
Abstract
Heterogeneous nuclear ribonucleoproteins (HNRNPs) represent a large family of RNA-binding proteins consisting of more than 20 members and have attracted great attention with their distinctive roles in cancer progression by regulating RNA splicing, transcription, and translation. Nevertheless, the cancer-specific modulation of HNRNPs has not been fully elucidated. The research of LC-MS/MS technology has documented that HNRNPs were widely and significantly targeted by different post-translational modifications (PTMs), which have emerged as core regulators in shaping protein functions and are involved in multiple physiological processes. Accumulating studies have highlighted that several PTMs are involved in the mechanisms of HNRNPs regulation in cancer and may be suitable therapeutic targets. In this review, we summarize the existing evidence describing how PTMs modulate HNRNPs functions on gene regulation and the involvement of their dysregulation in cancer, which will help shed insights on their clinical impacts as well as possible therapeutic tools targeting PTMs on HNRNPs.
Similar content being viewed by others

Facts
-
The HNRNP protein family forms a structurally conserved and functionally highly correlated protein subgroup that is involved in many important cellular life activities.
-
HNRNPs contribute to pathogenesis and progression of tumors by facilitating cancer cell proliferation, migration, metabolic dysregulation, and other related factors.
-
With the development of the LC-MS/MS technology, the HNRNPs proteins were identified to target by widespread and multiple modifications.
-
Protein modifications can mediate multiple biological functions of HNRNPs such as structure, subcellular localization, and protein interactions, and then regulate cancer development.
Open questions
-
How many types of PTMs are identified mapped to HNRNP family in cancer?
-
What roles do protein modifications play in the functions of HNRNPs and cancer progression?
-
Do protein modifications have practical application prospects in the diagnosis and treatment of cancers?
Introduction
Cancer is a severely detrimental disease to human health and imposes a significant burden on human society [1]. Over the years, we have committed to unravel the mechanisms of cancer progression and seek innovative therapeutic strategies. However, the refractory, recurrent, and augmented survival capacities of tumor cells continue to pose significant challenges in cancer therapy for the future. It is noteworthy that the HNRNP family composed of a series of RNA-binding proteins (RBPs) participates in regulation of crucial biological processes, including RNA splicing, translation, and mRNA localization [2, 3]. Studies have suggested that most HNRNPs negatively regulate splicing function, and they combine with cis-acting elements intronic splicing silencer (ISS) or exonic splicing silencer (ESS) to inhibit the activity of adjacent splicing sites or the ability of splicing bodies to recognize adjacent splicing sites [4]. These RBPs were shown to be created out of about 20 types of HNRNPs, named from A to U, and the expression levels of these HNRNPs are higher in most cancer cells than corresponding normal cells. Additionally, the respective members of the HNRNP family contribute to pathogenesis and progression of tumors by facilitating cancer cell proliferation, migration, metabolic dysregulation, inflammation, and other related factors [4,5,6,7]. The intensive studies of the HNRNPs regulation in cancer will help us find the potential therapeutic targets of cancers.
Post-translational modifications (PTMs) covalently bond small molecular groups on the amino acid side chain of proteins with broad function distributions, which contain diverse types such as phosphorylation, ubiquitination, glycosylation, and acylations including succinylation, hydroxyisobutyrylation, nitrosylation, lactylation [8,9,10,11]. These PTMs can mediate the protein functions in multiple cellular biological processes and subsequently influence physiological processes [11]. With the development of the high-resolution liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) [9], the HNRNP family was identified as targets of widespread and multiple modifications, including the phosphorylation in serine and threonine residues, arginine methylation, as well as SUMOylation and glycosylation processes. These modifications can induce alterations in the structure, subcellular localization, and protein interactions, thereby impacting the cancer development [12,13,14]. This review offers a broad overview of the regulation of HNRNPs by PTMs and the involvement of their dysregulation in cancer.
HNRNP family
The HNRNP complexes were initially identified as composed of A1, A2/B1, C1, and C2 in human HeLa cells, which were designated as core HNRNP particles [15]. These HNRNPs have high affinity and specificity for pre-mRNA, forming the fundamental 40S HNRNP-pre-mRNA complexes. In addition to core HNRNP particles, the HNRNP family was identified as composed of multiple RBPs and referred to as minor HNRNP. Minor HNRNP members have relatively low binding ability to pre-mRNA [8]. The protein molecular weights of these HNRNP family members range from 34 to 120 kDa, and names from A1 to U [16]. The HNRNPs bind to ESSs or ISSs of pre-mRNA, thereby stabilizing mRNA and affecting mRNA localization, which facilitates the generation of inflammatory splicing subtypes or increase resistance-related splicing subtypes [17,18,19].
The majority of HNRNPs have three fundamental structural domains-the RNA recognition motif (RRM), the Arginine-Glycine-Glycine (RGG), and the KH domain (KH). Several HNRNPs also contain quasi-RNA recognition motif (qRRM), nuclear localization signal (NLS), nucleo-plasmic shuttle domain (NS), and Zinc Finger (ZnF) domains. Moreover, a significant proportion of HNRNPs exhibit glycine-rich residues at their C-terminal region. In eukaryotic organisms, RNA Polymerase II mediates the release of mRNA transcripts [20]. Precise splicing on pre-mRNA leads to the generation of mature mRNA through alternative splicing (AS), which contributes to the remarkable diversity within the human proteome [21]. AS is specifically regulated by spliceosomes and other RNA-binding proteins, which act as splicing factors, such as serine and arginine-rich (SR) proteins, the HNRNP family proteins, and a diverse array of proteins featuring RRM, KH, and basic leucine zipper (bZIP) structures. It is widely demonstrated that SR and HNRNP proteins exhibit antagonistic roles, and SR proteins are splicing activators, while HNRNP proteins are splicing repressors. The splicing process is precisely regulated by the interaction between SRs and HNRNPs with a concentration-dependent manner [4, 22].
PTMs in cancer
PTM refers to the covalent attachment of molecular groups to amino acid side chains in proteins, which leads to the alteration or modulation of protein conformational structure as well as their activities and physicochemical properties (Fig.1). Ultimately, these modifications exert a profound impact on crucial cellular processes including cell cycle, transcription, metabolism, immunity, and autophagy. Consequently, they play an indispensable role in cell survival, proliferation, migration, differentiation, and apoptosis [23]. Currently, the techniques for characterizing and analyzing PTMs are relatively mature. The LC-MS/MS technique was first used in the early 2000s to identify chemically modified peptides, an analytical method that ionizes samples, separates them by ionic mass charge ratio, and measures the abundance of ions. The process begins with the enrichment of proteins using specific antibodies, with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) serving as the final step of preparation; The target protein on the gel is then isolated and digested with endonuclease; Finally, PTMs was characterized and analyzed by LC-MS/MS [24, 25]. The protein regulatory network shows remarkable diversity, including over 600 identified types of PTMs. The well-established classic modifications include phosphorylation, methylation, acetylation, ubiquitination, and SUMOylation. Additionally, specific cysteine modifications such as oxidation, nitrosylation, glutathionylation, and sulfhydration significantly contribute to protein regulation. Furthermore, glycosylation plays a prominent role in modifying numerous membrane proteins including O-GlcNAc and N-GlcNAc [10]. Recently, acylation modifications such as lactylation, butyrylation, succinylation, 3-hydroxybutyrylation, and propionylation, have also been found to occur in protein lysine residues [26,27,28].

(1) classical modifications that are abundant in cells and play key roles in cellular energy metabolism, lipid metabolism, ketone body metabolism, and amino acid metabolism. (2) Modifications present only in cysteine. When cells are subjected to oxidative stress, oxidation modification on cysteine may occur as a response. (3) Membrane proteins glycosylation plays an important role in the protection, stability, and barrier of cell membranes. (4) Novel acylation modifications are involved in epigenetic regulation, which plays a role in DNA damage repair, histone acylation modification, chromatin packaging, and so on.
Most classical PTMs function in the regulation of cellular energy metabolism, lipid metabolism, ketone body metabolism, and amino acid metabolism. Moreover, the PTMs such as methylation and acetylation also mediate the functions of nucleosome proteins, which are critical regulators of DNA damage repair, chromosome stability, and epigenetic inheritance. Additionally, glycosylation modifications are identified in proteins distributed in cell membranes, which play roles in cell protection, stability, and barrier.
The types and roles of PTM in tumor progression are broad. For example, protein phosphorylation is prevalent in many cancers. A typical example is phosphorylation at Ser37 of Pyruvate kinase 2(PKM2) in triple-negative breast cancer (TNBC), which is a biomarker for TNBC. Apostolidi et al. found that cyclin-dependent kinase (CDK) inhibitors and PKM activators can bind to Ser37 and affect the protein structure at the phosphorylation site, which can effectively inhibit cancer progression [29]. Recently, Peng et al. also found that PKM activators can play a similar therapeutic role in colorectal cancer [30]. Another typical example is the phosphorylation of the key molecule mTOR. Different kinases lead to different types of activation of mTOR, thereby activating different substrate proteins, including AKT, S6K, p53, etc., which has great potential value in the discussion of the development process of cancer [31]. Ubiquitination is involved in the development of cancer by promoting protein degradation. Ubiquitin-specific protease 7 (USP7) is a deubiquitinase that can deubiquitinate substrates by binding to them. Mouse double minute 2 homolog (MDM2) is a typical USP7 substrate, and specific USP7 inhibitors can promote the degradation of MDM2 by inhibiting the function of USP7, and ultimately activate the p53 pathway. This small molecule inhibitor has shown promising clinical performance [32]. In addition, proteolytic targeting chimera (PROTAC) technology is becoming increasingly mature. This technology links an E3 ubiquitin ligase ligand to a target protein-ligand through a linker, thereby bringing them into proximity and facilitating target protein degradation via the ubiquitin-proteasome pathway [33]. This approach can significantly enhance the druggability of difficult drug targets, making it crucial to advance the improvement and development of PROTAC drug technology. For example, Specific small ubiquitin-like modifier proteases 1 (SENP1), a deSUMOylase, can effectively deSUMOylate c-Myc and reduce c-Myc polyubiquitination and promote monoubiquitination. Finally, it enhances the stability of oncoprotein c-Myc. SENP1 is highly expressed in a variety of cancers, which has a broad stabilizing effect on c-Myc [34]. This study goes against the conventional wisdom that SUMOylation can stabilize proteins, it is possible that SUMOylation also could promote protein degradation, which requires more data to support it. In summary, we can roughly divide the role of PTM in cancer into two broad categories. On the one hand, in cancer, PTM on target proteins can directly regulate cancer signaling pathways. For example, the enzyme proteins of the glycometabolic pathway or lipid metabolic pathway have different activities before and after PTM. PKM2 is highly O-GlcNAc modified, which disrupts the stability of PKM2 tetramer and promotes PKM2 nuclear translocation. This modification is indispensable for inducing metabolic reprogramming of tumor cells and Warburg effect [35]. Glutaminase is another important metabolic enzyme. Wang et al. found that HDAC4 can effectively deacetylate glutaminase, which accelerates the catalytic decomposition of glutamine to glutamate, promotes the TCA pathway of tumor cells, and stimulate tumor metabolism [36]. In addition, acetyl-coA acetyltransferase 1(ACAT1) is a fatty acid metabolizing enzyme, and Tyr407 phosphorylation of ACAT1 promotes the stability of its tetramer morphology and accelerates tumor metabolism [37]. However, on the other hand, PTM of immune regulatory molecules can regulate tumor development by controlling tumor immune evasion or autoimmunity. The most typical example is that PD-1 has a variety of post-translational modifications: glycosylation, phosphorylation, and ubiquitination. PTM therapy targeting PD-1 in T cells may have a good effect of enhancing anti-tumor immunity [38].
In recent years, several reports have described promising therapeutic strategies for targeting changes in the level of post-translational modification of proteins in cancer signaling pathways, such as phosphorylated proteomics techniques used to uncover breast cancer patient populations characterized by phosphokinase activity, which may provide precision medicine and personalized treatment options [39]. Li et al analyzed the protein and phosphorylation levels of 10 tumors (including pancreatic cancer, non-small cell lung cancer, breast cancer, etc.) using proteomic phosphorylation technology, and analyzed the regulation of PTM in pan-cancer with large-scale multi-omics data by drawing multi-omics molecular maps [40]. The ability of multi-omics technology to link PTM to cancer occurrence and progression provides an effective way to expand our understanding of cancer and reveal the driving factors of cancer. Based on the important role of PTM in cancer and the lack of summary of PTM in the HNRNP family in previous studies, this review will summarize the association of the HNRNP family with cancers and mainly elucidate the underlying mechanisms of HNRNPs mediated by PTMs, as well as their potential implications in cancer therapy.
Regulation of HNRNPs by PTMs
HNRNP A/B (A0, A1, A2/B1, A3)
The HNRNPA/B family is composed of A0, A1, A2/B1, A3 and usually contains two RRMs and a long glycine-rich chain at the C-terminus. The phosphorylation in HNRNP A0 at Ser84, which is activated by mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK-2) in mammalian macrophages. The phosphorylated A protein specifically binds to the AU-rich elements (AREs) of mRNA, thereby facilitating post-transcriptional regulation of key macrophage cytokines including Tumor Necrosis Factor α (TNF-α), cyclooxygenase-2 (COX-2) and macrophage inflammatory protein-1 (MIP-2) [41]. Cannell et al. found that the phosphorylated HNRNP A0 at Ser84 activated by MAPKAPK-2 showed more apparent cytoplasmic localization compared to the non-phosphorylated HNRNP A0 in lung cancer, and increased the stability of mRNA p27/growth arrest and DNA damage-inducible α (p27/Gadd45α) when DNA was impaired. In addition, the efficiency of platinum-based chemotherapy in lung cancer mouse model was enhanced through phosphorylation of HNRNP A0 by MAPKAPK-2 [42]. (Table 1A).
Similar to A0, HNRNP A1 exhibits a high affinity for AREs [43]. The ubiquitination of Lys3 in A1 mediates the alternative splicing of Arhgap1. This process has been proven to promote hematopoietic function in mouse models, suggesting a promising target for the treatment of myeloid malignancy [17]. Phosphorylation at Ser4/6 residue of the A1 protein, which is activated by S6K2 kinase in colorectal cancer, controls the cytoplasmic transport and translation of BCL-x(L) and x-linked inhibitor of apoptosis (XIAP). Furthermore, phosphorylation at Ser6 can modulate PKM splicing, thereby exerting an impact on glucose metabolism reprogramming in cancer cells. The mRNA binding ability of A1 protein is jointly controlled by phosphorylation modifications at Ser4/6 and SUMOylation at the Lys183 [44, 45]. SAE2-mediated SUMOylation of A1-Lys113 in pancreatic cancer holds promise as a potential therapeutic target for KRAS-associated lymph node metastasis. Meanwhile, SUMOylation of Lys113, based on Ubc9 activation in bladder cancer, facilitates the recognition of LncRNA-ELNAT1 by the endosomal sorting complex required for transport (ESCRT) and promotes its packaging into extracellular vesicles (EVs), which ultimately induces lymphatic vessel assembly and generation [46, 47]. In glioblastoma, the phosphorylation of HNRNP A1 at Ser199 hampers the function of the IRES trans-acting factor (ITAF) on cyclin D1 and c-myc mRNA, while the methylation of HNRNP A1 Arg218/225 sustains internal ribosome entry site-binding (IRES-binding) and preserves cyclin D1 or c-MYC IRES activity. The mutual influence of both HNRNP A1 Ser199 phosphorylation and HNRNP A1 Arg218/225 methylation establishes a regulatory network [48, 49]. Zinc finger protein 91 (ZFP91) is an E3 ligase. Chen et al reported that ZFP91 can promote ubiquitination of HNRNP A1 at Lys8, which leads to degradation of A1 protein and down-regulation of its expression in HCC cells. The mechanism is to inhibit metabolic reprogramming, proliferation, and migration of hepatocellular carcinoma through alternative splicing of PKM [50].
The HNRNP A3 interacts with the cis-acting element A2 recognition element (A2RE) and participates in the transport of A2RE-related mRNA. Importantly, HNRNP A3 exhibits the capability of shuttling between the nucleus and cytoplasm [51]. However, the precise epigenetic mechanisms underlying the role of HNRNP A3 in cancer remain relatively unexplored, providing significant opportunities for further comprehensive studies.
In the HNRNP A/B subfamily, HNRNP A2/B1 binds to A2RE to induce subcellular localization of various pre-mRNAs within the cell. The translation products of these pre-mRNAs include myelin basic protein (MBP), myelin-associated oligodendrocyte basic protein (MOBP), and microtubule associated protein (MAP), which facilitates the translocation of pre-mRNAs from the nucleus to the cytoplasm and promotes their translation [52, 53]. SUMOylation of A2/B1 is a decisive regulator by facilitating the selective packaging of miRNAs into exosomes and indicates how miRNAs are integrated into exosomes and exported to cells to produce remote regulation [54, 55]. For example, recent studies by Guo et al have shown that SUMOylation of HNRNP A2/B1 can promote the exosomal packaging and sorting pathway of miR-204-3p under hypoxia conditions. Through the signal transducer and activator of transcription 3 (STAT3) pathway, angiogenesis in the microenvironment of glioblastoma is promoted, thereby alleviating the survival pressure of cancer cells under hypoxia conditions and deepening the deterioration of cancer cells [56]. In non-small cell lung cancer, circTLCD4-RWDD3, a circular RNA, binds to HNRNP A2/B1 and promotes SUMOylation of the Lys108 of HNRNP A2/B1 by ubiquitin-conjugating enzyme 9 (UBC9). And then, the SUMO interaction motif (SIM) in ALG-2-Interacting Protein X (ALIX) identified the recruitment program that activates ESCRT-III after SUMOylation. Ev-packaged circTLCD4-RWDD3 is taken up by human lymphatic endothelial cells to promote the transcription of lymphangiogenesis factor Prospero homeobox 1 (PROX1) and promote lymphangiogenesis and lymphatic metastasis in non-small cell lung cancer [57]. Zhang et al. found that the cullin-associated and neddylation dissociated 1 (CAND1), which has an abnormally high expression level in liver cancer cells, can inhibit F-box protein 11 (FBXO11) activated HNRNP A2/B1 ubiquitination, thereby inhibiting the degradation of HNRNP A2/B1. Therefore, targeting the CAND1-SCFFBXO11-HNRNP A2/B1 axis may be a potential therapeutic strategy for liver cancer [58]. Notably, in renal cell carcinoma, the ubiquitination in HNRNP A2/B1 at Lys274/305 residues is regulated by von hippel-lindau α (VHLα). Additionally, HNRNP A2/B1 also mediates the alternative splicing process of VHL. These findings suggest a potential feedback loop involving VHLα-HNRNP A2/B1 may be involved in the anti-tumor mechanisms [59]. The recent investigations have unveiled that HNRNP A2/B1 can specifically interact with m6A [60], which can interact with MIR100HG and control the activity of the Wnt signaling pathway through recognition of m6A sites on transcription factor-7-like-2 (TCF7L2) mRNA in colorectal cancer [61].
HNRNPC, D, and E
Apart from HNRNP A2/B1, another core particle responsible for 40S HNRNP nucleation is HNRNP C [62]. The coding region of the HNRNP C gene contains a NLS at positions 155-161 amino acids, flanked by an upstream RRM and a downstream basic zipper-like leucine motif (bZLM) [63, 64]. The HNRNP C regulates the ability of translation in a stability- and cell cycle-dependent manner by interacting with the poly-U tail located within the 3′ and 5′-UTR of the mRNA [65, 66]. Moreover, HNRNP C categorizes nascent transcripts according to their length and subsequently facilitates their transportation to the cytoplasm [67]. Recent researches by Sayaka Dantsuji et al. indicate that HNRNP C interacts with the cap-binding complex (CBC) to enhance the binding of CBC to transcription and export (TREX) and weakens the binding of CBC to phosphorylated adapter for RNA export (PHAX), which facilitates the nuclear-cytoplasmic transport of the transcripts exceeding 300 nucleotides [64].
In terms of protein modifications, uPA-mediated phosphorylation in HNRNP C Tyr57 could stabilize urokinase-type plasminogen activator receptor (uPAR) mRNA in lung cancer and is essential for facilitating the interaction between the C protein and the 3’UTR region [68]. When cells are stimulated with low concentrations of H2O2, which can induce cellular oxidative stress, the Ser225-Ser228 and Ser240 residues in HNRNP C can be rapidly phosphorylated. However, the functional and phenotypic meanings of phosphorylation at these residues in cancers remain to be studied in depth [69, 70]. The HNRNP CL is a branch of the HNRNP C subfamily, and their amino acid sequence and structure are highly similar. Recently, Cao et al. interpreted SUMOylation of HNRNP CL2 to promote the progression of glioblastoma. The SUMOylation of Lys175 in HNRNP CL2 is facilitated by Ubiquitin-like modifier-activating enzyme 2 (UBA2, a subunit of E1). The SUMOylation of HNRNP CL2 enhances its stability, and it is subsequently anchored to forkhead box D1 (FOXD1) mRNA to promote FOXD1 expression. FOXD1, an oncogene upregulated in cancers [71], promotes the transcriptional expression of DKK1 and malignant proliferation and migration of glioma cells [72].
Similar to the HNRNP A/B family, HNRNP D also consists of two RRMs, a C-terminal glycine-rich region, and an RGG box. The similarity between HNRNP D and HNRNP A0 is that both of them have specific affinity for AREs [73]. Further investigation of HNRNP D showed that it has four distinct isoforms: p37, p40, p42, and p45, and each of them exhibits unique functions that mediate different phenotypic outcomes [74]. In these isoforms, only p40 and p45 of HNRNP D possess phosphorylation at three specific amino acid residues: Ser83, Ser87, and Thr91. The selective phosphorylation at these sites significantly modulates the binding affinity of HNRNP D with ARE elements [75, 76]. The HNRNP D, especially p40 and p45 isoforms, could promote Akt phosphorylation at Thr308, Thr450, and Ser473. Meanwhile, mTORC2 can activate the AKT-mTOR pathway and induce HNRNP D phosphorylation [77]. Furthermore, ubiquitin-induced degradation of the p40 isoform is dependent on autophosphorylation of Ser83 and Ser87 [78]. Additionally, LC-MS/MS analysis revealed the existence of four methylated Arg residues in the C-terminal RGG box of HNRNP D, which play a crucial role in maintaining the functional activity of HNRNP D RGG box, thereby leading to the inhibition of vascular endothelial growth factor (VEGF) expression [79].
The HNRNP E comprises at least four isoforms known as Poly(rC)-binding protein (PCBP), namely E1 to E4 [80]. All four isoforms have three typical type I KH and contain a GXXG loop. However, Only E1 and E2 were expressed stably in various cells and tissues, while E3 and E4 showed a trend of low expression in some tissues or developmental stages [81, 82]. Notably, E3 and E4 have not been classified as part of the HNRNP family due to their localization in the cytoplasm [83]. The phosphorylation of PCBP1 at Ser43 is activated by Akt2 kinase, which drives TGF-β-induced epithelial to Epithelial-Mesenchymal Transition (EMT) in NSCLC and breast cancer [84, 85]. In the cytoplasm, phosphorylation of PCBP1 at Thr60 and Thr127 are activated by Pak1, reduces its mRNA binding affinity, thereby leading to the mRNA translation suppression. In the nucleus, PCBP1 phosphorylation at Thr60 and Thr127 not only governs transcriptional regulation by binding to specific gene promoters but also controls the alternative splicing process of Caper α (a U2 snRNP auxiliary factor-related protein) [18].
Furthermore, the PCBP1 protein is widely recognized as a tumor suppressor. For instance, depletion of PCBP1 leads to activation of phosphatase of regenerating liver-3 (PRL-3) and the AKT signaling pathway, thereby promoting tumorigenesis. Moreover, PCBP1 binds to the 3’UTR region of P27 transcript through its KH1 domain, which promotes the stability of P27 and inhibits tumorigenesis as well as cell cycle progression [86]. However, the PCBP2 has dual function in cancer. Janecki et al. found that PCBP2 promotes the stability and translation efficiency of p53-mRNA by binding to the p53 transcript, and inhibits the progression of colorectal cancer [87]. On the contrary, in gastric cancer, PCBP2 interacts with the 3′UTR region of CDK2 transcript, thereby accelerating the cell cycle of gastric cancer cells and promoting tumorigenesis. Moreover, the oncogenic role of PCBP2 has been verified in various malignancies including glioblastoma and breast cancer [88,89,90].
Alterations in the tyrosine kinase activity of the p210-BCR/ABL oncoprotein drive the development of chronic myeloid lymphoma (CML). Moreover, upregulation of p210-BCR/ABL expression is a prerequisite for PCBP2-induced anti-differentiation signals. BCR/ABL and MAPKERK1/2 kinases can activate PCBP2, leading to phosphorylation at Ser173, Ser189, Ser272, and Thr213. The phosphorylation in these residues increases the stability of PCBP2 protein and enhances its affinity for the 5′UTR of CCAAT/enhancer-binding protein α (C/EBPα) mRNA. After deletion of PCBP2 phosphorylation, this affinity is also reduced, which promotes C/EBPα expression efficiency and G-CSFR-driven neutrophil maturation, initiating differentiation of chronic myeloid leukemia blastoblastic crisis (CML-BC) progenitor cells [91].
HNRNPF and H
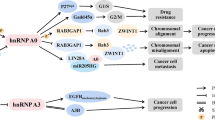
The HNRNP F and HNRNP H exhibit a significant structural similarity, and both of them harbor three qRRMs and two glycine-rich domains. The RRMs of HNRNP F and HNRNP H are not completely conserved, which are classified as qRRMs [92]. Currently, the HNRNP F/H family consists of HNRNP F, HNRNP H1, HNRNP H2, and G-rich RNA sequence binding factor 1 (GRSF1). Subcellular localization analyses have revealed that HNRNP F, HNRNP H1, and HNRNP H2 mainly localize in nucleus, while GRSF1 localizes to the mitochondria. GRSF1 possesses a unique glycine-rich region in its N-terminal structure, which may contribute to its mitochondrial localization and gene regulation mediated by GRSF1 [93]. The specific crosstalk between acetylation and ubiquitination (KAc/Ub) in HNRNP F at Lys98 and Lys224 is especially important for Bcl-x(L)/x(S) splicing control in breast cancer. This crosstalk was also found at residue Lys167 in HNRNP H [19] (Fig. 2).

The top of this picture represents the extracellular environment. From the top to bottom are the cell membrane, the cytoplasmic matrix, and the nucleus. At the bottom are the mechanisms affected by the HNRNPs. The proteins in purple are kinases or upstream regulatory enzymes, HNRNPs are in light blue, downstream transcripts are in gray, and the final mechanism is in black boxes. P Phosphorylation; AC Acetylation; Me Methylation; Ub Ubiquitination; SUMO Sumoylation.
HNRNPG, I, and L
Only HNRNP G in HNRNPs was confirmed to be targeted by glycosylation modification, which was designated as RNA-binding motif protein X-linked (RBMX) due to the location of its open reading frame (ORF) on X chromosome. Similarly, the homologous counterpart on the Y chromosome is called RBMY [94, 95]. In developmental and biological investigations related to RBMY, it has been established that RBMY plays a crucial role in the ontogeny and viability of males [96]. The primary structures of RBMX and RBMY show a high degree of homology, and both of them contain a single RNA recognition motif (RRM). In comparison to RBMX, the downstream sequences in RBMY include four repeated copies of serine-arginine-glycine-tyrosine (SRGY) motifs, while RBMX only holds one copy of the SRGY motif. RBMX is strongly associated with mitosis, and it has been shown that lacking RBMX exhibits significant impairment in sister chromatid cohesion and homologous chromosome segregation and results in mitotic disruption [97, 98]. Several studies have shown that the RBMX gene is a tumor suppressor in many cancers, including endometrial cancer and papillary thyroid cancer [99, 100]. In a recent report, the Ser216 residue of RBMX was identified as a phosphorylation substrate for TGF-β activated kinase 1 (TAK1), however, its precise role in cancer has not been reported [101]. Interestingly, RBMY, the predicted homolog of RBMX, has been identified as a potential driver gene for HCC in males [102]. Recent research has confirmed that the PIM1 kinase is responsible for the phosphorylation of RBMY, and the cytoplasmic accumulation of phosphorylated RBMY promotes HCC metastasis [103].
The HNRNP I, also known as polypyrimidine tract binding protein 1 (PTBP1), contains four RRMs, and these four RRMs in HNRNP I show significant similarity to the RRMs in HNRNP L. Moreover, HNRNP L exhibits significant homology with HNRNP I [104]. Distinguished from other nucleocytoplasmic shuttling HNRNPs, the shuttling of HNRNP I from nucleus to cytoplasm is independent from mRNA binding, which depends on its N-terminal NS domain to participate in the translation of mRNA in the nucleoplasm [104,105,106]. At the post-transcriptional level, PTBP1 is downregulated in response to cellular oxidative stress, which induces alternative splicing of soluble guanylyl cyclase (sGC) and dimerizes PTBP1 for degrading [107]. In addition, PTBP1 modulates the stability of the myeloid cell leukemia 1 (MCL1) transcript and promotes the accumulation of MCL1 in the cytoplasm, which inhibits the apoptosis of tumor cells [108]. The PTBP1 downregulation can increase the expression level of cell division cycle 42-v2 (CDC42-v2), which consequently inhibits tumorigenesis [109]. Moreover, PTBP1 can repress the expression of autophagy-related genes (ATGs), thereby facilitating tumor growth and metastasis in colorectal cancer [110].
Protein kinase A (PKA) induces the phosphorylation modification of endogenous HNRNP I at Ser16 residue, which located in the NS domain of the primary structure, and Ser16 phosphorylation promoted the accumulation of HNRNP I in cytoplasm. On the contrary, the transport of non-phosphorylated HNRNP I from the nucleus to the cytoplasm was greatly reduced, leading to its nuclear accumulation [111]. In wild-type endothelial cells, Lkb1 activates phosphorylation of Thr138 residues of PTBP1. Dephosphorylation of PTBP1 Thr138 leads to the removal of Scf exon 6, resulting in decreased Scf secretion. At the same time, phosphorylated PTBP1 is destroyed to rescue the differentiation process of classical dendritic cells (DCs) generated from macrophage-DC precursors (MDPs) through common DC precursor (CDP), and maintains DC count to inhibit tumorigenesis [112].
The four highly conserved RRMs in HNRNP L are indispensable for splicing regulation. The HNRNP L binds to nascent transcripts and inhibits the inclusion of exons. The HNRNP L contains a glycine-rich sequence in the N-terminal and a proline-rich sequence between RRM2 and RRM3 [113]. Previous studies have shown that HNRNP L, HNRNP I, and HNRNP E2 regulate splicing by binding to ESS. Notably, HNRNP L, a potent splicing repressor, exhibits stronger binding to ESS than the other two proteins [114]. The regulation of splicing is through the exon or intron retention [114, 115]. The HNRNP L is indispensable for the activation of H3 histone Lys36 methylation [116]. In NSCLC, HNRNP L is a potential suppressor of the p53 pathway, and HNRNP L silence induces the apoptosis in lung cancer cells by mediating the function of p53 [117]. Furthermore, Akt-mediated phosphorylation of HNRNP L at Ser52 hinders its interaction with HNRNP U on caspase-9 mRNA, thereby modulating the ratio of caspase-9a to caspase-9b splice variants, which inhibits cell apoptosis and promotes NSCLC progression [118, 119]. Phosphorylation at Tyr359 in HNRNP L is activated under hypoxia conditions, which promotes the development and progression of malignant tumor. Normally, HNRNP L predominantly localizes in the nucleus. Excessive accumulation of phosphorylated HNRNP L in the cytoplasm promotes VEGFA expression by weakening the binding of miR-297 with VEGFA, which leads to the increase of angiogenesis and leukocyte. Finally, there will be an increase in chronic inflammatory diseases, various cancers, or atherosclerosis [120,121,122]. Furthermore, the calcium signaling pathway is implicated in promoting tumor development. In this review, calcium/calmodulin-dependent protein Kinase IV (CaMKIV) phosphorylates the Ser544 residue of HNRNP L, which augments the affinity for the CaRRE element at the 3′ splice site of mRNA [123, 124]. F-box protein 16 (FBXO16) is an E3 ubiquitin ligase that can promote ubiquitin-proteasome degradation of L protein through ubiquitination of HNRNP L, and ultimately prevent the malignant development of ovarian cancer [125].
HNRNPK
KH homeodomains were initially identified in HNRNP K, which include three KH domains that primarily recognize and bind to specific RNA and DNA molecules [126, 127]. A classical NLS sequence is located upstream of KH1, which could maintain the stability and cellular localization of HNRNP K [128]. Additionally, a nucleo-cytoplasmic shuttling domain, also known as KNS, has been identified between the KH2 and KH3 regions, which is similar to the NS [129]. The HNRNP K specifically binds to polyC-rich or cytosine-rich pre-mRNAs, which enables the precise splicing of pre-mRNA transcripts [130]. Currently, many studies have shown that the overexpression and abnormal cytoplasmic localization of HNRNP K are potential prognostic indicators for multiple malignancies [131,132,133,134,135]. In terms of transcriptional regulation, HNRNP K was able to bind to the CT element region within the c-MYC gene promoter, which promotes recruitment of RNA polymerase II and enhances transcription of the c-MYC gene. Both HNRNP K and HNRNP E1/E2 can motivate ribosomal site activity, thereby promoting translation of c-MYC mRNA [136, 137]. However, deletion of chromosome 9 results in the loss of one single copy of the HNRNP K gene and plays a carcinogenic role by inhibiting p53-dependent p21 expression and promoting tumor development in patients with hematologic malignancies, which indicates that HNRNP K shows oncogenicity not only in various solid tumor but also in hematologic malignancies such as leukemia [138].
The Skp1-Cullin-1-F-box (SCF) E3 ligase primarily participates in the process of ubiquitin degradation. The Fbox family is a core component of SCF E3 ligases and substrate recognition molecules [139]. In melanoma, the Fbxo4 component of the SCF E3 ligase mediates the K63 ubiquitination of HNRNP K to suppress the activity of K protein, which could suppress cancer cell migration and proliferation. Moreover, the SCFFbxo4-HNRNP K-c-Myc axis controls the carcinogenicity and mRNA binding capacity of HNRNP K [140]. (Table 1B)S100B, a member of the acidic Ca2+-binding protein family, could regulate many cellular processes including cell proliferation, differentiation, Ca2+ homeostasis, protein phosphorylation, and other essential signaling pathways [141]. S100B can promote the cytoplasmic localization of HNRNP K. Natarajan et al. constructed a THP1-HNRNP KK219I/S284D/353D model in normal human monocytes, which was a model without HNRNP K Lys219 methylation and Ser284/Ser353 phosphorylation. With this model, they found S100B-mediated cytoplasmic translocation of HNRNP K [142]. However, it is not known whether this feature has an inhibitory effect on cancer development in malignant cells. In nasopharyngeal carcinoma and colorectal cancer, phosphorylation of HNRNP K at Ser284 and Ser353 by MAPKERK1/2 kinase enhances the stability of the K protein and facilitates cytoplasmic transport of HNRNP K [143,144,145].
Heat shock factor 1 (HSF1) is a transcription factor that is activated in cells under heat shock conditions or related stresses, which specifically binds with heat shock elements (HSE) located in nucleus. LC-MS/MS identification indicates that Cys132 of HNRNP K contains thiol oxidation modification, which enhances the interaction between HNRNP K and HSF1 and inhibits the binding of HSF1 with HSE [146]. HSF1-targeting has long been a therapeutic strategy for neurodegenerative diseases and various cancers. Therefore, studying the inhibitory mechanism of HNRNP K could potentially contribute to the development of anticancer drugs [146, 147].
Cyproterone acetate (CPA), a steroidal anti-androgen with progestin-like activity, is commonly used in the clinical treatment of advanced prostate cancer. In vivo, CPA mediates AKT hyperphosphorylation of HNRNP K at Ser116 and increases its tight binding to androgen receptor (AR), which delays prostate cancer progression [148, 149].
Circ-GALNT16 is associated with a good prognosis of colorectal cancer. It can enhance the transcription and expression of the HNRNP K-p53-p21 axis by binding to the KH3 domain of HNRNP K and promoting SUMOylation at the Lys422 site in colorectal cancer [150]. This study is similar to many previous studies depicting the effect of K422-SUMOylation on DNA damage repair [151, 152]. The methylation of HNRNP K at Arg296/299 could inhibit the phosphorylation of HNRNP K at Ser302 mediated by PKCdelta, and thus, PKC-mediated apoptosis is inhibited [153], which has significant implications for understanding how cancer cells evade apoptosis. In triple-negative breast cancer, Aurora-A kinase phosphorylates HNRNP K at Ser379, inhibiting cancer cell migration. Meanwhile, the HNRNP K phosphorylation can be inhibited by Arg methylation in the KI region. However, when Ser379 phosphorylation was lost, the migration ability of cancer cells increased through HNRNP K-β-catenin-MMP12 axis accompanied by the HNRNP K Ser379 phosphorylation deletion [13]. Zhu et al. found that ubiquitination of HNRNP K is also associated with the β-catenin pathway. PROX1 is abnormally high expressed in breast cancer. PROX1 binds to HNRNP K and inhibits its ubiquitination, thereby promoting K protein stability and ultimately activating the HNRNP K-β-Catenin-Wnt axis to promote breast cancer development [154]. Han et al. discovered a novel lncRNA--syndecan-binding protein 2-antisense RNA 1 (SDCBP2-AS1), which can inhibit the SUMOylation and promote the ubiquitination of HNRNP K by binding HNRNP K in gastric cancer cells. After down-regulating SDCBP2-AS1 expression level in vitro, HNRNP K ubiquitination level decreased successively, and then promoted the activity of downstream target genes regulated by HNRNP K/β-catenin, leading to the development and metastasis of gastric cancer [155]. These studies suggest that HNRNP K/β-catenin may be a potential anti-tumor therapeutic target.
HNRNPM, P, Q, R and U
There are three RRMs in the HNRNP M protein. The N-terminal of the protein contains a unique py-NLS sequence (NLS sequence ends with a conserved proline (P) and a tyrosine (Y)), which is distinct from the classical NLS sequence [156, 157]. The M protein shows a strong binding affinity to both poly-G and poly-U RNA homopolymers [158]. In innate immunity, HNRNP M binds to IL-6 mRNA and represses the splicing process. However, when Ser574 in the M protein is phosphorylated by P38-dependent MAPK, the splicing activity of HNRNP M is significantly weakened, which is beneficial for IL-6 responding to viral infection or clearance of cancer cells in innate immune process [159].
The HNRNP P2, also known as translocated in liposarcoma (TLS) or fused in sarcoma (FUS), contains a highly conserved Serine-Tyrosine-Glycine-Glutamine (SYGQ) motif at the N-terminal and single RRM. There is an NLS sequence in the C-terminus of HNRNP P2, and an NES sequence is included in the RRM. Moreover, FUS contains multiple RGG motifs and an RGG-ZnF-RGG motif in the C-terminal [160]. Epidermal growth factor receptor (EGFR) kinase phosphorylates two tyrosine sites in FUS, Tyr6 and Tyr296. Phosphorylation of these two sites facilitates the nuclear translocation of FUS and the interaction of FUS with integrin α-4, which may provide a potential new therapeutic target for fibrotic diseases [161]. Recently studies have shown that Casein kinase 1δ/ε exhibits the ability to phosphorylate many serine sites in FUS, especially Ser182 and Ser183. The phosphorylation of serine residues significantly strengthens the solubility of the FUS protein in patients with neurodegenerative disorders, which may have implications for targeted therapy of neurodegenerative diseases [162]. The c-Jun protein, a member of the Jun family, is an integral component of the transcription activator protein1 (AP-1) [163]. Normally, protein degradation or hydrolysis is facilitated by the ubiquitin-proteasome pathway, which is associated with the 26S proteasome [164]. However, FUS hydrolysis is induced by c-Jun rather than through the ubiquitin-proteasome degradation pathway and is enhanced by HNRNP A1. BCR-ABL, an antiapoptotic gene with high tyrosine kinase activity, enhances the stability of FUS through PKCβII-mediated phosphorylation of FUS at Ser256 [165]. Acetylation in FUS at Lys315, Lys316, and Lys510 activated by P300/ CBP plays a regulatory role in FUS subcellular localization and mRNA binding [166]. In glioblastoma, lncRNA-RMST enhances UBC9-mediated SUMOylation at Lys333, which significantly enhances the stability of the FUS protein. The interaction between SUMOylated FUS and HNRNP D promotes the stability of autophagy-related 4D cysteine peptidase (ATG4D), which inhibits mitochondrial autophagy and suppresses glioblastoma development [167]. The methylation of arginine in the C-terminal NLS of FUS could also control its nucleocytoplasmic transportation [168]. Similarly, ABL kinase activates the phosphorylation of the C-terminus Tyr526 residue, which promotes the nucleocytoplasmic transport of FUS [169]. Currently, the research focus of HNRNP P2 is mainly in neurodegenerative diseases; however, it is noteworthy that FUS also shows oncogenic properties [162, 167]. Therefore, the role of HNRNP P2 in cancer still has great potential to be studied.
The HNRNP Q, also known as GRY-RBP, NSAP1, or SYNCRIP, has an acidic amino acid in the N-terminal region, which has three consecutive RRMs, and one of them contains a NLS at the C terminus, which regulates the subcellular localization of the Q protein. In addition, the C terminus contains an RGG box [170]. At present, although the molecular mechanism of HNRNP Q regulated by PTMs in malignancies has not been accurately reported, the variation of HNRNP Q at the protein level is of great significance for the induction of neurological diseases such as autism spectrum disorders (ASD) and neurodevelopmental disorders (NDD) [171].
The HNRNP R is similar to HNRNP Q, which induces diseases such as neurodevelopmental disorders, while relevant studies in cancer are still insufficient [172]. Several tyrosine phosphorylated residues on R protein have been identified in sarcoma cell lines and human tumor cells [173], but the function of these modified residues remains to be studied.
The HNRNP U, also known as SAF-A, has at least 4 conserved structural domains: SAP, SPRY, AAA+, and RGG. In these domains, the SAP domain shows DNA-binding activity, the AAA+ domain drives various cellular activities mediated by ATPase, and the RGG domain is an RNA-binding domain and rich in Arg-Gly-Gly motifs [174,175,176]. The phosphorylation of protein U at Ser59 activated by DNA-dependent protein kinase (DNA-PK) can mediate DNA damage. Moreover, Ser59 is phosphorylated by polo-like kinase 1 (PLK1) rather than DNA-PK during mitosis, which promotes key biological processes such as chromosome alignment and segregation during cell division [177,178,179]. Additionally, HNRNP U is implicated in the regulation of PTMs of many specific proteins. For example, Kim et al. revealed that HNRNP U binds transcription factor IIH (TFIIH) and inhibits the TFIIH-mediated phosphorylation of the C-terminal domain (CTD) of Pol II [180].
Functional prediction of novel PTMs in the HNRNPs
Recently, novel acylation modifications, including lactylation, butyrylation, succinylation, and 3-hydroxybutyrylation [26, 27], have been confirmed to be mapped to various proteins with a broad function distribution. In cancer cells, after identifying these novel modifications in members of the HNRNP family using LC-MS/MS technology, their potential function can be assessed. Firstly, it is possible to determine whether the amino acid residue at the modified site is evolutionarily conserved in multiple species because highly conserved positions are likely to be functionally significant. Subsequently, it is important to determine whether the modification site is located in a domain commonly found in the HNRNP family, such as RRM, qRRM, RGG, bZLM, or NLS, and the potential impact on protein structure or function could be roughly inferred, as well as alternative splicing or other signaling pathways that may be affected eventually. Finally, based on the speculation and hypothesis of its function, and combining the results of LC-MS/MS, we can classify it into two models: (1). The level of modification is significantly up-regulated or down-regulated. (2). The level of modification changes but is not significant. For the former, we can construct a cell model by first knocking out the endogenous target proteins in the cell, and then creating two cell models: one rescuing the wild-type and the other rescuing the modified site mutants. Subsequently, the function of this modification site in cancer can be explored using these cell models through in vitro experiments. Ultimately, the actual function and mechanism of this site can be elucidated, and its druggability and targetability can be verified. For proteins whose modification levels have changed but are not significant, experimenters could analyze their overall action network through bioinformatics methods and explore changes in the overall modification level in cancer, and finally explain the overall changes in the cancer microenvironment.
The potential applications of HNRNPs PTMs in cancer therapy
The major limitations of traditional cancer treatments such as chemotherapy and radiation therapy are that they kill not only cancer cells but also normal cells in human body [181, 182], which has prompted extensive research efforts in developing targeted anti-cancer therapies that selectively attack cancer cells while sparing healthy ones. Therefore, it is more and more important to design targeted anti-tumor drugs and implement them in clinical treatments. We summarize the selected drugs targeting HNRNPs protein PTMs, some of which have advanced to clinical trials with promising results (Table 2). The value of a single agent for the activating enzymes of PTMs is gradually diminishing, and combination therapy has been necessary in clinical trials.
Targeting tyrosine kinases
Tyrosine kinases play a pivotal role in cellular signal transduction pathways, regulating multiple functions such as cell growth, differentiation, and apoptosis [183], which are broadly divided into two main groups: receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (NRTKs). RTKs are localized mainly on cell membranes and perform functions by interacting with ligands. Such as vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) as we mentioned earlier. Conversely, most NRTKs are localized in the cytoplasm, such as Abl and Src kinases [184]. The activities of tyrosine kinases are associated with tumorigenesis and tumor progression, such as promoting cell proliferation, migration, and inhibiting apoptosis [183].
Tyrosine kinase inhibitors (Tkis) can inhibit tumor cell signal transduction by targeting tyrosine kinase activity. A previous example is that targeting MEK can inhibit cancer through HNRNP molecules and eventually target CML therapy.
Among the factors driving CML, upregulation of p210 BCR-ABL can activate the MEK-MAPKERK1/2-HNRNP E2 (phosphorylation) pathway and promote the EMT process. CI-1040 is a MEK-targeted inhibitor. After treatment of CML cells with CI-1040, the expression of MAPKERK1/2 is inhibited, and then the binding of HNRNP E2 and C/EBPmRNA is reduced by down-regulating the phosphorylation level of HNRNP E2, thus inhibiting the progression of CML [91].
In addition, Tkis that have been used in the clinic are relatively mature, and these Tkis include drugs such as Brigatinib, Imatinib, and Sorafenib. Sorafenib, a multitargeted kinase inhibitor, directly blocks tumor cell proliferation by disrupting the RAF/MEK/ERK signaling cascade. It also inhibits tumor angiogenesis indirectly by targeting VEGF and PDGF. Clinically, it is a targeted agent for the treatment of advanced liver and kidney cancer [185, 186]. However, drug resistance and tumor recurrence are still important factors that restrict the use of Tkis. It is still necessary to explore the mechanism of human resistance to Tkis and tumor recurrence and seek better treatment strategies.
Targeting the PI3K-AKT-mTOR signaling pathway
The PI3K-AKT-mTOR (phosphocarnosine 3-kinase-AKT-mammalian target of rapamycin) pathway is a crucial signaling pathway that regulates essential processes including cell growth, differentiation, inflammation, and metabolism [187, 188]. Phosphocarnosine 3-kinase (PI3K), a protein family consisting of lipoenzymes, has a major substrate--AKT. When AKT is activated by PI3K, it exhibits kinase activity by mediating many cellular phenotypes such as survival and proliferation [188, 189]. A number of studies have revealed the potential dysregulation of PI3K-AKT-mTOR pathway in tumors. At the same time, gene mutations in this pathway can frequently enhance the proliferation and migration of tumor cells. Targeting mTOR with inhibitors such as rapamycin and other small molecules has shown promising therapeutic effects in many clinical trials [189,190,191]. In our previous discussion, phosphorylation at Ser199 of HNRNP A1 was activated by AKT, and rapamycin was able to activate cyclin D1/c-myc IRES by targeting the AKT mTOR-HNRNP A1 (phosphorylation) signaling axis. This indicates that the phosphorylation level of HNRNP A1 protein and the activity of cyclin D1/c-myc IRES can determine the sensitivity and resistance of tumor cells to rapamycin [48]. PP242 (Torkinib), another inhibitor targeting mTOR, which can also promote the activity of cyclin D1/c-myc IRES. GSK3235025 is a potent inhibitor of PRMT5. The methylation of HNRNP A1 protein Arg-218/225 mediated by PRMT5 is functionally antagonistic to Ser199 phosphorylation, and GSK3235025 can reduce the activity of cyclin D1/c-myc IRES. The combination of GSK3235025 and PP242 has a remarkable effect on GBM in vitro [49]. LI et al. showed that the positive feedback regulation formed by AKT and mTORC2 can be inhibited by Torin 1 (another mTOR-targeted inhibitor), thereby inhibiting the expression of AKT and ultimately inhibiting the phosphorylation of AUF1 in liver cancer cells, while AUF1 can promote the membrane localization and phosphorylation of AKT. Therefore, AUF1-targeting activators may promote cancers in which the mTORC2-AKT pathway is upregulated, and it is valuable to further investigate whether they can be combined with Torin-1 drugs to treat such cancers [77]. AKTi-1/2 is a quinoxoline compound. Ngoc T. Vu et al.'s study found that AKTi-1/2 can control the variable splicing of Caspase9 by inhibiting the phosphorylation of HNRNP L and promoting the up-regulation of the expression of Caspase9a subtype, thus promoting the apoptosis of lung cancer cells [119].
PI3K, AKT, and mTOR regulate each other to drive the development of cancer. Several inhibitors such as rapamycin, PP242, and AKTi-1/2 regulate the phosphorylation level of HNRNP family molecules by targeting the PI3K-AKT-mTOR axis, thereby inhibiting the proliferation, migration, and survival of cancer cells. In addition, many inhibitors targeting the PI3K-Akt-mtor axis have been developed, PI3K inhibitors that have been put into clinical use include AMG319, Alpelisib, Copanlisib, etc. Akt inhibitors include Afuresertib, Ipatasertib. The mTOR inhibitors include Temsirolimus, Everolimus, etc [192]. At present, the effect of single inhibitor is often weak, and it can easily lead to drug resistance. The combination of drugs to inhibit the PI3K-Akt-mTOR axis remains a hot research topic, but the potential side effects of combining these specific inhibitors still warrant careful consideration. Additionally, PI3K-AKT-mTOR axis not only plays a crucial role in cancer cells but also has important functions in normal cells. Achieving truly specific targeted therapy and effective drug combinations remains a challenge.
Targeting cyclin kinase
Multiple complex processes work together to regulate the cell cycle, and these processes are controlled by multiple protein factors. CDKs are the core kinase family that regulates the normal progression of the cell cycle and their functions are regulated by cell cycle checkpoints. Targeting the regulation of cell cycle and checkpoint mechanism in tumor cells has become an important potential direction for anticancer therapy [193,194,195]. In glioblastoma, the intricate interaction between phosphorylation and methylation of HNRNP A1 results in modified activity of cyclin D1 [48, 49]. The development of drugs that selectively suppress the activity of cyclin D1 may potentially block or control this altered activity, perhaps offering promising prospects for the treatment of glioblastoma. In NSCLC, activation of HNRNP A0 by MK2 stabilizes Gadd45α and p27 mRNA, which promotes checkpoints and hinders the cell cycle progression [42].
We found that these cell cycle factors are regulated by post-translational modified HNRNPs molecules, and the design of inhibitors targeting these modified HNRNPs may be a feasible approach to targeted therapy.
Targeting strategies for SUMOylation
SUMOylation has the function of stabilizing protein molecules and plays a key role in tumor EMT, metastasis, treatment resistance, and anti-tumor immune response. Meanwhile, the SUMOylation of HNRNP family members can enhance the stability of signaling pathways and effectively enhance cell activity [196]. Targeting SUMO E1 activating enzymes, E2 conjugating enzymes, or E3 ligases can block the SUMOylation cascade and suppress the stability of specific oncogenic molecules. TAK-981, a small molecule SUMO E1 inhibitor. Guo et al’s research has interpreted the potent anti-tumor ability of TAK-981, which can effectively inhibit the SUMOylation of HNRNP A2/B1 and inhibit the malignant trend of GBM. However, its poor penetration of the blood-brain barrier in humans remains a limiting problem [56].
TAK-981 activates the IFN signaling pathway in immune cells and has been shown to promote the innate immune response and the activation of innate immune cells, including NK cells, macrophages, dendritic cells, and T cells [197]. TAK-981 is the first E1 SUMO activation enzyme inhibitor to enter clinical trials. Compared to traditional SUMOylation inhibitors like ML-792 and ML-93, TAK-981 can achieve higher efficacy at lower doses [198]. In the future, drug studies targeting SUMOylation will help us understand the mechanism of SUMOylation in cancer progression, which will provide innovative strategies for cancer treatment. In malignant tumors, inhibition of upstream molecules that activate SUMOylation may be an indirect direction to target carcinogenic HNRNPs-SUMO, but the specificity of drugs and the resistance of cancers are still limited problems.
Hormonotherapy
A final treatment option that must be mentioned is hormonotherapy, which is often considered an adjuvant therapy for radiotherapy and surgery. Hormone therapy for prostate cancer is relatively well-established. In the previous discussion, cyproterone acetate (CPA) is a steroid anti-androgen. CPA mediates the hyperphosphorylation of HNRNP K in vivo and increases the tight binding of HNRNP K to androgen receptor (AR), which is often used in the clinical treatment of advanced prostate cancer [148]. In addition, several hormone therapy drugs are gradually used in the clinic, such as abiraterone, enzalutamide, and apalutamide. However, many common adverse reactions still limit the widespread use of these drugs.
Conclusion and prospects
Developing cancer treatment strategies based on PTMs of HNRNPs molecules still remains significant challenge. Firstly, the diverse phenotypes of cancer cells caused by the crosstalk of various PTMs are complex. PTMs are increasingly recognized as only a small part of the protein network regulation. Precise targeting within the highly dynamic and complex post-translational modification process is a key focus of targeted therapy. Secondly, besides common modifications, further researches are needed to investigate the regulatory mechanisms of novel and unknown modifications. Moreover, in preclinical studies of various cancer treatments, there is a lack of efficacy and safety in targeting these PTMs. In conclusion, developing effective therapeutic strategies directed against HNRNPs-based PTMs requires a comprehensive understanding of the complex regulatory networks involved in multiple cancers, while minimizing damage to normal human cells.
In this context, we have elucidated not only the different modifications of HNRNPs in cancers (Fig. 3) but also the different mechanisms caused by them. Additionally, we anticipate that the discovery of new post-translational modifications (PTMs) and regulatory pathways will provide new insights into cancer development, thereby offering innovative approaches for targeted cancer therapy.

As shown in this figure, the outer circle represents each cancer, and the inner circle is the PTMs type possessed by the HNRNPs corresponding to each cancer. On the right, we add an explanatory text to explain the abbreviations (P, Ub, Ac, Me, Sumo).
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. 2018. Report No.: 0007-92351542-4863.
Bradley RK, Anczuków O. RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer. 2023;23:135–55.
Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135:851–67.
Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. WIREs RNA. 2011;3:1–12.
Tian XY, Li J, Liu TH, Li DN, Wang JJ, Zhang H, et al. The overexpression of AUF1 in colorectal cancer predicts a poor prognosis and promotes cancer progression by activating ERK and AKT pathways. Cancer Med. 2020;9:8612–23.
Li H, Liu J, Shen S, Dai D, Cheng S, Dong X, et al. Pan‐cancer analysis of alternative splicing regulator heterogeneous nuclear ribonucleoproteins (hnRNPs) family and their prognostic potential. J Cell Mol Med. 2020;24:11111–9.
Han N, Li W, Zhang M. The function of the RNA-binding protein hnRNP in cancer metastasis. J Cancer Res Ther. 2013;9:S129–S134.
Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1’s multifunctional regulatory roles. RNA. 2010;16:1449–62.
Venne AS, Kollipara L, Zahedi RP. The next level of complexity: crosstalk of posttranslational modifications. Proteomics. 2014;14:513–24.
Zhong Q, Xiao X, Qiu Y, Xu Z, Chen C, Chong B et al. Protein posttranslational modifications in health and diseases: Functions, regulatory mechanisms, and therapeutic implications. MedComm 2023;4:e261.
Shang S, Liu J, Hua F. Protein acylation: mechanisms, biological functions and therapeutic targets. Signal Transduct Target Ther. 2022;7:396.
Dreyfuss G, Matunis MJ, Pinolroma S, Burd CG. HNRNP proteins and the biogenesis of messenger-RNA. Annu Rev Biochem. 1993;62:289–321.
Xu Y, Wu W, Han Q, Wang Y, Li C, Zhang P, et al. Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol. 2019;9:180239.
Gorlach M, Burd CG, Portman DS, Dreyfuss G. The HNRNP proteins. Mol Biol Rep. 1993;18:73–78.
Beyer AL, Christensen ME, Walker BW, Lestourgeon WM. Identification and characterization of packaging proteins of core 40S HNRNP particles. Cell. 1977;11:127–38.
Pinolroma S, Choi YD, Matunis MJ, Dreyfuss G. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 1988;2:215–27.
Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S, et al. Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat Immunol. 2016;18:236–45.
Meng Q, Rayala SK, Gururaj AE, Talukder AH, O’Malley BW, Kumar R. Signaling-dependent and coordinated regulation of transcription, splicing, and translation resides in a single coregulator, PCBP1. Proc Natl Acad Sci USA. 2007;104:5866–71.
Koumbadinga GA, Mahmood N, Lei L, Kan Y, Cao W, Lobo VG, et al. Increased stability of heterogeneous ribonucleoproteins by a deacetylase inhibitor. Biochim et Biophys Acta (BBA). 2015;1849:1095–103.
Dreyfuss G. Structure and function of nuclear and cytoplasmic ribonucleoprotein-particles. Annu Rev Cell Biol. 1986;2:459–98.
Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63.
Kędzierska H, Piekiełko-Witkowska A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017;396:53–65.
Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666–72.
Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207.
Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–61.
Fu Y, Yu J, Li F, Ge S. Oncometabolites drive tumorigenesis by enhancing protein acylation: from chromosomal remodelling to nonhistone modification. J Exp Clin Cancer Res. 2022;41:144.
Wang H, Yang L, Liu M, Luo J. Protein post-translational modifications in the regulation of cancer hallmarks. Cancer Gene Ther. 2022;30:529–47.
Walsh CT, Garneau‐Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed. 2005;44:7342–72.
Apostolidi M, Vathiotis IA, Muthusamy V, Gaule P, Gassaway BM, Rimm DL, et al. Targeting pyruvate kinase M2 phosphorylation reverses aggressive cancer phenotypes. Cancer Res. 2021;81:4346–59.
Peng C, Yang P, Zhang D, Jin C, Peng W, Wang T, et al. KHK-A promotes fructose-dependent colorectal cancer liver metastasis by facilitating the phosphorylation and translocation of PKM2. Acta Pharm Sin B. 2024;14:2959–76.
Battaglioni S, Benjamin D, Wälchli M, Maier T, Hall MN. mTOR substrate phosphorylation in growth control. Cell. 2022;185:1814–36.
Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481–6.
Li JW, Zheng G, Kaye FJ, Wu L. PROTAC therapy as a new targeted therapy for lung cancer. Mol Ther. 2023;31:647–56.
Sun X-X, Chen Y, Su Y, Wang X, Chauhan KM, Liang J, et al. SUMO protease SENP1 deSUMOylates and stabilizes c-Myc. Proc Natl Acad Sci. 2018;115:10983–8.
Wang Y, Liu J, Jin X, Zhang D, Li D, Hao F, et al. O-GlcNAcylation destabilizes the active tetrameric PKM2 to promote the Warburg effect. Proc Natl Acad Sci USA. 2017;114:13732–7.
Wang T, Lu Z, Han T, Wang Y, Gan M, Wang JB. Deacetylation of Glutaminase by HDAC4 contributes to Lung Cancer Tumorigenesis. Int J Biol Sci. 2022;18:4452–65.
Farrell AS, Pelz C, Wang XY, Daniel CJ, Wang ZP, Su YL, et al. Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol Cell Biol. 2013;33:2930–49.
Lee TA, Tsai EY, Liu SH, Hsu Hung SD, Chang SJ, Chao CH, et al. Post-translational modification of PD-1: potential targets for cancer immunotherapy. Cancer Res. 2024;84:800–7.
Mouron S, Bueno MJ, Lluch A, Manso L, Calvo I, Cortes J, et al. Phosphoproteomic analysis of neoadjuvant breast cancer suggests that increased sensitivity to paclitaxel is driven by CDK4 and filamin A. Nat Commun. 2022;13:7529.
Li Y, Porta-Pardo E, Tokheim C, Bailey MH, Yaron TM, Stathias V, et al. Pan-cancer proteogenomics connects oncogenic drivers to functional states. Cell. 2023;186:3921–+.
Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. Embo J. 2002;21:6505–14.
Cannell Ian G, Merrick Karl A, Morandell S, Zhu C-Q, Braun Christian J, Grant Robert A, et al. A pleiotropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53 -defective tumors to chemotherapy. Cancer Cell. 2015;28:623–37.
Hamilton BJ, Nagy E, Malter JS, Arrick BA, Rigby WF. Association of heterogeneous nuclear ribonucleoprotein A1 and C proteins with reiterated AUUUA sequences. J Biol Chem. 1993;268:8881–7.
Sun Y, Luo M, Chang G, Ren W, Wu K, Li X, et al. Phosphorylation of Ser6 in hnRNPA1 by S6K2 regulates glucose metabolism and cell growth in colorectal cancer. Oncol Lett. 2017;14:7323–31.
Roy R, Durie D, Li H, Liu B-Q, Skehel JM, Mauri F, et al. hnRNPA1 couples nuclear export and translation of specific mRNAs downstream of FGF-2/S6K2 signalling. Nucleic Acids Res. 2014;42:12483–97.
Pirlog R, Calin GA. KRAS mutations as essential promoters of lymphangiogenesis via extracellular vesicles in pancreatic cancer. J Clin Investig. 2022;132:e161454.
Chen C, Zheng H, Luo Y, Kong Y, An M, Li Y, et al. SUMOylation promotes extracellular vesicle–mediated transmission of lncRNA ELNAT1 and lymph node metastasis in bladder cancer. J Clin Investig. 2021;131:e146431.
Jo OD, Martin J, Bernath A, Masri J, Lichtenstein A, Gera J. Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J Biol Chem. 2008;283:23274–87.
Holmes B, Benavides-Serrato A, Saunders JT, Landon KA, Schreck AJ, Nishimura RN, et al. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J Neuro-Oncol. 2019;145:11–22.
Chen D, Wang Y, Lu R, Jiang X, Chen X, Meng N, et al. E3 ligase ZFP91 inhibits Hepatocellular Carcinoma Metabolism Reprogramming by regulating PKM splicing. Theranostics. 2020;10:8558–72.
Ma ASW, Moran-Jones K, Shan J, Munro TP, Snee MJ, Hoek KS, et al. Heterogeneous nuclear ribonucleoprotein A3, a novel RNA trafficking response element-binding protein. J Biol Chem. 2002;277:18010–20.
Munro TP, Magee RJ, Kidd GJ, Carson JH, Barbarese E, Smith LM, et al. Mutational analysis of a heterogeneous nuclear ribonucleoprotein A2 response element for RNA trafficking. J Biol Chem. 1999;274:34389–95.
Brumwell C. Intracellular trafficking of HnRNP A2 in oligodendrocytes. Exp Cell Res. 2002;279:310–20.
Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980.
Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. 2014;28:3–13.
Guo QD, Fan Y, Wang QT, Li BY, Qiu W, Qi YH, et al. Glioblastoma upregulates SUMOylation of hnRNP A2/B1 to eliminate the tumor suppressor miR-204-3p, accelerating angiogenesis under hypoxia. Cell Death Dis. 2023;14:147.
Diao X, Guo C, Zheng H, Zhao K, Luo Y, An M, et al. SUMOylation-triggered ALIX activation modulates extracellular vesicles circTLCD4-RWDD3 to promote lymphatic metastasis of non-small cell lung cancer. Signal Transduct Target Ther. 2023;8:426.
Zhang H, Xia P, Yang Z, Liu J, Zhu Y, Huang Z, et al. Cullin-associated and neddylation-dissociated 1 regulate reprogramming of lipid metabolism through SKP1-Cullin-1-F-box(FBXO11) -mediated heterogeneous nuclear ribonucleoprotein A2/B1 ubiquitination and promote hepatocellular carcinoma. Clin Transl Med. 2023;13:e1443.
Liu Y, Zhang H, Li X, Zhang C, Huang H. Identification of anti-tumoral feedback loop between VHLα and hnRNPA2B1 in renal cancer. Cell Death Dis. 2020;11:688.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
Liu H, Li D, Sun L, Qin H, Fan A, Meng L, et al. Interaction of lncRNA MIR100HG with hnRNPA2B1 facilitates m6A-dependent stabilization of TCF7L2 mRNA and colorectal cancer progression. Mol Cancer. 2022;21:74.
Huang M, Rech JE, Northington SJ, Flicker PF, Mayeda A, Krainer AR, et al. The C-Protein tetramer binds 230 TO 240 nucleotides of premessenger RNA and nucleates the assembly of 40S heterogeneous nuclear ribonucleoprotein-particles. Mol Cell Biol. 1994;14:518–33.
McAfee JG, ShahiedMilam L, Soltaninassab SR, LeStourgeon WM. A major determinant of hnRNP C protein binding to RNA is a novel bZIP-like RNA binding domain. Rna. 1996;2:1139–52.
Dantsuji S, Ohno M, Taniguchi I. The hnRNP C tetramer binds to CBC on mRNA and impedes PHAX recruitment for the classification of RNA polymerase II transcripts. Nucleic Acids Res. 2023;51:1393–408.
Shetty S. Regulation of urokinase receptor mRNA stability by hnRNP C in lung epithelial cells. Mol Cell Biochem. 2005;272:107–18.
Kim JH, Paek KY, Choi KB, Kim TD, Hahm BS, Kim KT, et al. Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Mol Cell Biol. 2003;23:708–20.
McCloskey A, Taniguchi I, Shinmyozu K, Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science. 2012;335:1643–6.
Velusamy T, Shetty P, Bhandary YP, Liu M-C, Shetty S. Posttranscriptional regulation of urokinase receptor expression by heterogeneous nuclear ribonuclear protein C. Biochemistry. 2008;47:6508–17.
Kattapuram T, Yang S, Maki JL, Stone JR. Protein kinase CK1α regulates mRNA binding by heterogeneous nuclear ribonucleoprotein C in response to physiologic levels of hydrogen peroxide. J Biol Chem. 2005;280:15340–7.
Stone JR, Maki JL, Collins T. Basal and hydrogen peroxide stimulated sites of phosphorylation in heterogeneous nuclear ribonucleoprotein C1/C2. Biochemistry. 2003;42:1301–8.
Cai K, Chen S, Zhu C, Li L, Yu C, He Z, et al. FOXD1 facilitates pancreatic cancer cell proliferation, invasion, and metastasis by regulating GLUT1-mediated aerobic glycolysis. Cell Death Dis. 2022;13:765.
Cao S, Wang D, Wang P, Liu Y, Dong W, Ruan X, et al. SUMOylation of RALY promotes vasculogenic mimicry in glioma cells via the FOXD1/DKK1 pathway. Cell Biol Toxicol. 2023;39:3323–40.
Kajita Y, Nakayama J-i, Aizawa M, Ishikawa F. The UUAG-specific RNA binding protein, heterogeneous nuclear ribonucleoprotein D0. J Biol Chem. 1995;270:22167–75.
Sarkar B, Lu J-Y, Schneider RJ. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J Biol Chem. 2003;278:20700–7.
Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, et al. Phosphorylation of p40AUF1 regulates binding to A + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J Biol Chem. 2003;278:33039–48.
Shen Z-J, Malter J. Regulation of AU-rich element RNA binding proteins by phosphorylation and the prolyl isomerase Pin1. Biomolecules. 2015;5:412–34.
Li M-L, Ragupathi A, Patel N, Hernandez T, Magsino J, Werlen G, et al. The RNA-binding protein AUF1 facilitates Akt phosphorylation at the membrane. J Biol Chem. 2022;298:102437.
Li M-L, Defren J, Brewer G. Hsp27 and F-Box protein β-TrCP promote degradation of mRNA decay factor AUF1. Mol Cell Biol. 2023;33:2315–26.
Fellows A, Deng B, Mierke DF, Robey RB, Nichols RC. Peptides modeled on the RGG domain of AUF1/hnRNP-D regulate 3′ UTR-dependent gene expression. Int Immunopharmacol. 2013;17:132–41.
Makeyev AV, Liebhaber SA. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. Rna. 2002;8:265–78.
Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem. 2013;288:17791–802.
Ghanem LR, Kromer A, Silverman IM, Chatterji P, Traxler E, Penzo-Mendez A, et al. The poly(C) binding protein Pcbp2 and its retrotransposed derivative Pcbp1 are independently essential to mouse development. Mol Cell Biol. 2016;36:304–19.
Chkheidze AN, Liebhaber SA. A novel set of nuclear localization signals determine distributions of the αCP RNA-binding proteins. Mol Cell Biol. 2023;23:8405–15.
Hussey George S, Chaudhury A, Dawson Andrea E, Lindner Daniel J, Knudsen Charlotte R, Wilce Matthew CJ, et al. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell. 2011;41:419–31.
Xue XY, Wang X, Liu YX, Teng GG, Wang Y, Zang XF, et al. SchA-p85-FAK complex dictates isoform-specific activation of Akt2 and subsequent PCBP1-mediated post-transcriptional regulation of TGFβ-mediated epithelial to mesenchymal transition in human lung cancer cell line A549. Tumor Biol. 2014;35:7853–9.
Wang H, Vardy LA, Tan CP, Loo JM, Guo K, Li J, et al. PCBP1 suppresses the translation of metastasis-associated PRL-3 phosphatase. Cancer Cell. 2010;18:52–62.
Janecki DM, Swiatkowska A, Szpotkowska J, Urbanowicz A, Kabacinska M, Szpotkowski K, et al. Poly(C)-binding protein 2 regulates the p53 expression via interactions with the 5′-terminal region of p53 mRNA. Int J Mol Sci. 2021;22:13306.
Chen C, Lei J, Zheng Q, Tan S, Ding K, Yu C. Poly(rC) binding protein 2 (PCBP2) promotes the viability of human gastric cancer cells by regulating CDK2. FEBS Open Bio. 2018;8:764–73.
Han W, Xin Z, Zhao Z, Bao W, Lin X, Yin B, et al. RNA-binding protein PCBP2 modulates glioma growth by regulating FHL3. J Clin Investig. 2013;123:2103–18.
Wang XN, Guo QY, Wang H, Yuan XD, Wang BJ, Lobie PE, et al. PCBP2 posttranscriptional modifications induce breast cancer progression via upregulation of UFD1 and NT5E. Mol Cancer Res. 2021;19:86–98.
Chang JS, Santhanam R, Trotta R, Neviani P, Eiring AM, Briercheck E, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2–dependent suppression of C/EBPα-driven myeloid differentiation. Blood. 2007;110:994–1003.
Dominguez C, Fisette J-F, Chabot B, Allain FHT. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat Struct Mol Biol. 2010;17:853–61.
Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EricA. The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab. 2013;17:386–98.
Delbridge ML, Lingenfelter PA, Disteche CM, Graves JAM. The candidate spermatogenesis gene RBMY has a homologue on the human X chromosome. Nat Genet. 1999;22:223–4.
Soulard M, Dellavalle V, Siomi MC, Pinolroma S, Codogno P, Bauvy C, et al. HNRNP-G—sequence and characterization of a glycosylated RNA-binding protein. Nucleic Acids Res. 1993;21:4210–7.
Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho T-J, et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature. 2014;508:494–9.
Cho Y, Ideue T, Nagayama M, Araki N, Tani T. RBMX is a component of the centromere noncoding RNP complex involved in cohesion regulation. Genes Cells. 2018;23:172–84.
Matsunaga S, Takata H, Morimoto A, Hayashihara K, Higashi T, Akatsuchi K, et al. RBMX: a regulator for maintenance and centromeric protection of sister chromatid cohesion. Cell Rep. 2012;1:299–308.
Antonello ZA, Hsu N, Bhasin M, Roti G, Joshi M, Van Hummelen P, et al. Vemurafenib-resistance via de novo RBM genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with BRAFV600E. Oncotarget. 2017;8:84743–60.
Hirschfeld M, Ouyang YQ, Jaeger M, Erbes T, Orlowska-Volk M, zur Hausen A, et al. HNRNP G and HTRA2-BETA1 regulate estrogen receptor alpha expression with potential impact on endometrial cancer. BMC Cancer. 2015;15:86.
Levin RS, Hertz NT, Burlingame AL, Shokat KM, Mukherjee S Innate immunity kinase TAK1 phosphorylates Rab1 on a hotspot for posttranslational modifications by host and pathogen. Proceedings of the National Academy of Sciences 2016;113.
Chua HH, Tsuei DJ, Lee PH, Jeng YM, Lu J, Wu JF, et al. RBMY, a novel inhibitor of glycogen synthase kinase 3β, increases tumor stemness and predicts poor prognosis of hepatocellular carcinoma. Hepatology. 2015;62:1480–96.
Chua H-H, Chang M-H, Chen Y-H, Tsuei D-J, Jeng Y-M, Lee P-H, et al. PIM1-induced cytoplasmic expression of RBMY mediates hepatocellular carcinoma metastasis. Cell Mol Gastroenterol Hepatol. 2023;15:121–52.
Ghetti A, Pinolroma S, Michael WM, Morandi C, Dreyfuss G. hnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992;20:3671–8.
Kamath RV, Leary DJ, Huang S. Nucleocytoplasmic shuttling of polypyrimidine tract-binding protein is uncoupled from RNA export. Mol Biol Cell. 2001;12:3808–20.
Keppetipola N, Sharma S, Li Q, Black DL. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol. 2012;47:360–78.
Bonini MG, Cote GJ, Zhu W, Thomas A, Martin E, Murad F, et al. Hydrogen peroxide alters splicing of soluble guanylyl cyclase and selectively modulates expression of splicing regulators in human cancer cells. PLoS One. 2012;7:e41099.
Cui J, Placzek WJ. PTBP1 modulation of MCL1 expression regulates cellular apoptosis induced by antitubulin chemotherapeutics. Cell Death Differ. 2016;23:1681–90.
He X, Yuan C, Yang J. Regulation and functional significance of CDC42 alternative splicing in ovarian cancer. Oncotarget. 2015;6:29651–63.
Jo YK, Roh SA, Lee H, Park NY, Choi ES, Oh J-H, et al. Polypyrimidine tract-binding protein 1-mediated down-regulation of ATG10 facilitates metastasis of colorectal cancer cells. Cancer Lett. 2017;385:21–27.
Xie JY, Lee JA, Kress TL, Mowry KL, Black DL. Protein kinase A phosphorylation modulates transport of the polypyrimidine tract-binding protein. Proc Natl Acad Sci USA. 2003;100:8776–81.
Zhao Q, Han Y-M, Song P, Liu Z, Yuan Z, Zou M-H. Endothelial cell-specific expression of serine/threonine kinase 11 modulates dendritic cell differentiation. Nat Commun. 2022;13:648.
Shankarling G, Lynch Kristen W. Minimal functional domains of paralogues hnRNP L and hnRNP LL exhibit mechanistic differences in exonic splicing repression. Biochem J. 2013;453:271–9.
Rothrock CR, House AE, Lynch KW. HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J. 2005;24:2792–802.
House AE, Lynch KW. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol. 2006;13:937–44.
Yuan W, Xie J, Long C, Erdjument-Bromage H, Ding X, Zheng Y, et al. Heterogeneous nuclear ribonucleoprotein L is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem. 2009;284:15701–7.
Siebring‐van Olst E, Blijlevens M, de Menezes RX, van der Meulen‐Muileman IH, Smit EF, van Beusechem VW. A genome‐wide siRNA screen for regulators of tumor suppressor p53 activity in human non‐small cell lung cancer cells identifies components of the RNA splicing machinery as targets for anticancer treatment. Mol Oncol. 2017;11:534–51.
Goehe RW, Shultz JC, Murudkar C, Usanovic S, Lamour NF, Massey DH, et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Investig. 2010;120:3923–39.
Vu NT, Park MA, Shultz JC, Goehe RW, Hoeferlin LA, Shultz MD, et al. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J Biol Chem. 2013;288:8575–84.
Yao P, Wu J, Lindner D, Fox PL. Interplay between miR-574-3p and hnRNP L regulates VEGFA mRNA translation and tumorigenesis. Nucleic Acids Res. 2017;45:7950–64.
Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504.
Mabeta P, Steenkamp V. The VEGF/VEGFR axis revisited: implications for cancer therapy. Int J Mol Sci. 2022;23:15585.
Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287:31666–73.
Liu G, Razanau A, Hai Y, Yu J, Sohail M, Lobo VG, et al. A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J Biol Chem. 2012;287:22709–16.
Ji M, Zhao Z, Li Y, Xu P, Shi J, Li Z, et al. FBXO16-mediated hnRNPL ubiquitination and degradation plays a tumor suppressor role in ovarian cancer. Cell Death Dis. 2021;12:758.
Grishin NV. KH domain: one motif, two folds. Nucleic Acids Res. 2001;29:638–43.
Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The premessenger RNA-BINDING K-PROTEIN contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–8.
Hutchins EJ, Belrose JL, Szaro BG. A novel role for the nuclear localization signal in regulating hnRNP K protein stability in vivo. Biochem Biophys Res Commun. 2016;478:772–6.
Michael WM, Eder PS, Dreyfuss G. The K nuclear shuttling domain: a novel signal for nuclear import and nuclear export in the hnRNP K protein. Embo J. 1997;16:3587–98.
Matunis MJ, Michael WM, Dreyfuss G. Characterization and primary structure of the poly(C)-binding heterogeneous nuclear ribonucleoprotein complex K protein. Mol Cell Biol. 1992;12:164–71.
Carpenter B, McKay M, Dundas SR, Lawrie LC, Telfer C, Murray GI. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer. 2006;95:921–7.
Roychoudhury P, Chaudhuri K. Evidence for heterogeneous nuclear ribonucleoprotein K overexpression in oral squamous cell carcinoma. Br J Cancer. 2007;97:574–5.
Wen F, Shen A, Shanas R, Bhattacharyya A, Lian F, Hostetter G, et al. Higher expression of the heterogeneous nuclear ribonucleoprotein K in melanoma. Ann Surg Oncol. 2010;17:2619–27.
Aitken MJL, Malaney P, Zhang XR, Herbrich SM, Chan L, Benitez O, et al. Heterogeneous nuclear ribonucleoprotein K is overexpressed in acute myeloid leukemia and causes myeloproliferation in mice via altered Runx1 splicing. Nar Cancer. 2022;4:zcac39.
Barboro P, Salvi S, Rubagotti A, Boccardo S, Spina B, Truini M, et al. Prostate cancer: prognostic significance of the association of heterogeneous nuclear ribonucleoprotein K and androgen receptor expression. Int J Oncol. 2014;44:1589–98.
Michelotti EF, Michelotti GA, Aronsohn AI, Levens D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol Cell Biol. 1996;16:2350–60.
Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene. 2003;22:8012–20.
Gallardo M, Lee HunJ, Zhang X, Bueso-Ramos C, Pageon Laura R, McArthur M, et al. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015;28:486–99.
Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–51.
Mucha B, Qie S, Bajpai S, Tarallo V, Diehl JN, Tedeschi F, et al. Tumor suppressor mediated ubiquitylation of hnRNPK is a barrier to oncogenic translation. Nat Commun. 2022;13:6614.
Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, et al. S100B’s double life: intracellular regulator and extracellular signal. Biochim et Biophys Acta. 2009;1793:1008–22.
Natarajan K, Sundaramoorthy A, Shanmugam N. HnRNPK and lysine specific histone demethylase-1 regulates IP-10 mRNA stability in monocytes. Eur J Pharmacol. 2022;920:174683.
Chen LC, Liu HP, Li HP, Hsueh C, Yu JS, Liang CL, et al. Thymidine phosphorylase mRNA stability and protein levels are increased through ERK-mediated cytoplasmic accumulation of hnRNP K in nasopharyngeal carcinoma cells. Oncogene. 2009;28:1904–15.
Habelhah H, Shah K, Huang L, Ostareck-Lederer A, Burlingame AL, Shokat KM, et al. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat Cell Biol. 2001;3:325–30.
Grassilli E, Pisano F, Cialdella A, Bonomo S, Missaglia C, Cerrito MG, et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene. 2016;35:4368–78.
Kim H-J, Lee J-J, Cho J-H, Jeong J, Park AY, Kang W, et al. Heterogeneous nuclear ribonucleoprotein K inhibits heat shock-induced transcriptional activity of heat shock factor 1. J Biol Chem. 2017;292:12801–12.
Abravaya K, Myers MP, Murphy SP, Morimoto RI. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 1992;6:1153–64.
Barboro P, Borzi L, Repaci E, Ferrari N, Balbi C. Androgen receptor activity is affected by both nuclear matrix localization and the phosphorylation status of the heterogeneous nuclear ribonucleoprotein K in anti-androgen-treated LNCaP cells. Plos One. 2013;8:e79212.
Schroder FH. Cyproterone acetate—mechanism of action and clinical effectiveness in prostate cancer treatment. Cancer. 1993;72:3810–5.
Peng C, Tan Y, Yang P, Jin K, Zhang C, Peng W, et al. Circ-GALNT16 restrains colorectal cancer progression by enhancing the SUMOylation of hnRNPK. J Exp Clin Cancer Res. 2021;40:272.
Pelisch F, Pozzi B, Risso G, Muñoz MJ, Srebrow A. DNA damage-induced heterogeneous nuclear ribonucleoprotein K SUMOylation regulates p53 transcriptional activation. J Biol Chem. 2012;287:30789–99.
Lee SW, Lee MH, Park JH, Kang SH, Yoo HM, Ka SH, et al. SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J. 2012;31:4441–52.
Yang J-H, Chiou Y-Y, Fu S-L, Shih IY, Weng T-H, Lin W-J, et al. Arginine methylation of hnRNPK negatively modulates apoptosis upon DNA damage through local regulation of phosphorylation. Nucleic Acids Res. 2014;42:9908–24.
Zhu L, Tian Q, Gao H, Wu K, Wang B, Ge G, et al. PROX1 promotes breast cancer invasion and metastasis through WNT/beta-catenin pathway via interacting with hnRNPK. Int J Biol Sci. 2022;18:2032–46.
Han J, Nie M, Chen C, Cheng X, Guo T, Huangfu L, et al. SDCBP-AS1 destabilizes beta-catenin by regulating ubiquitination and SUMOylation of hnRNP K to suppress gastric tumorigenicity and metastasis. Cancer Commun. 2022;42:1141–61.
Lee BJ, Cansizoglu AE, Süel KE, Louis TH, Zhang Z, Chook YM. Rules for nuclear localization sequence recognition by Karyopherinβ2. Cell. 2006;126:543–58.
Cansizoglu AE, Lee BJ, Zhang ZC, Fontoura BMA, Chook YM. Structure-based design of a pathway-specific nuclear import inhibitor. Nat Struct Mol Biol. 2007;14:452–4.
Datar KV, Dreyfuss G, Swanson MS. The human hnRNP M proteins: identification of a methionine/arginine-rich repeat motif in ribonucleoproteins. Nucleic Acids Res. 1993;21:439–46.
West KO, Scott HM, Torres-Odio S, West AP, Patrick KL, Watson RO. The splicing factor hnRNP M is a critical regulator of innate immune gene expression in macrophages. Cell Rep. 2019;29:1594–1609.e1595.
Chen C, Ding X, Akram N, Xue S, Luo S-Z. Fused in sarcoma: properties, self-assembly and correlation with neurodegenerative diseases. Molecules. 2019;24:1622.
Chiusa M, Hu W, Zienkiewicz J, Chen X, Zhang M-Z, Harris RC, et al. EGF receptor-mediated FUS phosphorylation promotes its nuclear translocation and fibrotic signaling. J Cell Biol. 2020;219:e20200110.
Kishino Y, Matsukawa K, Matsumoto T, Miyazaki R, Wakabayashi T, Nonaka T, et al. Casein kinase 1δ/ε phosphorylates fused in sarcoma (FUS) and ameliorates FUS-mediated neurodegeneration. J Biol Chem. 2022;298:102191.
Jochum W, Passegué E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–12.
Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. Structure and function of the 26S proteasome. Annu Rev Biochem. 2018;87:697–724.
Perrotti D, Iervolino A, Cesi V, Cirinná M, Lombardini S, Grassilli E, et al. BCR-ABL prevents c-Jun-mediated and proteasome-dependent FUS (TLS) proteolysis through a protein kinase CβII-dependent pathway. Mol Cell Biol. 2000;20:6159–69.
Arenas A, Chen J, Kuang L, Barnett KR, Kasarskis EJ, Gal J, et al. Lysine acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS. Hum Mol Genet. 2020;29:2684–97.
Liu C, Peng Z, Li P, Fu H, Feng J, Zhang Y, et al. lncRNA RMST suppressed GBM cell mitophagy through enhancing FUS SUMOylation. Mol Ther Nucleic Acids. 2020;19:1198–208.
Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, et al. Arginine methylation next to the PY-NLS modulates transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–75.
Motaln H, Čerček U, Yamoah A, Tripathi P, Aronica E, Goswami A, et al. Abl kinase-mediated FUS Tyr526 phosphorylation alters nucleocytoplasmic FUS localization in FTLD-FUS. Brain. 2023;146:4088–104.
Quaresma AJC, Oyama S, Barbosa JARG, Kobarg J. The acidic domain of hnRNPQ (NSAP1) has structural similarity to Barstar and binds to Apobec1. Biochem Biophys Res Commun. 2006;350:288–97.
Semino F, Schröter J, Willemsen MH, Bast T, Biskup S, Beck‐Woedl S, et al. Further evidence for de novo variants in SYNCRIP as the cause of a neurodevelopmental disorder. Hum Mutat. 2021;42:1094–1100.
Briese M, Saal-Bauernschubert L, Ji C, Moradi M, Ghanawi H, Uhl M, et al. hnRNP R and its main interactor, the noncoding RNA 7SK, coregulate the axonal transcriptome of motoneurons. Proceedings of the National Academy of Sciences 2018;115.
Bai Y, Li J, Fang B, Edwards A, Zhang G, Bui M, et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. 2012;72:2501–11.
Aravind L, Koonin EV. SAP—a putative DNA-binding motif involved in chromosomal organization. Trends Biochem Sci. 2000;25:112–4.
Nozawa R-S, Boteva L, Soares DC, Naughton C, Dun AR, Buckle A, et al. SAF-A regulates interphase chromosome structure through oligomerization with chromatin-associated RNAs. Cell. 2017;169:1214–1227.e1218.
Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of Aaa+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114.
Britton S, Froment C, Frit P, Monsarrat B, Salles B, Calsou P. Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle. 2014;8:3717–22.
Douglas P, Ye R, Morrice N, Britton S, Trinkle-Mulcahy L, Lees-Miller SP. Phosphorylation of SAF-A/hnRNP-U serine 59 by polo-like kinase 1 is required for mitosis. Mol Cell Biol. 2023;35:2699–713.
Berglund FM, Clarke PR. hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem Biophys Res Commun. 2009;381:59–64.
Kim MK, Nikodem VM. hnRNP U inhibits carboxy-terminal domain phosphorylation by TFIIH and represses RNA polymerase II elongation. Mol Cell Biol. 1999;19:6833–44.
Ogawara D, Fukuda M, Ueno S, Ohue Y, Takemoto S, Mizoguchi K, et al. Drug fever after cancer chemotherapy is most commonly observed on posttreatment days 3 and 4. Support Care Cancer. 2015;24:615–9.
Aapro MS, Bohlius J, Cameron DA, Lago LD, Donnelly JP, Kearney N, et al. 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer. 2011;47:8–32.
Esteban-Villarrubia J, Soto-Castillo JJ, Pozas J, San Román-Gil M, Orejana-Martín I, Torres-Jiménez J, et al. Tyrosine kinase receptors in oncology. Int J Mol Sci. 2020;21:8529.
Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–98.
Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang Y-s. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17:36.
Yang Y, Li S, Wang Y, Zhao Y, Li Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct Target Ther. 2022;7:329.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44.
Burke JE. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol Cell. 2018;71:653–73.
Elmenier FM, Lasheen DS, Abouzid KAM. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur J Med Chem. 2019;183:111718.
Wright SCE, Vasilevski N, Serra V, Rodon J, Eichhorn PJA. Mechanisms of resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021;13:1538.
Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22:138.
Yu L, Wei J, Liu P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol. 2022;85:69–94.
Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079–93.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66.
Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15:122.
Han Z-J, Feng Y-H, Gu B-H, Li Y-M, Chen H. The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol. 2018;52:1081–94.
Lightcap ES, Yu P, Grossman S, Song K, Khattar M, Xega K, et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci Transl Med. 2021;13:eaba7791.
Langston SP, Grossman S, England D, Afroze R, Bence N, Bowman D, et al. Discovery of TAK-981, a first-in-class inhibitor of SUMO-activating enzyme for the treatment of cancer. J Med Chem. 2021;64:2501–20.
Luo YM, Li ZH, Kong Y, He W, Zheng HH, An MJ, et al. KRAS mutant driven SUMOylation controls extracellular vesicle transmission to trigger lymphangiogenesis in pancreatic cancer. J Clin Investig. 2022;132:e157644.
Xue X, Wang X, Liu Y, Teng G, Wang Y, Zang X, et al. SchA–p85–FAK complex dictates isoform-specific activation of Akt2 and subsequent PCBP1-mediated post-transcriptional regulation of TGFβ-mediated epithelial to mesenchymal transition in human lung cancer cell line A549. Tumor Biol. 2014;35:7853–9.
Lobaccaro J-MA, Barboro P, Borzì L, Repaci E, Ferrari N, Balbi C. Androgen receptor activity is affected by both nuclear matrix localization and the phosphorylation status of the heterogeneous nuclear ribonucleoprotein K in anti-androgen-treated LNCaP cells. PLoS One. 2013;8:e79212.
He D, Huang C, Zhou Q, Liu D, Xiong L, Xiang H, et al. HnRNPK/miR-223/FBXW7 feedback cascade promotes pancreatic cancer cell growth and invasion. Oncotarget. 2017;8:20165–78.
Messias AC, Harnisch C, Ostareck-Lederer A, Sattler M, Ostareck DH. The DICE-binding activity of KH domain 3 of hnRNP K is affected by c-Src-mediated tyrosine phosphorylation. J Mol Biol. 2006;361:470–81.
Acknowledgements
This work was supported by grants to GW from the National Natural Science Foundation of China (82273252), Key Research and Development Program of Shandong (2021CXGC011101), and to YD from the China Postdoctoral Science Foundation (2023M732073).
Author information
Authors and Affiliations
Contributions
BHL collected the related papers and drafted the manuscript. MXW: writing, review, and editing final manuscript, FG and YSW participated in the design of the review. YMD and GWW initiated the study and revised and finalized the manuscript. All authors have read and approved the final manuscript. BHL and MXW contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, B., Wen, M., Gao, F. et al. Regulation of HNRNP family by post-translational modifications in cancer. Cell Death Discov. 10, 427 (2024). https://doi.org/10.1038/s41420-024-02198-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-024-02198-7
This article is cited by
-
Multiomics analysis identifies the prognostic significance and biological roles of the HNRNP family in lung adenocarcinoma
Discover Oncology (2026)
-
Research advances in epigenetic modifications and post-translational modifications in endothelial-mesenchymal transition
Epigenetics & Chromatin (2025)
-
Role of m6A RNA methylation regulators in pancreatic cancer: interactions and potential implications
Cancer Cell International (2025)
-
The paradox of hnRNPK: both absence and excess impair skeletal muscle function in mice
Skeletal Muscle (2025)
