, Nangaku M, Ohashi Y, et al. Multifactorial intervention has a significant effect on diabetic kidney disease in patients with type 2 diabetes. Kidney Int. 2021;99:256–66.
Flaxel CJ, Adelman RA, Bailey ST, Fawzi A, Lim JI, Vemulakonda GA, et al. Diabetic retinopathy preferred practice pattern®. Ophthalmology. 2020;127:66–145. Erratum in: Ophthalmology. 2020;127(9):1279.
Cheng YW, Liu J, Finkel T. Mitohormesis. Cell Metab. 2023;35:1872–86.
Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–43.
He Y, Ji Z, Gong Y, Fan L, Xu P, Chen X, et al. Numb/Parkin-directed mitochondrial fitness governs cancer cell fate via metabolic regulation of histone lactylation. Cell Rep. 2023;42:112033.
Tang Y, Huang Y, Wan Z, Zhou B, Wu Z. Mitochondrial quality control links two seemingly unrelated neurodegenerative diseases. Autophagy. 2022;18:2495–7.
Li B, Liu F, Chen X, Chen T, Zhang J, Liu Y, et al. FARS2 deficiency causes cardiomyopathy by disrupting mitochondrial homeostasis and the mitochondrial quality control system. Circulation. 2024;149:1268–84.
Li JM, Li X, Chan LWC, Hu R, Zheng T, Li H, et al. Lipotoxicity-polarised macrophage-derived exosomes regulate mitochondrial fitness through Miro1-mediated mitophagy inhibition and contribute to type 2 diabetes development in mice. Diabetologia. 2023;66:2368–86.
Van Huynh T, Rethi L, Rethi L, Chen CH, Chen YJ, Kao YH. The complex interplay between imbalanced mitochondrial dynamics and metabolic disorders in type 2 diabetes. Cells. 2023;12:1223.
Blagov AV, Summerhill VI, Sukhorukov VN, Popov MA, Grechko AV, Orekhov AN. Type 1 diabetes mellitus: Inflammation, mitophagy, and mitochondrial function. Mitochondrion. 2023;72:11–21.
Liu L, Li Y, Wang J, Zhang D, Wu H, Li W, et al. Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021;22:e50629.
Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–38.
Brown EL, Foletta VC, Wright CR, Sepulveda PV, Konstantopoulos N, Sanigorski A, et al. PGC-1α and PGC-1β increase protein synthesis via ERRα in C2C12 myotubes. Front Physiol. 2018;9:1336.
Alam C, Hoque MT, Sangha V, Bendayan R. Nuclear respiratory factor 1 (NRF-1) upregulates the expression and function of reduced folate carrier (RFC) at the blood-brain barrier. FASEB J. 2020;34:10516–30.
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24.
Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–41.
Vercauteren K, Pasko RA, Gleyzer N, Marino VM, Scarpulla RC. PGC-1-related coactivator: immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol Cell Biol. 2006;26:7409–19.
Vercauteren K, Gleyzer N, Scarpulla RC. PGC-1-related coactivator complexes with HCF-1 and NRF-2beta in mediating NRF-2(GABP)-dependent respiratory gene expression. J Biol Chem. 2008;283:12102–11.
Qian X, Li X, Shi Z, Bai X, Xia Y, Zheng Y, et al. KDM3A senses oxygen availability to regulate PGC-1α-mediated mitochondrial biogenesis. Mol Cell. 2019;76:885–895.e7.
Busch JD, Fielden LF, Pfanner N, Wiedemann N. Mitochondrial protein transport: versatility of translocases and mechanisms. Mol Cell. 2023;83:890–910.
Liu Q, Chang CE, Wooldredge AC, Fong B, Kennedy BK, Zhou C. Tom70-based transcriptional regulation of mitochondrial biogenesis and aging. Elife. 2022;11:e75658.
Liu Y, Fu T, Li G, Li B, Luo G, Li N, et al. Mitochondrial transfer between cell crosstalk - An emerging role in mitochondrial quality control. Ageing Res Rev. 2023;91:102038.
Borcherding N, Brestoff JR. The power and potential of mitochondria transfer. Nature. 2023;623:283–91.
Jiang Y, Krantz S, Qin X, Li S, Gunasekara H, Kim YM, et al. Caveolin-1 controls mitochondrial damage and ROS production by regulating fission - fusion dynamics and mitophagy. Redox Biol. 2022;52:102304.
Henschke S, Nolte H, Magoley J, Kleele T, Brandt C, Hausen AC, et al. Food perception promotes phosphorylation of MFFS131 and mitochondrial fragmentation in liver. Science. 2024;384:438–46.
Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature. 2017;542:372–6.
Zhou J, Zhang H, Zhong K, Tao L, Lin Y, Xie G, et al. N6-methyladenosine facilitates mitochondrial fusion of colorectal cancer cells via induction of GSH synthesis and stabilization of OPA1 mRNA. Natl Sci Rev. 2024;11:nwae039.
Liu X, Weaver D, Shirihai O, Hajnóczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009;28:3074–89.
Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, et al. CerS6-derived sphingolipids interact with mff and promote mitochondrial fragmentation in obesity. Cell. 2019;177:1536–1552.e23.
Ren J, Liu J, Zhang J, Hu X, Cui Y, Wei X, et al. Dynamin-related protein 1 binding partners MiD49 and MiD51 increased mitochondrial fission in vitro and atherosclerosis in high-fat-diet-fed ApoE-/- mice. Int J Mol Sci. 2023;25:244.
Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–73.
Yu Y, Peng XD, Qian XJ, Zhang KM, Huang X, Chen YH, et al. Fis1 phosphorylation by Met promotes mitochondrial fission and hepatocellular carcinoma metastasis. Signal Transduct Target Ther. 2021;6:401.
Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. 2016;212:531–44.
Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–22.
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–85.
Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–33.
Terešak P, Lapao A, Subic N, Boya P, Elazar Z, Simonsen A. Regulation of PRKN-independent mitophagy. Autophagy. 2022;18:24–39.
Sagar S, Faizan MI, Chaudhary N, Singh V, Singh P, Gheware A, et al. Obesity impairs cardiolipin-dependent mitophagy and therapeutic intercellular mitochondrial transfer ability of mesenchymal stem cells. Cell Death Dis. 2023;14:324.
Yan C, Gong L, Chen L, Xu M, Abou-Hamdan H, Tang M, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy. 2020;16:419–34.
Di Rienzo M, Romagnoli A, Ciccosanti F, Refolo G, Consalvi V, Arena G, et al. AMBRA1 regulates mitophagy by interacting with ATAD3A and promoting PINK1 stability. Autophagy. 2022;18:1752–62.
Yi J, Wang HL, Lu G, Zhang H, Wang L, Li ZY, et al. Spautin-1 promotes PINK1-PRKN-dependent mitophagy and improves associative learning capability in an alzheimer disease animal model. Autophagy. 2024;20:2655–76.
Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–7.
Kruppa AJ, Buss F. Actin cages isolate damaged mitochondria during mitophagy. Autophagy. 2018;14:1644–5.
Towers CG, Wodetzki DK, Thorburn J, Smith KR, Caino MC, Thorburn A, et al. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev Cell. 2021;56:2029–2042.e5.
Wilson ZN, Balasubramaniam SS, Wong S, Schuler MH, Wopat MJ, Hughes AL. Mitochondrial-derived compartments remove surplus proteins from the outer mitochondrial membrane. J Cell Biol. 2024;223:e202307036.
Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–56.
Peng T, Xie Y, Sheng H, Lian Y, Xie N. Mitochondrial-derived vesicles: Gatekeepers of mitochondrial response to oxidative stress. Free Radic Biol Med. 2022;188:185–93.
König T, Nolte H, Aaltonen MJ, Tatsuta T, Krols M, Stroh T, et al. MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat Cell Biol. 2021;23:1271–86.
Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896–2910.e13.
Prashar A, Bussi C, Fearns A, Capurro MI, Gao X, Sesaki H, et al. Lysosomes drive the piecemeal removal of mitochondrial inner membrane. Nature. 2024;632:1110–7.
Kaufman BA, Li C, Soleimanpour SA. Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Mol Asp Med. 2015;42:91–104.
Zhao Q, Luo T, Gao F, Fu Y, Li B, Shao X, et al. GRP75 regulates mitochondrial-supercomplex turnover to modulate insulin sensitivity. Diabetes. 2022;71:233–48.
Liu B, Hua D, Shen L, Li T, Tao Z, Fu C, et al. NPC1 is required for postnatal islet β cell differentiation by maintaining mitochondria turnover. Theranostics. 2024;14:2058–74.
Jacovetti C, Donnelly C, Menoud V, Suleiman M, Cosentino C, Sobel J, et al. The mitochondrial tRNA-derived fragment, mt-tRF-LeuTAA, couples mitochondrial metabolism to insulin secretion. Mol Metab. 2024;84:101955.
Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005;48:282–9.
Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat Commun. 2019;10:2474.
Song D, Yang Q, Jiang X, Shan A, Nan J, Lei Y, et al. YY1 deficiency in β-cells leads to mitochondrial dysfunction and diabetes in mice. Metabolism. 2020;112:154353.
Corkey BE, Deeney JT, Yaney GC, Tornheim K, Prentki M. The role of long-chain fatty acyl-CoA esters in beta-cell signal transduction. J Nutr. 2000;130:299S–304S.
Tang Y, Majewska M, Leß B, Mehmeti I, Wollnitzke P, Semleit N, et al. The fate of intracellular S1P regulates lipid droplet turnover and lipotoxicity in pancreatic beta-cells. J Lipid Res. 2024;65:100587.
Plötz T, von Hanstein AS, Krümmel B, Laporte A, Mehmeti I, Lenzen S. Structure-toxicity relationships of saturated and unsaturated free fatty acids for elucidating the lipotoxic effects in human EndoC-βH1 beta-cells. Biochim Biophys Acta Mol Basis Dis. 2019;1865:165525.
von Hanstein AS, Tsikas D, Lenzen S, Jörns A, Plötz T. Potentiation of lipotoxicity in human EndoC-βH1 β-cells by glucose is dependent on the structure of free fatty acids. Mol Nutr Food Res. 2023;67:e2200582.
Alberici LC, Oliveira HCF. Mitochondrial adaptive responses to hypertriglyceridemia and bioactive lipids. Antioxid Redox Signal. 2022;36:953–68.
Nishida Y, Nawaz A, Kado T, Takikawa A, Igarashi Y, Onogi Y, et al. Astaxanthin stimulates mitochondrial biogenesis in insulin resistant muscle via activation of AMPK pathway. J Cachexia Sarcopenia Muscle. 2020;11:241–58.
Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Muñoz JP, Sala D, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109:5523–8.
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40.
Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–11.
Audzeyenka I, Rachubik P, Rogacka D, Saleem MA, Piwkowska A. Insulin induces bioenergetic changes and alters mitochondrial dynamics in podocytes. J Endocrinol. 2024;261:e230357.
Pearson G, Soleimanpour SA. A ubiquitin-dependent mitophagy complex maintains mitochondrial function and insulin secretion in beta cells. Autophagy. 2018;14:1160–1.
Pearson G, Chai B, Vozheiko T, Liu X, Kandarpa M, Piper RC, et al. Clec16a, Nrdp1, and USP8 form a ubiquitin-dependent tripartite complex that regulates β-cell mitophagy. Diabetes. 2018;67:265–77.
Axelrod CL, Fealy CE, Erickson ML, Davuluri G, Fujioka H, Dantas WS, et al. Lipids activate skeletal muscle mitochondrial fission and quality control networks to induce insulin resistance in humans. Metabolism. 2021;121:154803.
Audzeyenka I, Rachubik P, Typiak M, Kulesza T, Topolewska A, Rogacka D, et al. Hyperglycemia alters mitochondrial respiration efficiency and mitophagy in human podocytes. Exp Cell Res. 2021;407:112758.
Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–14.
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–71.
Besse-Patin A, Jeromson S, Levesque-Damphousse P, Secco B, Laplante M, Estall JL. PGC1A regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc Natl Acad Sci USA. 2019;116:4285–90.
Choi J, Ravipati A, Nimmagadda V, Schubert M, Castellani RJ, Russell JW. Potential roles of PINK1 for increased PGC-1α-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion. 2014;18:41–8.
Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–8.
Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–50.
Diaz-Morales N, Rovira-Llopis S, Bañuls C, Escribano-Lopez I, de Marañon AM, Lopez-Domenech S, et al. Are Mitochondrial Fusion and Fission Impaired in Leukocytes of Type 2 Diabetic Patients?. Antioxid Redox Signal. 2016;25:108–15.
Tur J, Pereira-Lopes S, Vico T, Marín EA, Muñoz JP, Hernández-Alvarez M, et al. Mitofusin 2 in macrophages links mitochondrial ROS production, cytokine release, phagocytosis, autophagy, and bactericidal activity. Cell Rep. 2020;32:108079.
Lin HY, Weng SW, Chang YH, Su YJ, Chang CM, Tsai CJ, et al. The causal role of mitochondrial dynamics in regulating insulin resistance in diabetes: link through mitochondrial reactive oxygen species. Oxid Med Cell Longev. 2018;2018:7514383.
He F, Huang Y, Song Z, Zhou HJ, Zhang H, Perry RJ, et al. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J Exp Med. 2021;218:e20201416.
Wu H, Wang Y, Li W, Chen H, Du L, Liu D, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019;15:1882–98.
Walker EM, Pearson GL, Lawlor N, Stendahl AM, Lietzke A, Sidarala V, et al. Retrograde mitochondrial signaling governs the identity and maturity of metabolic tissues. Science. 2025;388:eadf2034.
Ding W, Yang X, Lai K, Jiang Y, Liu Y. The potential of therapeutic strategies targeting mitochondrial biogenesis for the treatment of insulin resistance and type 2 diabetes mellitus. Arch Pharm Res. 2024;47:219–48.
Mugabo Y, Zhao S, Lamontagne J, Al-Mass A, Peyot ML, Corkey BE, et al. Metabolic fate of glucose and candidate signaling and excess-fuel detoxification pathways in pancreatic β-cells. J Biol Chem. 2017;292:7407–22.
Assali EA, Shlomo D, Zeng J, Taddeo EP, Trudeau KM, Erion KA, et al. Nanoparticle-mediated lysosomal reacidification restores mitochondrial turnover and function in β cells under lipotoxicity. FASEB J. 2019;33:4154–65.
Oberhauser L, Granziera S, Colom A, Goujon A, Lavallard V, Matile S, et al. Palmitate and oleate modify membrane fluidity and kinase activities of INS-1E β-cells alongside altered metabolism-secretion coupling. Biochim Biophys Acta Mol Cell Res. 2020;1867:118619.
Rönn T, Ofori JK, Perfilyev A, Hamilton A, Pircs K, Eichelmann F, et al. Genes with epigenetic alterations in human pancreatic islets impact mitochondrial function, insulin secretion, and type 2 diabetes. Nat Commun. 2023;14:8040.
Las G, Oliveira MF, Shirihai OS. Emerging roles of β-cell mitochondria in type-2-diabetes. Mol Asp Med. 2020;71:100843.
Baumel-Alterzon S, Katz LS, Lambertini L, Tse I, Heidery F, Garcia-Ocaña A, et al. NRF2 is required for neonatal mouse beta cell growth by maintaining redox balance and promoting mitochondrial biogenesis and function. Diabetologia. 2024;67:547–60.
Mohan R, Jo S, Lockridge A, Ferrington DA, Murray K, Eschenlauer A, et al. OGT regulates mitochondrial biogenesis and function via diabetes susceptibility gene Pdx1. Diabetes. 2021;70:2608–25.
Hong Y, Tak H, Kim C, Kang H, Ji E, Ahn S, et al. RNA binding protein HuD contributes to β-cell dysfunction by impairing mitochondria dynamics. Cell Death Differ. 2020;27:1633–43.
Li F, Munsey TS, Sivaprasadarao A. TRPM2-mediated rise in mitochondrial Zn2+ promotes palmitate-induced mitochondrial fission and pancreatic β-cell death in rodents. Cell Death Differ. 2017;24:1999–2012.
Cobo-Vuilleumier N, Rodríguez-Fernandez S, López-Noriega L, Lorenzo PI, Franco JM, Lachaud CC, et al. LRH-1/NR5A2 targets mitochondrial dynamics to reprogram type 1 diabetes macrophages and dendritic cells into an immune tolerance phenotype. Clin Transl Med. 2024;14:e70134.
Hernández MG, Aguilar AG, Burillo J, Oca RG, Manca MA, Novials A, et al. Pancreatic β cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis. 2018;9:481.
Park K, Sonn SK, Seo S, Kim J, Hur KY, Oh GT, et al. Impaired TFEB activation and mitophagy as a cause of PPP3/calcineurin inhibitor-induced pancreatic β-cell dysfunction. Autophagy. 2023;19:1444–58.
Kaniuka O, Deregowska A, Bandura Y, Sabadashka M, Chala D, Kulachkovskyi O, et al. Upregulation of GRP78 is accompanied by decreased antioxidant response and mitophagy promotion in streptozotocin-induced type 1 diabetes in rats. Biochim Biophys Acta Mol Basis Dis. 2025;1871:167531.
Besse-Patin A, Jeromson S, Levesque-Damphousse P, Secco B, Laplante M, Estall JL. Pgc1a regulates the irs1: Irs2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc Natl Acad Sci USA. 2019;116:4285–90.
Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. 2012;32:309–319.
Su Z, Nie Y, Huang X, Zhu Y, Feng B, Tang L, et al. Mitophagy in hepatic insulin resistance: therapeutic potential and concerns. Front Pharm. 2019;10:1193.
Zheng L, Rao Z, Wu J, Ma X, Jiang Z, Xiao W. Resistance exercise improves glycolipid metabolism and mitochondrial biogenesis in skeletal muscle of T2DM mice via miR-30d-5p/SIRT1/PGC-1α axis. Int J Mol Sci. 2024;25:12416.
da Silva Rosa SC, Martens MD, Field JT, Nguyen L, Kereliuk SM, Hai Y, et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy. 2021;17:2257–72.
Zhao W, Zhao B, Meng X, Li B, Wang Y, Yu F, et al. The regulation of MFG-E8 on the mitophagy in diabetic sarcopenia via the HSPA1L-Parkin pathway and the effect of D-pinitol. J Cachexia Sarcopenia Muscle. 2024;15:934–48.
Dantas WS, Heintz EC, Zunica ERM, Mey JT, Erickson ML, Belmont KP, et al. Deubiquitinating enzymes regulate skeletal muscle mitochondrial quality control and insulin sensitivity in patients with type 2 diabetes. J Cachexia Sarcopenia Muscle. 2025;16:e13763.
Ito R, Xie S, Tumenjargal M, Sugahara Y, Yang C, Takahashi H, et al. Mitochondrial biogenesis in white adipose tissue mediated by JMJD1A-PGC-1 axis limits age-related metabolic disease. iScience. 2024;27:109398.
Xia W, Veeragandham P, Cao Y, Xu Y, Rhyne TE, Qian J, et al. Obesity causes mitochondrial fragmentation and dysfunction in white adipocytes due to RalA activation. Nat Metab. 2024;6:273–89.
Hu D, Tan M, Lu D, Kleiboeker B, Liu X, Park H, et al. TMEM135 links peroxisomes to the regulation of brown fat mitochondrial fission and energy homeostasis. Nat Commun. 2023;14:6099.
Wang G, Meyer JG, Cai W, Softic S, Li ME, Verdin E, et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol Cell. 2019;74:844–857.e7.
Widlansky ME, Hill RB. Mitochondrial regulation of diabetic vascular disease: an emerging opportunity. Transl Res. 2018;202:83–98.
Paltauf-Doburzynska J, Malli R, Graier WF. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J Cardiovasc Pharm. 2004;44:423–36.
Shi Y, Fan S, Wang D, Huyan T, Chen J, Chen J, et al. FOXO1 inhibition potentiates endothelial angiogenic functions in diabetes via suppression of ROCK1/Drp1-mediated mitochondrial fission. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2481–94.
Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. 2011;15:1517–30.
Kizhakekuttu TJ, Wang J, Dharmashankar K, Ying R, Gutterman DD, Vita JA, et al. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2012;32:2531–9.
Sun D, Wang J, Toan S, Muid D, Li R, Chang X, et al. Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: focus on mitochondrial quality surveillance. Angiogenesis. 2022;25:307–29.
Jin L, Geng L, Ying L, Shu L, Ye K, Yang R, et al. FGF21-sirtuin 3 axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation. 2022;146:1537–57.
Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM Jr, Klein JB, et al. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2004;287:E896–E905.
Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124:1360–71.
Zou R, Tao J, He J, Wang C, Tan S, Xia Y, et al. PGAM5-mediated PHB2 dephosphorylation contributes to diabetic cardiomyopathy by disrupting mitochondrial quality surveillance. Res (Wash D C). 2022;2022:0001.
Chen Y, Huang J, Zhou H, Lin J, Tao J. Pgam5 aggravates hyperglycemia-induced myocardial dysfunction through disrupting Phb2-dependent mitochondrial dynamics. Int J Med Sci. 2024;21:1194–203.
Tao L, Huang X, Xu M, Yang L, Hua F. MiR-144 protects the heart from hyperglycemia-induced injury by regulating mitochondrial biogenesis and cardiomyocyte apoptosis. FASEB J. 2020;34:2173–97.
Wang Y, Gao P, Wei C, Li H, Zhang L, Zhao Y, et al. Calcium sensing receptor protects high glucose-induced energy metabolism disorder via blocking gp78-ubiquitin proteasome pathway. Cell Death Dis. 2017;8:e2799. Erratum in: Cell Death Dis. 2020;11(9):784.
Luo J, Hu S, Liu J, Shi L, Luo L, Li W, et al. Cardiac-specific PFKFB3 overexpression prevents diabetic cardiomyopathy via enhancing OPA1 stabilization mediated by K6-linked ubiquitination. Cell Mol Life Sci. 2024;81:228.
Chen Y, Li S, Guan B, Yan X, Huang C, Du Y, et al. MAP4K4 exacerbates cardiac microvascular injury in diabetes by facilitating S-nitrosylation modification of Drp1. Cardiovasc Diabetol. 2024;23:164.
Feng X, Wang S, Yang X, Lin J, Man W, Dong Y, et al. Mst1 knockout alleviates mitochondrial fission and mitigates left ventricular remodeling in the development of diabetic cardiomyopathy. Front Cell Dev Biol. 2021;8:628842.
Wang S, Zhao Z, Fan Y, Zhang M, Feng X, Lin J, et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1905–14.
Shu S, Cui H, Liu Z, Zhang H, Yang Y, Chen X, et al. Suppression of RCAN1 alleviated lipid accumulation and mitochondrial fission in diabetic cardiomyopathy. Metabolism. 2024;158:155977.
Liu ZY, Lin LC, Liu ZY, Song K, Tu B, Sun H, et al. N6-Methyladenosine-mediated phase separation suppresses NOTCH1 expression and promotes mitochondrial fission in diabetic cardiac fibrosis. Cardiovasc Diabetol. 2024;23:347.
Mu J, Zhang D, Tian Y, Xie Z, Zou MH. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J Mol Cell Cardiol. 2020;149:1–14.
Ma T, Huang X, Zheng H, Huang G, Li W, Liu X, et al. SFRP2 improves mitochondrial dynamics and mitochondrial biogenesis, oxidative stress, and apoptosis in diabetic cardiomyopathy. Oxid Med Cell Longev. 2021;2021:9265016.
Zheng H, Li W, Huang G, Zhu H, Wen W, Liu X, et al. Secreted frizzled-related protein 2 ameliorates diabetic cardiomyopathy by activating mitophagy. Biochim Biophys Acta Mol Basis Dis. 2024;1870:166989.
Wu QR, Zheng DL, Liu PM, Yang H, Li LA, Kuang SJ, et al. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis. 2021;12:216.
Liu C, Xu X, Sun G, Song C, Jiang S, Sun P, et al. Targeting DUSP26 to drive cardiac mitochondrial dynamics via FAK-ERK signaling in diabetic cardiomyopathy. Free Radic Biol Med. 2024;225:856–70.
Zhao M, Shen Z, Zheng Z, Xu Y, Zhang J, Liu J, et al. Cardiomyocyte LGR6 alleviates ferroptosis in diabetic cardiomyopathy via regulating mitochondrial biogenesis. Metabolism. 2024;159:155979.
Chang P, Zhang X, Zhang J, Wang J, Wang X, Li M, et al. BNP protects against diabetic cardiomyopathy by promoting Opa1-mediated mitochondrial fusion via activating the PKG-STAT3 pathway. Redox Biol. 2023;62:102702.
Zhou L, Su W, Wang Y, Zhang Y, Xia Z, Lei S. FOXO1 reduces STAT3 activation and causes impaired mitochondrial quality control in diabetic cardiomyopathy. Diab Obes Metab. 2024;26:732–44.
Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. 2019;9:3687–706.
Zhang Y, Zou R, Abudureyimu M, Liu Q, Ma J, Xu H, et al. Mitochondrial aldehyde dehydrogenase rescues against diabetic cardiomyopathy through GSK3β-mediated preservation of mitochondrial integrity and Parkin-mediated mitophagy. J Mol Cell Biol. 2024;15:mjad056.
Cleveland KH, Schnellmann RG. Pharmacological targeting of mitochondria in diabetic kidney disease. Pharm Rev. 2023;75:250–62.
Yao L, Liang X, Qiao Y, Chen B, Wang P, Liu Z. Mitochondrial dysfunction in diabetic tubulopathy. Metabolism. 2022;131:155195.
Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. 2017;23:753–62.
Song Y, Yu H, Sun Q, Pei F, Xia Q, Gao Z, et al. Grape seed proanthocyanidin extract targets p66Shc to regulate mitochondrial biogenesis and dynamics in diabetic kidney disease. Front Pharm. 2023;13:1035755.
Qu H, Gong X, Liu X, Zhang R, Wang Y, Huang B, et al. Deficiency of mitochondrial glycerol 3-phosphate dehydrogenase exacerbates podocyte injury and the progression of diabetic kidney disease. Diabetes. 2021;70:1372–87.
Feng J, Chen Z, Ma Y, Yang X, Zhu Z, Zhang Z, et al. AKAP1 contributes to impaired mtDNA replication and mitochondrial dysfunction in podocytes of diabetic kidney disease. Int J Biol Sci. 2022;18:4026–42.
Chen Y, Yang Y, Liu Z, He L. Adiponectin promotes repair of renal tubular epithelial cells by regulating mitochondrial biogenesis and function. Metabolism. 2022;128:154959.
Zhou D, Zhou M, Wang Z, Fu Y, Jia M, Wang X, et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019;10:524.
Lv Z, Wang Z, Hu J, Su H, Liu B, Lang Y, et al. LncRNA PVT1 induces mitochondrial dysfunction of podocytes via TRIM56 in diabetic kidney disease. Cell Death Dis. 2024;15:697.
Ma N, Xu C, Wang Y, Cui K, Kuang H. Telomerase reverse transcriptase protects against diabetic kidney disease by promoting AMPK/PGC-1a-regulated mitochondrial energy homeostasis. Chem Biol Interact. 2024;403:111238.
Jiang N, Zhao H, Han Y, Li L, Xiong S, Zeng L, et al. HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. 2020;53:e12909.
Hao Y, Fan Y, Feng J, Zhu Z, Luo Z, Hu H, et al. ALCAT1-mediated abnormal cardiolipin remodelling promotes mitochondrial injury in podocytes in diabetic kidney disease. Cell Commun Signal. 2024;22:26.
Li L, Long J, Mise K, Poungavrin N, Lorenzi PL, Mahmud I, et al. The transcription factor ChREBP links mitochondrial lipidomes to mitochondrial morphology and progression of diabetic kidney disease. J Biol Chem. 2023;299:105185.
Kang JS, Cho NJ, Lee SW, Lee JG, Lee JH, Yi J, et al. RIPK3 causes mitochondrial dysfunction and albuminuria in diabetic podocytopathy through PGAM5-Drp1 signaling. Metabolism. 2024;159:155982.
Mise K, Long J, Galvan DL, Ye Z, Fan G, Sharma R, et al. NDUFS4 regulates cristae remodeling in diabetic kidney disease. Nat Commun. 2024;15:1965.
Yuan D, Li H, Dai W, Zhou X, Zhou W, He L. IGF2BP3-stabilized CAMK1 regulates the mitochondrial dynamics of renal tubule to alleviate diabetic nephropathy. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167022.
Han F, Wu S, Dong Y, Liu Y, Sun B, Chen L. Aberrant expression of NEDD4L disrupts mitochondrial homeostasis by downregulating CaMKKβ in diabetic kidney disease. J Transl Med. 2024;22:465.
Zhang Z, Zhou F, Lu M, Zhang D, Zhang X, Xu S, et al. WTAP-mediated m6A modification of TRIM22 promotes diabetic nephropathy by inducing mitochondrial dysfunction via ubiquitination of OPA1. Redox Rep. 2024;29:2404794.
Li Y, Li X, Yang Y, Li F, Chen Q, Zhao Z, et al. Hepatocyte growth factor attenuates high glucose-disturbed mitochondrial dynamics in podocytes by decreasing ARF6-dependent DRP1 translocation. Biochim Biophys Acta Mol Cell Res. 2024;1871:119623.
Huang C, Yi H, Shi Y, Cao Q, Shi Y, Cheng D, et al. KCa3.1 mediates dysregulation of mitochondrial quality control in diabetic kidney disease. Front Cell Dev Biol. 2021;9:573814.
Gao P, Yang M, Chen X, Xiong S, Liu J, Sun L. DsbA-L deficiency exacerbates mitochondrial dysfunction of tubular cells in diabetic kidney disease. Clin Sci (Lond). 2020;134:677–94.
Yang M, Zhang Q, Luo S, Han Y, Zhao H, Jiang N, et al. DsbA-L alleviates tubular injury in diabetic nephropathy by activating mitophagy through maintenance of MAM integrity. Clin Sci (Lond). 2023;137:931–45.
Liu L, Bai F, Song H, Xiao R, Wang Y, Yang H, et al. Upregulation of TIPE1 in tubular epithelial cell aggravates diabetic nephropathy by disrupting PHB2 mediated mitophagy. Redox Biol. 2022;50:102260.
Ji H, Zhao Y, Ma X, Wu L, Guo F, Huang F, et al. Upregulation of UHRF1 promotes PINK1-mediated mitophagy to alleviates ferroptosis in diabetic nephropathy. Inflammation. 2024;47:718–32.
Chen H, Zhang H, Li AM, Liu YT, Liu Y, Zhang W, et al. VDR regulates mitochondrial function as a protective mechanism against renal tubular cell injury in diabetic rats. Redox Biol. 2024;70:103062.
Li C, Li L, Yang M, Yang J, Zhao C, Han Y, et al. PACS-2 ameliorates tubular injury by facilitating endoplasmic reticulum-mitochondria contact and mitophagy in diabetic nephropathy. Diabetes. 2022;71:1034–50.
Ye S, Zhang M, Zheng X, Li S, Fan Y, Wang Y, et al. YAP1 preserves tubular mitochondrial quality control to mitigate diabetic kidney disease. Redox Biol. 2024;78:103435.
Liu CC, Ji JL, Wang Z, Zhang XJ, Ding L, Zhang Y, et al. TRPC6-calpain-1 axis promotes tubulointerstitial inflammation by inhibiting mitophagy in diabetic kidney disease. Kidney Int Rep. 2024;9:3301–17.
Zhou P, Wang N, Lu S, Xiong J, Zhang Y, Jiang Q, et al. Dihydrolipoamide S-acetyltransferase activation alleviates diabetic kidney disease via AMPK-autophagy axis and mitochondrial protection. Transl Res. 2024;274:81–100.
Ma F, Li H, Huo H, Han Q, Liao J, Zhang H, et al. N-acetyl-L-cysteine alleviates FUNDC1-mediated mitophagy by regulating mitochondrial dynamics in type 1 diabetic nephropathy canine. Life Sci. 2023;313:121278.
Sachdeva MM. Retinal neurodegeneration in diabetes: an emerging concept in diabetic retinopathy. Curr Diab Rep. 2021;21:65.
Kim D, Sankaramoorthy A, Roy S. Downregulation of Drp1 and Fis1 inhibits mitochondrial fission and prevents high glucose-induced apoptosis in retinal endothelial cells. Cells. 2020;9:1662.
Kowluru RA, Alka K. Mitochondrial quality control and metabolic memory phenomenon associated with continued progression of diabetic retinopathy. Int J Mol Sci. 2023;24:8076.
Duraisamy AJ, Mohammad G, Kowluru RA. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1617–26.
Alka K, Kumar J, Kowluru RA. Impaired mitochondrial dynamics and removal of the damaged mitochondria in diabetic retinopathy. Front Endocrinol (Lausanne). 2023;14:1160155.
Chen M, Zhang Q, Wang S, Zheng F. Inhibition of diabetes-induced Drp1 deSUMOylation prevents retinal vascular lesions associated with diabetic retinopathy. Exp Eye Res. 2023;226:109334.
Ravi R, Nagarajan H, Muralikumar S, Vetrivel U, Subramaniam Rajesh B. Unveiling the therapeutic potential of a mutated paraoxonase 2 in diabetic retinopathy: defying glycation, mitigating oxidative stress, ER stress and inflammation. Int J Biol Macromol. 2024;258:128899.
Zhang MY, Zhu L, Zheng X, Xie TH, Wang W, Zou J, et al. TGR5 activation ameliorates mitochondrial homeostasis via regulating the PKCδ/Drp1-HK2 signaling in diabetic retinopathy. Front Cell Dev Biol. 2022;9:759421.
Qian S, Qian Y, Huo D, Wang S, Qian Q. Tanshinone IIa protects retinal endothelial cells against mitochondrial fission induced by methylglyoxal through glyoxalase 1. Eur J Pharm. 2019;857:172419.
Xie J, Cui Y, Chen X, Yu H, Chen J, Huang T, et al. VDAC1 regulates mitophagy in NLRP3 inflammasome activation in retinal capillary endothelial cells under high-glucose conditions. Exp Eye Res. 2021;209:108640.
Yang W, Xia F, Mei F, Shi S, Robichaux WG 3rd, Lin W, et al. Upregulation of Epac1 promotes pericyte loss by inducing mitochondrial fission, reactive oxygen species production, and apoptosis. Invest Ophthalmol Vis Sci. 2023;64:34.
Aloysius Dhivya M, Sulochana KN, Bharathi Devi SR. High glucose induced inflammation is inhibited by copper chelation via rescuing mitochondrial fusion protein 2 in retinal pigment epithelial cells. Cell Signal. 2022;92:110244.
Zhang S, Chen S, Sun D, Li S, Sun J, Gu Q, et al. TIN2-mediated reduction of mitophagy induces RPE senescence under high glucose. Cell Signal. 2024;119:111188.
Yang W, Qiu C, Lv H, Zhang Z, Yao T, Huang L, et al. Sirt3 protects retinal pigment epithelial cells from high glucose-induced injury by promoting mitophagy through the AMPK/mTOR/ULK1 pathway. Transl Vis Sci Technol. 2024;13:19.
Huang L, Yao T, Chen J, Zhang Z, Yang W, Gao X, et al. Effect of Sirt3 on retinal pigment epithelial cells in high glucose through Foxo3a/ PINK1-Parkin pathway mediated mitophagy. Exp Eye Res. 2022;218:109015.
Peng H, Li H, Ma B, Sun X, Chen B. DJ-1 regulates mitochondrial function and promotes retinal ganglion cell survival under high glucose-induced oxidative stress. Front Pharm. 2024;15:1455439.
Devi TS, Somayajulu M, Kowluru RA, Singh LP. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis. 2017;8:e2777.
Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic neuropathy. Nat Rev Dis Prim. 2019;5:42.
Chen R, Shi J, Yin Q, Li X, Sheng Y, Han J, et al. Morphological and pathological characteristics of brain in diabetic encephalopathy. J Alzheimers Dis. 2018;65:15–28.
Maranta F, Cianfanelli L, Cianflone D. Glycaemic control and vascular complications in diabetes mellitus type 2. Adv Exp Med Biol. 2021;1307:129–52.
Santos RX, Correia SC, Alves MG, Oliveira PF, Cardoso S, Carvalho C, et al. Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes. Mol Cell Biochem. 2014;394:13–22.
Carvalho C, Correia SC, Seiça R, Moreira PI. WWOX inhibition by Zfra1-31 restores mitochondrial homeostasis and viability of neuronal cells exposed to high glucose. Cell Mol Life Sci. 2022;79:487.
Cheng ZY, Hu YH, Xia QP, Wang C, He L. DRD1 agonist A-68930 improves mitochondrial dysfunction and cognitive deficits in a streptozotocin-induced mouse model. Brain Res Bull. 2021;175:136–49.
Lu LL, Liu LZ, Li L, Hu YY, Xian XH, Li WB. Sodium butyrate improves cognitive dysfunction in high-fat diet/ streptozotocin-induced type 2 diabetic mice by ameliorating hippocampal mitochondrial damage through regulating AMPK/PGC-1α pathway. Neuropharmacology. 2024;261:110139.
Han X, Huang S, Zhuang Z, Zhang X, Xie M, Lou N, et al. Phosphatidate phosphatase Lipin1 involves in diabetic encephalopathy pathogenesis via regulating synaptic mitochondrial dynamics. Redox Biol. 2024;69:102996.
Tang W, Yan C, He S, Du M, Cheng B, Deng B, et al. Neuron-targeted overexpression of caveolin-1 alleviates diabetes-associated cognitive dysfunction via regulating mitochondrial fission-mitophagy axis. Cell Commun Signal. 2023;21:357.
Park G, Lee JY, Han HM, An HS, Jin Z, Jeong EA, et al. Ablation of dynamin-related protein 1 promotes diabetes-induced synaptic injury in the hippocampus. Cell Death Dis. 2021;12:445.
George DS, Hackelberg S, Jayaraj ND, Ren D, Edassery SL, Rathwell CA, et al. Mitochondrial calcium uniporter deletion prevents painful diabetic neuropathy by restoring mitochondrial morphology and dynamics. Pain. 2022;163:560–78.
Xu S, Gao Z, Jiang L, Li J, Qin Y, Zhang D, et al. High glucose- or AGE-induced oxidative stress inhibits hippocampal neuronal mitophagy through the Keap1-Nrf2-PHB2 pathway in diabetic encephalopathy. Sci Rep. 2024;14:24044.
Jin H, Zhu Y, Li Y, Ding X, Ma W, Han X, et al. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis. 2019;24:511–28.
Cho JH, Chae CW, Lim JR, Jung YH, Han SJ, Yoon JH, et al. Sodium butyrate ameliorates high glucose-suppressed neuronal mitophagy by restoring PRKN expression via inhibiting the RELA-HDAC8 complex. Autophagy. 2024;20:1505–22.
Khan I, Preeti K, Kumar R, Kumar Khatri D, Bala Singh S. Piceatannol promotes neuroprotection by inducing mitophagy and mitobiogenesis in the experimental diabetic peripheral neuropathy and hyperglycemia-induced neurotoxicity. Int Immunopharmacol. 2023;116:109793.
Yang J, Yu Z, Jiang Y, Zhang Z, Tian Y, Cai J, et al. SIRT3 alleviates painful diabetic neuropathy by mediating the FoxO3a-PINK1-Parkin signaling pathway to activate mitophagy. CNS Neurosci Ther. 2024;30:e14703.
Yuan P, Song F, Zhu P, Fan K, Liao Q, Huang L, et al. Poly (ADP-ribose) polymerase 1-mediated defective mitophagy contributes to painful diabetic neuropathy in the db/db model. J Neurochem. 2022;162:276–89.
Masini M, Anello M, Bugliani M, Marselli L, Filipponi F, Boggi U, et al. Prevention by metformin of alterations induced by chronic exposure to high glucose in human islet beta cells is associated with preserved ATP/ADP ratio. Diab Res Clin Pract. 2014;104:163–70.
Sekar P, Hsiao G, Hsu SH, Huang DY, Lin WW, Chan CM. Metformin inhibits methylglyoxal-induced retinal pigment epithelial cell death and retinopathy via AMPK-dependent mechanisms: Reversing mitochondrial dysfunction and upregulating glyoxalase 1. Redox Biol. 2023;64:102786.
Han YC, Tang SQ, Liu YT, Li AM, Zhan M, Yang M, et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021;12:925.
Zhao Y, Sun M. Metformin rescues Parkin protein expression and mitophagy in high glucose-challenged human renal epithelial cells by inhibiting NF-κB via PP2A activation. Life Sci. 2020;246:117382.
Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66:193–205.
de Marañón AM, Díaz-Pozo P, Canet F, Díaz-Morales N, Abad-Jiménez Z, López-Domènech S, et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 2022;53:102342.
Guo Y, Jiang H, Wang M, Ma Y, Zhang J, Jing L. Metformin alleviates cerebral ischemia/reperfusion injury aggravated by hyperglycemia via regulating AMPK/ULK1/PINK1/Parkin pathway-mediated mitophagy and apoptosis. Chem Biol Interact. 2023;384:110723.
Zhang X, Zhang Z, Yang Y, Suo Y, Liu R, Qiu J, et al. Alogliptin prevents diastolic dysfunction and preserves left ventricular mitochondrial function in diabetic rabbits. Cardiovasc Diabetol. 2018;17:160.
Zhang X, Zhang Z, Zhao Y, Jiang N, Qiu J, Yang Y, et al. Alogliptin, a dipeptidyl peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J Am Heart Assoc. 2017;6:e005945.
Zhang Q, He L, Dong Y, Fei Y, Wen J, Li X, et al. Sitagliptin ameliorates renal tubular injury in diabetic kidney disease via STAT3-dependent mitochondrial homeostasis through SDF-1α/CXCR4 pathway. FASEB J. 2020;34:7500–19.
Liu H, Xiang H, Zhao S, Sang H, Lv F, Chen R, et al. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J Cell Mol Med. 2019;23:798–810.
Yang Q, Ai W, Nie L, Yan C, Wu S. Vildagliptin reduces myocardial ischemia-induced arrhythmogenesis via modulating inflammatory responses and promoting expression of genes regulating mitochondrial biogenesis in rats with type-II diabetes. J Inter Card Electrophysiol. 2020;59:517–26.
Pham TK, Nguyen THT, Yi JM, Kim GS, Yun HR, Kim HK, et al. Evogliptin, a DPP-4 inhibitor, prevents diabetic cardiomyopathy by alleviating cardiac lipotoxicity in db/db mice. Exp Mol Med. 2023;55:767–78.
Liu X, Xu C, Xu L, Li X, Sun H, Xue M, et al. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism. 2020;111:154334.
Lee YH, Kim SH, Kang JM, Heo JH, Kim DJ, Park SH, et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am J Physiol Ren Physiol. 2019;317:F767–F780.
Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–46. Erratum in: Redox Biol. 2024; 71:103083.
Li N, Zhu QX, Li GZ, Wang T, Zhou H. Empagliflozin ameliorates diabetic cardiomyopathy probably via activating AMPK/PGC-1α and inhibiting the RhoA/ROCK pathway. World J Diab. 2023;14:1862–76.
Yang C, Xiao C, Ding Z, Zhai X, Liu J, Yu M. Canagliflozin Mitigates Diabetic Cardiomyopathy through Enhanced PINK1-Parkin Mitophagy. Int J Mol Sci. 2024;25:7008.
Parichatikanond W, Pandey S, Mangmool S. Exendin-4 exhibits cardioprotective effects against high glucose-induced mitochondrial abnormalities: Potential role of GLP-1 receptor and mTOR signaling. Biochem Pharm. 2024;229:116552.
Chen K, Jin HJ, Wu ZH, Zhang BF, Wu J, Huang ZY, et al. Glucagon-like peptide-1 receptor agonist exendin 4 ameliorates diabetes-associated vascular calcification by regulating mitophagy through the AMPK signaling pathway. Mol Med. 2024;30:58.
Mostafa Tork O, Ahmed Rashed L, Bakr Sadek N, Abdel-Tawab MS. Targeting Altered Mitochondrial Biogenesis in the Brain of Diabetic Rats: Potential Effect of Pioglitazone and Exendin-4. Rep Biochem Mol Biol. 2019;8:287–300.
Zhang Y, Wang S, Chen X, Wang Z, Wang X, Zhou Q, et al. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol Cell Endocrinol. 2022;545:111560.
Fang WJ, Wang CJ, He Y, Zhou YL, Peng XD, Liu SK. Resveratrol alleviates diabetic cardiomyopathy in rats by improving mitochondrial function through PGC-1α deacetylation. Acta Pharm Sin. 2018;39:59–73.
Diao J, Zhao H, You P, You H, Wu H, Shou X, et al. Rosmarinic acid ameliorated cardiac dysfunction and mitochondrial injury in diabetic cardiomyopathy mice via activation of the SIRT1/PGC-1α pathway. Biochem Biophys Res Commun. 2021;546:29–34.
Li L, Zou J, Zhou M, Li H, Zhou T, Liu X, et al. Phenylsulfate-induced oxidative stress and mitochondrial dysfunction in podocytes are ameliorated by Astragaloside IV activation of the SIRT1/PGC1α /Nrf1 signaling pathway. Biomed Pharmacother. 2024;177:117008.
Liu X, Wang W, Song G, Wei X, Zeng Y, Han P, et al. Astragaloside IV ameliorates diabetic nephropathy by modulating the mitochondrial quality control network. PLoS One. 2017;12:e0182558.
Su J, Gao C, Xie L, Fan Y, Shen Y, Huang Q, et al. Astragaloside II ameliorated podocyte injury and mitochondrial dysfunction in streptozotocin-induced diabetic rats. Front Pharm. 2021;12:638422.
Xue H, Li P, Luo Y, Wu C, Liu Y, Qin X, et al. Salidroside stimulates the Sirt1/PGC-1α axis and ameliorates diabetic nephropathy in mice. Phytomedicine. 2019;54:240–7.
Li Y, Wei X, Liu SL, Zhao Y, Jin S, Yang XY. Salidroside protects cardiac function in mice with diabetic cardiomyopathy via activation of mitochondrial biogenesis and SIRT3. Phytother Res. 2021;35:4579–91.
Sun MY, Ye HJ, Zheng C, Jin ZJ, Yuan Y, Weng HB. Astragalin ameliorates renal injury in diabetic mice by modulating mitochondrial quality control via AMPK-dependent PGC1α pathway. Acta Pharm Sin. 2023;44:1676–86.
Liu C, Cao Q, Chen Y, Chen X, Zhu Y, Zhang Z, et al. Rhein protects retinal Müller cells from high glucose-induced injury via activating the AMPK/Sirt1/PGC-1α pathway. J Recept Signal Transduct Res. 2023;43:62–71.
Chen F, Zhang HY, He D, Rao CM, Xu B. Cardioprotective effect of gynostemma pentaphyllum against streptozotocin induced cardiac toxicity in rats via alteration of AMPK/Nrf2/HO-1 pathway. J Oleo Sci. 2022;71:991–1002.
Wu QJ, Zhang TN, Chen HH, Yu XF, Lv JL, Liu YY, et al. The sirtuin family in health and disease. Signal Transduct Target Ther. 2022;7:402.
Ni T, Lin N, Huang X, Lu W, Sun Z, Zhang J, et al. Icariin ameliorates diabetic cardiomyopathy through apelin/Sirt3 signalling to improve mitochondrial dysfunction. Front Pharm. 2020;11:256.
Ding X, Zhao H, Qiao C. Icariin protects podocytes from NLRP3 activation by Sesn2-induced mitophagy through the Keap1-Nrf2/HO-1 axis in diabetic nephropathy. Phytomedicine. 2022;99:154005.
Qin X, Jiang M, Zhao Y, Gong J, Su H, Yuan F, et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br J Pharm. 2020;177:3646–61.
Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics. 2019;9:1698–713.
Sun S, Yang S, Cheng Y, Fang T, Qu J, Tian L, et al. Jinlida granules alleviate podocyte apoptosis and mitochondrial dysfunction via the AMPK/PGC 1α pathway in diabetic nephropathy. Int J Mol Med. 2025;55:26.
Li L, Chen X, Su C, Wang Q, Li R, Jiao W, et al. Si-Miao-Yong-An decoction preserves cardiac function and regulates GLC/AMPK/NF-κB and GLC/PPARα/PGC-1α pathways in diabetic mice. Biomed Pharmacother. 2020;132:110817.
Wang Y, Yu L, Li Y, Cha S, Shi L, Wang J, et al. Supplemented Gegen Qinlian Decoction Formula attenuates podocyte mitochondrial fission and renal fibrosis in diabetic kidney disease by inhibiting TNF-α-mediated necroptosis, compared with empagliflozin. J Ethnopharmacol. 2024;334:118572.
Liu C, Han Y, Gu X, Li M, Du Y, Feng N, et al. Paeonol promotes Opa1-mediated mitochondrial fusion via activating the CK2α-Stat3 pathway in diabetic cardiomyopathy. Redox Biol. 2021;46:102098.
Fu F, Liu C, Shi R, Li M, Zhang M, Du Y, et al. Punicalagin protects against diabetic cardiomyopathy by promoting Opa1-mediated mitochondrial fusion via regulating PTP1B-Stat3 pathway. Antioxid Redox Signal. 2021;35:618–41.
Gong M, Guo Y, Dong H, Wu F, He Q, Gong J, et al. Modified Hu-lu-ba-wan protects diabetic glomerular podocytes via promoting PKM2-mediated mitochondrial dynamic homeostasis. Phytomedicine. 2024;123:155247.
Shen Y, Chen W, Lin K, Zhang H, Guo X, An X, et al. Notoginsenoside Fc, a novel renoprotective agent, ameliorates glomerular endothelial cells pyroptosis and mitochondrial dysfunction in diabetic nephropathy through regulating HMGCS2 pathway. Phytomedicine. 2024;126:155445.
Ji Y, Zhang X, Chen J, Song S, Fang S, Wang Z, et al. Asiatic acid attenuates tubular injury in diabetic kidney disease by regulating mitochondrial dynamics via the Nrf-2 pathway. Phytomedicine. 2023;109:154552.
Yan M, Liu S, Zeng W, Guo Q, Mei Y, Shao X, et al. The Chinese herbal medicine Fufang Zhenzhu Tiaozhi ameliorates diabetic cardiomyopathy by regulating cardiac abnormal lipid metabolism and mitochondrial dynamics in diabetic mice. Biomed Pharmacother. 2023;164:114919.
Zhong Y, Liu J, Sun D, Guo T, Yao Y, Xia X, et al. Dioscin relieves diabetic nephropathy via suppressing oxidative stress and apoptosis, and improving mitochondrial quality and quantity control. Food Funct. 2022;13:3660–73.
Zhang P, Wu H, Lou H, Zhou J, Hao J, Lin H, et al. Baicalin attenuates diabetic cardiomyopathy in vivo and in vitro by inhibiting autophagy and cell death through SENP1/SIRT3 signaling pathway activation. Antioxid Redox Signal. 2024. Epub ahead of print.
Zhou P, Xie W, Meng X, Zhai Y, Dong X, Zhang X, et al. Notoginsenoside R1 ameliorates diabetic retinopathy through PINK1-dependent activation of mitophagy. Cells. 2019;8:213 Erratum in: Cells. 2020;9(2): E450.
Wang Y, He X, Xue M, Sun W, He Q, Jin J. Germacrone protects renal tubular cells against ferroptotic death and ROS release by re-activating mitophagy in diabetic nephropathy. Free Radic Res. 2023;57:413–29.
Liu X, Lu J, Liu S, Huang D, Chen M, Xiong G, et al. Huangqi-Danshen decoction alleviates diabetic nephropathy in db/db mice by inhibiting PINK1/Parkin-mediated mitophagy. Am J Transl Res. 2020;12:989–98.
Wu Q, Yan R, Yang H, Wang Y, Zhang C, Zhang J, et al. Qing-Re-Xiao-Zheng-Yi-Qi formula relieves kidney damage and activates mitophagy in diabetic kidney disease. Front Pharm. 2022;13:992597.
Li H, Wang Y, Su X, Wang Q, Zhang S, Sun W, et al. San-Huang-Yi-Shen capsule ameliorates diabetic kidney disease through inducing PINK1/parkin-mediated mitophagy and inhibiting the activation of NLRP3 signaling pathway. J Diab Res. 2022;2022:2640209.
Zheng Q, Zhang X, Guo J, Wang Y, Jiang Y, Li S, et al. JinChan YiShen TongLuo Formula ameliorate mitochondrial dysfunction and apoptosis in diabetic nephropathy through the HIF-1α-PINK1-Parkin pathway. J Ethnopharmacol. 2024;328:117863.
Zhu Z, Luan G, Peng S, Fang Y, Fang Q, Shen S, et al. Huangkui capsule attenuates diabetic kidney disease through the induction of mitophagy mediated by STING1/PINK1 signaling in tubular cells. Phytomedicine. 2023;119:154975.
Liu J, Zhang Y, Xu X, Dong X, Pan Y, Sun X, et al. Ginsenoside Ro prevents endothelial injury via promoting Epac1/AMPK- mediated mitochondria protection in early diabetic retinopathy. Pharm Res. 2025;211:107562.
Wu Y, Deng H, Sun J, Tang J, Li X, Xu Y. Poricoic acid A induces mitophagy to ameliorate podocyte injury in diabetic kidney disease via downregulating FUNDC1. J Biochem Mol Toxicol. 2023;37:e23503.
Wu JJ, Zhang SY, Mu L, Dong ZG, Zhang YJ. Heyingwuzi formulation alleviates diabetic retinopathy by promoting mitophagy via the HIF-1α/BNIP3/NIX axis. World J Diab. 2024;15:1317–39.
Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu YL, et al. Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: Role of SIRT6. J Pineal Res. 2021;70:e12698.
Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z, et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1α-SIRT3 signaling. Sci Rep. 2017;7:41337.
Rocha IRC, Perez-Reyes E, Chacur M. Effect of photobiomodulation on mitochondrial dynamics in peripheral nervous system in streptozotocin-induced type 1 diabetes in rats. Photochem Photobio Sci. 2021;20:293–301.
Zhao R, Su Z, Gu J, Zhao H, Bian L, Jiang Y, et al. Enhancing anti-CD3 mAb-mediated diabetes remission in autoimmune diabetes through regulation of dynamin-related protein 1(Drp1)-mediated mitochondrial dynamics in exhausted CD8+T-cell subpopulations. BMC Med. 2025;23:189.
Yao L, Liang X, Liu Y, Li B, Hong M, Wang X, et al. Non-steroidal mineralocorticoid receptor antagonist finerenone ameliorates mitochondrial dysfunction via PI3K/Akt/eNOS signaling pathway in diabetic tubulopathy. Redox Biol. 2023;68:102946.
Han X, Wang J, Li R, Huang M, Yue G, Guan L, et al. Placental mesenchymal stem cells alleviate podocyte injury in diabetic kidney disease by modulating mitophagy via the SIRT1-PGC-1alpha-TFAM pathway. Int J Mol Sci. 2023;24:4696 28.
Acknowledgements
We thank FigDraw (https://www.figdraw.com/) for providing the original material for the figures. We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service. The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Science and Technology Innovation Team Project of Hunan Province (No.2020RC4050), Hunan Province Traditional Chinese Medicine Research Program Project (No.E2022010), Hunan Province Health High-level Talent Leading Talent Program (No.20240304008), Hunan Provincial Postgraduate Research Innovation Program (No.CX20240750), Project of Innovative Research Group of Hunan Provincial Natural Science Foundation (No.2024JJ1007), Scientific Research in Traditional Chinese Medicine of Hunan Province (No.B2023057).
Author information
Authors and Affiliations
Contributions
YC: Investigation, Conceptualization, Writing–original draft. XL: Methodology, Writing-review and editing. YL (Yixuan Liu): Writing-review and editing, Methodology. YL (Yujia Li): Writing-review and editing. DL: Writing-review, methodology, and editing. ZM: Writing-review, methodology, and editing, Conceptualization. YD: Conceptualization, Funding acquisition, Methodology, Project administration. All authors contributed to writing the manuscript and read and approved its final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: Mauro Piacentini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Liu, X., Liu, Y. et al. Mitochondrial quality control in diabetes mellitus and complications: molecular mechanisms and therapeutic strategies. Cell Death Dis 16, 652 (2025). https://doi.org/10.1038/s41419-025-07936-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-025-07936-y
