
Abstract
Diabetic kidney disease (DKD) is characterized by kidney damage and abnormal renal energy metabolism, but the molecular mechanism of DKD is still unclear. In this study, we show that p16- positive senescent cells are an important regulator in the progression of DKD. The expression of p16 and senescence are increased in the kidneys of DM mice and DKD patients. To better understand the role of p16 in DKD, we induce type 1 diabetes in INK-ATTAC mice, a mouse model that allows the selective ablation of p16-expressing cells upon administration of the drug AP20187. We found that clearance of p16-positive cells, most of them are senescent cells, (1) decreased senescence and the expression of the components of the senescence-associated secretory phenotypes (SASPs), (2) restored kidney adenosine triphosphate (ATP) content, (3) decreased the expression of the key glycolytic genes to improve the metabolic reprogramming, (4) normalized the mitochondrial metabolism through AMPK and mTOR pathway, resulting in an amelioration of the progression of DKD. In addition, p16 mediated the blocking of the cell cycle is through the CDK4-Rb pathway in DKD kidneys. This study suggests that pharmacological deletion of p16-positive senescent cells may be a novel therapeutic strategy for DKD treatment.
Similar content being viewed by others


Introduction
Diabetic kidney disease (DKD), also called diabetic nephropathy, is a chronic kidney disease with serious complications of diabetes mellitus (DM), resulting in a high morbidity and mortality [1, 2]. DKD can be caused by high blood glucose, high blood pressure, or having diabetes for a long time. About 30–40% of diabetic patients will be complicated with DKD [3, 4], including glomerular hyperfiltration, progressive albuminuria, declining GFR, and ultimately leading to end-stage renal disease (ESRD) [5, 6]. Metabolic changes associated with diabetes lead to glomerular hypertrophy, glomerulosclerosis, and tubulointerstitial inflammation and fibrosis. Moreover, DKD patients often suffer from cardiovascular and neuropathy diseases, which sharply increases the risk of death [7, 8]. Although significant outcomes have been achieved in the pathogenesis and clinical therapy for DKD, there is a large residual risk of diabetic kidney disease onset and progression. Therefore, widespread innovation is urgently needed to improve health outcomes for patients with diabetic kidney disease. Achieving this goal requires further understanding molecular mechanism of DKD.
Recent attention has been focusing on the effect of senescence on lifespan and disease progression [9]. Senescence is a state of stable cell cycle arrest that occurs due to a variety of stimuli, such as DNA damage, oncogene activation, and telomere shortening [10]. Senescent cells are viable and metabolically active with other phenotypic alterations, playing roles in normal development, maintains tissue homeostasis, and limits tumor progression [11]. Senescent cells have several distinguishing characteristics, such as increased cell size and enzymatic activity of the lysosomal hydrolase senescence-associated β-galactosidase (SA-β-Gal), upregulation of prosurvival pathways to resist apoptosis, and the development of a “senescence-associated secretory phenotype (SASP)” [12]. The SASP is composed of a variety of soluble signaling factors, including pro-inflammatory cytokines, chemokines, and growth factors, insoluble extracellular matrix proteins, and non-protein components, which can be secreted into the surrounding microenvironment to affect neighboring cell biology and ultimately disrupting tissue structure and function [13]. Senescent cells are also characterized by an increase in the cyclin-dependent kinase inhibitor p16, which is one of the hallmarks of senescence [14]. It has been reported that purging p16-positive senescent cells in INK-ATTAC mice, a transgenic mouse model that allowed the removal of p16Ink4a-positive cells upon administration of a drug, AP20187, increased mean lifespan [9]. However, whether and how p16-positive senescent cells promote the progression of DKD remain unknown.
Recent studies support a key role of altered renal energy metabolism in the development of chronic kidney disease, including DKD [15, 16]. A range of metabolic abnormalities, including tricarboxylic acid circulation (TCA) disorders, altered fatty acid oxidation, and amino acid metabolic abnormalities, have been associated with kidney damage in DKD [17, 18]. However, the relationship between senescence and metabolic abnormalities in DKD and whether and how p16-positive senescent cells promote metabolic abnormalities and then DKD progression remains elusive. In this study, we address these questions by inducing diabetes in INK-ATTAC mice to determine the role of p16-positive senescent cells in kidney injury, glucolysis, and mitochondrial metabolic disorder in DKD.
Results
The expression of p16 is upregulated in the kidneys of DM mice and DKD patients
To understand the role of p16 in DKD, first, we found that the expression of p16 was increased in kidneys from DM mice compared with age-matched wild-type mice, as analyzed with Western blot (Fig. 1A), quantitative reverse-transcription PCR (qRT-PCR) (Fig. 1B) and immunohistochemistry staining (Fig. 1C). The expression of p16 was also increased in kidneys from DKD patients compared with normal human kidneys as analyzed with immunohistochemistry staining (Fig. 1D), which was predominantly concentrated in renal tubular epithelial cells (TECs). This distribution of p16 in proximal tubules was further validated by its co-localization with lotus tetragonolobus lectin (LTL), a proximal tubular marker (Supplemental Fig. S1).

A P16 protein and B mRNA levels in kidneys from diabetic (DM) mice and wild-type (WT) mice as analyzed through Western blot and qRT-PCR, respectively. ****p < 0.0001. n = 3. C Immunohistochemical staining of p16 in WT mice kidney tissues and DM mice kidney tissues. D Immunohistochemical staining of p16 in normal human kidney (NHK) tissues and kidneys from DKD patients. Scale bars: 100 μm.
Upregulation of p16 promotes senescence and increases the expression of the components of SASPs in the kidneys of DM mice and HK2 cells cultured in high glucose media
Upregulation of p16 has been used as one of the biomarkers of senescence [19]. In addition to an increase of p16-positive cells, we found that SA-β-gal positive cells was increased in the kidneys of DM mice as examined with the SA-β-gal staining (Fig. 2A). The acquisition of a senescence-associated secretory phenotype (SASP) is also a characteristic of senescent cells, and the secretion of SASPs can modify the cellular microenvironment and affect the function of neighboring cells [13]. We found that the expression of the most extensively characterized SASPs, such as TNF-α, IL-6, IL-1β, and MCP-1, was upregulated in kidneys from DM mice compared with age-matched wild-type control mice, as analyzed with qRT-PCR analysis (Fig. 2B).

A Representative images of senescence-associated SA-β-gal (red) co-stained with p16 (green) in the kidneys of WT mice and DM mice. Scale bar: 50 μm. B qRT-PCR analysis of SASPs (TNF-α, IL-6, IL-1β, and MCP-1) mRNA in WT and DM mice kidneys. (*p < 0.05, **p < 0.01 as determined by one-way ANOVA). n = 3. C Western blot analysis of the protein levels of p16 in HK2 cells cultured in normal glucose medium (NG) and high glucose medium (HG). ****p < 0.0001. D qRT-PCR analysis of p16 mRNA level in HK2 cells cultured in normal glucose medium and high glucose medium. ****p < 0.0001. n = 3. E Senescence-associated SA-β-gal staining increased in HK2 cells cultured in normal glucose medium, high glucose medium, and high glucose medium with p16 knockdown. Scale bar: 50 μm. F qRT-PCR analysis of SASPs (TNF-α, IL-6, IL-1β, and MCP-1) mRNA in HK2 cells cultured in normal glucose medium, high glucose medium, and high glucose medium with p16 knockdown. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, as determined by one-way ANOVA. n = 3.
To further understand the role of p16 in DKD, we cultured human kidney proximal tubular epithelial cells (HK2) with high glucose (25 mM) media to mimic a hyperglycemic (HG) condition in DKD. We found that treatment with high glucose (25 mM) increased the expression of p16 in HK2 cells compared to that in cells cultured in normal glucose (5 mM) media as analyzed with Western blot (Fig. 2C) and qRT-PCR (Fig. 2D). We also found that treatment with high glucose (25 mM) induced senescence of HK2 cells as seen by an increase of SA-β-gal staining (Fig. 2E) and the expression of SASPs, including TNF-α, IL-6, IL-1β, and MCP-1, compared to that in cells cultured in normal glucose media (Fig. 2F), whereas knockdown of p16 with siRNA decreased those effects in HK2 cells cultured in high glucose media. These results suggest that an upregulation of p16 may play a role in the progression of diabetic kidney disease through induction of senescence and the elevation of SASPs.
Clearance of p16-positive senescent cells delays the progression of diabetic kidney disease
Given that senescence and the expression of p16 were increased in kidneys of DM mice and DKD patients, it is reasonable to investigate whether clearance of p16-positive cells, most of them are senescent cells, decreases kidney injury and delays disease progression in DM mice. We induced type 1 diabetes in INK-ATTAC transgenic mice, a mouse model that allows the selective suicide gene-mediated ablation of highly p16-expressing cells upon administration of the drug AP20187 [9]. In brief, we treated 2-month-old INK-ATTAC male mice with low-dose streptozotocin (STZ) (60 mg/kg) and an equivalent volume of sodium citrate (vehicle control) by daily intraperitoneal injection for 5 days, which should induce persistent hyperglycemia in mice after 4 weeks, with the development of albuminuria. The DM INK-ATTAC transgenic mice were then treated intraperitoneally with AP20187 (3.3 mg/kg) and DMSO three times a week for 2 months, and kidneys were harvested one day after the last treatment with AP (Fig. 3A). Treatment with AP20187 cross-linked the ATTAC fusion protein, thereby activating its caspase-8 moiety to induce an apoptotic cell death, indicating as that the enhanced green fluorescence protein (EGFP) could not be detected after the induction with AP (Supplemental Fig. S2). We found that administration of AP20187 significantly decreased p16 level in the kidneys of DM INK-ATTAC mice compared to that in the kidneys from DMSO-injected age-matched DM INK-ATTAC mice, as analyzed with immunohistochemistry staining and Western blot analysis (Fig. 3B, C). Administration of AP20187 also decreased kidney weight/body weight ratios (KW/BW ratios), fasting blood glucose (Supplemental Fig. S3), urine microalbuminuria creatinine ratio (UACR), and serum creatinine (Fig. 3D, E) in DM INK-ATTAC mice compared to DMSO-treated DM INK-ATTAC mice. In addition, clearance of p16-positive cells with the treatment of AP20187 decreased glomerular matrix deposition, renal tubular atrophy and renal interstitial fibrosis in DM INK-ATTAC mice as examined by hematoxylin and eosin (H&E), periodic acid-Schiff (PAS) and Masson blue staining (Fig. 3F). The expression of kidney injury molecule-1 (KIM-1), a renal damage marker, and the expression of fibronectin and alpha-smooth muscle actin (α-SMA), two fibrotic markers, were also decreased in AP20187-treated DM INK-ATTAC kidneys compared to those in DMSO-treated DM INK-ATTAC kidneys (Fig. 3G). These results suggest that deletion of p16-positive senescent cells delays the progression of DKD.

A Experimental design and timeline in INK-ATTAC transgenic mice were studied for 5 months. B Immunohistochemistry analysis indicated that p16 was decreased in AP20187-treated DM INK-ATTAC mice kidneys compared to vehicle-treated DM INK-ATTAC mice kidneys. Scale bar: 100 μm. C Western blot analysis of p16 expression in kidneys from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. D, E Urine microalbuminuria-to-creatinine ratio (UACR) and serum creatinine in WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. Data represent the mean ± SEM for eight mice per group. F HE staining, PAS staining, and Masson trichrome staining in the kidneys from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. Scale bar: 100 μm. G Western blot analysis of the expression of fibronectin (Fn), a-SMA, and KIM-1 in WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice kidneys. *p < 0.05, **p < 0.01, and ***p < 0.001, as determined by one-way ANOVA.
Clearance of p16-positive senescent cells releases cell arrest and alleviates kidney injury in diabetic kidneys through the Rb-CDK4 pathway and the decrease of SASPs
Senescent cells provoke permanent cell cycle arrest by CDK inhibitors [20], which can also mediate renal injury in DKD [21]. To confirm whether clearance of p16-positive cells alleviates senescence and to understand underling mechanism, first, we found that deletion of p16-positive cells decreased SA-β-gal staining in AP20187-treated DM INK-ATTAC kidneys compared to that in DMSO-treated DM INK-ATTAC kidneys (Fig. 4A). Second, we found that deletion of p16-positive cells increased the expression of CDK4 and the phosphorylation of Rb (Fig. 4B), which could inhibit E2F, a transcription factor, possibly resulting in a downregulation of the expression of genes in the regulation of cell proliferation [13]. Last, we found that deletion of p16-positive cells decreased the levels of SASPs, including IL-1β、IL-6、MCP-1, and TNF-α, in AP20187-treated DM INK-ATTAC kidneys as analyzed with qRT-PCR analysis (Fig. 4C), which might contribute to the downregulation of KIM-1 and other factors to decrease renal injury in those kidneys. These results suggest that p16-positive cells might block the cell cycle through the CDK4-pRb pathway in senescent cells and neighboring cells and promote renal injury via SASPs.

A Representative images of senescence-associated SA-β-gal (red) co-stained with p16 (green) in kidneys from vehicle-treated DM INK-ATTAC mice and AP20187-treated DM INK-ATTAC mice. Scale bar: 50 μm. B Western blot analysis of the expression of CDK4, p-Rb, and Rb in WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice kidneys. The expression levels of CKD4 and p-Rb were decreased in kidneys from vehicle-treated DM INK-ATTAC mice compared to WT mice, and increased in AP20187-treated DM INK-ATTAC mice compared to vehicle-treated DM INK-ATTAC mice. C qRT-PCR analysis of SASPs (TNF-α, IL-6, IL-1β, and MCP-1) mRNA levels in kidneys from vehicle-treated DM INK-ATTAC mice and AP20187-treated DM INK-ATTAC mice. *p < 0.05, **p < 0.01, and ***p < 0.001, as determined by one-way ANOVA.
Clearance of p16-positive cells normalizes adenosine triphosphate (ATP) contents, glycolysis, and impaired mitochondrial metabolism in AP20187-treated DM INK-ATTAC kidneys
Studies have suggested a functional link between altered renal energy metabolism and senescent phenotype in DKD [22]. In addition, glycolysis and mitochondrial metabolism play an important role in energy metabolism, and their dysregulation contributes to DKD progression [23]. We found that ATP contents were decreased in DM mouse kidneys compared to that in age-matched wild-type mouse kidneys, whereas deletion of p16-positive cells increased ATP contents in AP20187-treated DM INK-ATTAC kidneys (Fig. 5A). We further found that the expression of the key glycolytic enzymes, Hk1, Ldha, Pkm2, and Ldhb, was increased in DM mouse kidneys compared to that in age-matched wild-type mouse kidneys, whereas deletion of p16-positive senescent cells decreased the expression of these key glycolytic genes in AP20187-treated DM INK-ATTAC kidneys (Fig. 5B). In addition, the expression of the genes involved in mitochondrial metabolism, including Pdk1, Mdh2, Sdha, Ndufa4, and Ndufs8, was also altered in DM mouse kidneys compared to that in age-matched WT mouse kidneys, which could be normalized in AP20187-treated DM INK-ATTAC kidneys (Fig. 5C). We also observed mitochondrial swelling and altered cristae in kidneys of DM mice compared to those in kidneys of wild-type mice, whereas clearance of p16-positive senescent cells partial normalized mitochondrial morphology and cristae density in AP20187-treated DM INK-ATTAC lidneys as examined with transmission electron microscopy (TEM) (Fig. 5D). These results support that deletion of p16-positive cells can normalize energy metabolism and mitochondrial morphology in DKD kidneys.

A ATP contents in kidneys from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. B qRT-PCR analysis of the mRNAs of the key glycolytic enzymes (Hk1, Ldha, Pkm2, and Ldhb) in kidneys of WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. C qRT-PCR analysis of the expression of genes involved in mitochondrial metabolism, including Pdk1, Mdh2, Sdha, Ndufa4, and Ndufs8, in kidneys from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. D Representative transmission electron microscopy (TEM) images showing mitochondrial morphology in kidney sections from WT mice, vehicle-treated DM INK-ATTAC mice, and AP20187-treated DM INK-ATTAC mice. Scale bar: 1 mm. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 was determined by one-way ANOVA. ATP adenosine triphosphate.
Knockdown of p16 restored glycolysis and metabolism in HK2 cells
The in vivo analysis suggests that p16-positive cells plays an important role in the metabolic reprogramming in DKD. To further support this notion, we knocked down p16 with siRNA in HK2 cells cultured in high glucose (25 mM) media and then tested its effect on glycolysis and mitochondrial metabolism in those cells with Seahorse analysis. We found that knockdown of p16 increased glycolysis, maximal glycolytic capacity, and glycolytic reserve in HK2 cells cultured in high glucose media compared to HK2 cells cultured in normal glucose media (Fig. 6A, B). Consistently, knockdown of p16 decreased the expression of the key glycolysis genes, Hk1, Ldha, Pkm2, and Ldh, in HK2 cells cultured in high glucose media compared with HK2 cells cultured with normal glucose media (Fig. 6C). In addition, we found that the oxygen consumption rate (OCR) and ATP level were decreased in HK2 cells cultured in high glucose media compared to HK2 cells cultured in normal glucose media, indicating that overall mitochondrial respiration (basal respiration, ATP-linked respiration, maximal respiration and reserve capacity) was decreased in those cells cultured in high glucose media, whereas knockdown of p16 restored the oxygen consumption rate and ATP level in HK2 cells cultured in normal glucose media (Fig. 6D–F). Furthermore, we found that the expression of genes involved in mitochondrial metabolism was altered in HK2 cells cultured with high glucose, which could be normalized in p16 knockdown HK2 cells cultured with high glucose media (Fig. 6G). The results support a direct role of the upregulation of p16 in the regulation of glycolysis and energy metabolism in tubular epithelial cells in DKD kidneys.

A Glycolytic activity of HK2 cells cultured in normal glucose medium, high glucose medium, and high glucose medium with p16 knockdown was detected by extracellular acidification rates (ECARs). B Glycolysis, glycolytic capacity, glycolytic reserve, and nonglycolytic acidification were calculated based on the glycolysis stress tests. C qRT-PCR analysis of the mRNAs of the key glycolytic enzymes (Hk1, Ldha, Pkm2, and Ldhb) in HK2 cells cultured in normal glucose media, high glucose media, and high glucose medium with p16 knockdown. D Oxygen consumption rate (OCR) of HK2 cells cultured in normal glucose medium (NG), high glucose medium (HG), and high glucose medium with p16 knockdown (HG+sip16) were quantified. E The basal respiration, ATP-linked respiration, max respiration capacity, and reserve capacity were calculated based on the mitochondrial stress tests. F Intracellular ATP contents in HK2 cells cultured in normal glucose medium, high glucose medium, and high glucose medium with p16 knockdown were directly quantified by the ATP determination kit. G qRT-PCR analysis of the expression of genes involved in mitochondrial metabolism, including Pdk1, Mdh2, Sdha, Ndufa4, and Ndufs8, in HK2 cells cultured in normal glucose medium, high glucose medium, and high glucose medium with p16 knockdown. (“ns” implies no significant changes, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 was determined by one-way ANOVA. ATP adenosine triphosphate.
Knockdown of p16 decreases mitochondrial metabolic disorder through the AMPK and mTOR pathways
Glycolytic metabolites have been reported to lead to mitochondrial metabolic disorder through AMPK and mTOR signaling [23]. We found that the phosphorylation of AMPK (active AMPK) was decreased in HK2 cells cultured with high glucose compared to that in HK2 cells cultured with normal glucose, whereas knockdown of p16 increased the phosphorylation of AMPK in HK2 cells cultured with high glucose (Fig. 7A). In addition, the phosphorylation of mTOR and its downstream targets, the ribosomal protein S6, were increased in HK2 cells cultured with high glucose compared with HK2 cells cultured with normal glucose, which could be decreased in p16 knockdown HK2 cells cultured with high glucose (Fig. 8A). We further found that the phosphorylation of AMPK was decreased and the phosphorylation of mTOR and S6 was increased in kidneys of DM mice compared with age-matched wild-type mice (Fig. 8B). Deletion of p16-positive senescent cells increased the phosphorylation of AMPK and decreased the phosphorylation of mTOR and S6 in AP20187-treated DM INK-ATTAC kidneys compared to those in vehicle-treated DM INK-ATTAC kidneys (Fig. 7B). These results suggest that p16-positive senescent cells may regulate mitochondrial metabolism disorder in DKD through AMPK and mTOR pathway.

A Western blot analysis of the expression of p-AMPK, AMPK, p-mTOR, mTOR, p-S6, and S6 in WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice kidneys. The expression level of p-AMPK was decreased in kidneys from DM mice compared to WT mice, and increased in kidneys from DM with AP treatment mice compared to vehicle-treated DM mice. The expression levels of p-mTOR and p-S6 were increased in kidneys from DM mice compared to WT mice, and decreased in kidneys from DM with AP treatment mice compared to DM mice. B Western blot analysis of the expression of p-AMPK, AMPK, p-mTOR, mTOR, p-S6, and S6 in HK2 cells cultured in normal glucose medium (NG), high glucose medium (HG), and high glucose medium with p16 knockdown (HG + sip16). The expression level of p-AMPK was decreased in HK2 cells cultured in high glucose medium compared to normal glucose medium, and increased in HK2 cells from high glucose medium with p16 knockdown compared to high glucose medium. The expression levels of p-mTOR and p-S6 were increased in HK2 cells cultured in high glucose medium compared to normal glucose medium and decreased in HK2 cells from high glucose medium with p16 knockdown compared to high glucose medium. “ns” implies not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 was determined by one-way ANOVA.

A Senescence-associated SA-β-gal staining in primary tubular epithelial cells (PTECs) from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. B Western blot analysis of the expression of p16, CDK4, p-Rb, Rb, and KIM-1 in PTECs from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. C qRT-PCR analysis of SASPs (TNF-α, IL-6, IL-1β, and MCP-1) mRNA levels in PTECs from vehicle-treated DM INK-ATTAC and AP20187-treated DM INK-ATTAC mice. D qRT-PCR analysis of the mRNAs of the key glycolytic enzymes (Hk1, Ldha, Pkm2, and Ldhb) in HK2 cells cultured in PTECs from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. E qRT-PCR analysis of the genes involved in mitochondrial metabolism, Pdk1, Mdh2, Sdha, Ndufa4, and Ndufs8, in PTECs from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mouse kidneys. F Western blot analysis of the expression of p-AMPK, AMPK, p-mTOR, mTOR, p-S6, and S6 in PTECs from WT, vehicle-treated DM INK-ATTAC, and AP20187-treated DM INK-ATTAC mice. “ns” implies not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 was determined by one-way ANOVA.
Deletion of p16-positive cells affects the neighboring cell biology and function in DKD
To further understand the effect of p16-positive cells on neighboring cell biology and function in DKD, we isolated the primary tubular epithelial cells (PTECs) from AP20187 and vehicle-treated DM INK-ATTAC kidneys. We found that (1) the percentage of SA-β-gal positive cells was higher and the levels of CDK4 and the phosphorylation of Rb were lower (Fig. 8A, B), (2) the expression of KIM-1 (Fig. 8B) and SASPs (Fig. 8C) was increased, (3) the expression of the key genes encoding glycolytic enzymes, Hk1, Ldha, Pkm2, and Ldhb, was increased (Fig. 8D), (4) the expression of genes involved in mitochondrial metabolism, including Pdk1, Mdh2, Sdha, Ndufa4, and Ndufs8, was altered (Fig. 8E), and (5) the phosphorylation of AMPK was decreased and the phosphorylation of mTOR and the phosphorylation of S6 was increased, in PTECs from DMSO-treated DM INK-ATTAC mouse kidneys compared to those in PTECs from AP20187-treated DM INK-ATTAC mouse kidneys (Fig. 8F). These results suggest that after the deletion of p16-positive cells from the diabetic mouse kidneys, the effect of p16-positive cells on PTECs was released.
Discussion
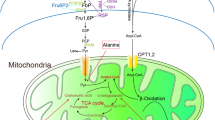
DKD is a leading cause of kidney failure in the world, representing ~44% of all cases of kidney failure in the United States [24, 25], and new interventions are needed to reduce the health burden of DKD [26]. In this study, we show that p16-positive senescent cells are a key regulator of DKD. The expression of p16 was upregulated in DKD kidneys and HK2 cells cultured in high glucose media, resulting in an increase of senescence and the expression of SASPs, including TNF-α, IL-6, IL-1β, and MCP-1, to decrease the production of ATP and promote glycolysis and mitochondrial metabolic disorder. Clearance of p16-positive cells delayed the progression of diabetic kidney disease in STZ-induced INK-ATTAC transgenic mice, restored the levels of ATP, decreased the expression of the common SASPs, decreased glycolysis, and improved metabolic reprogramming of mitochondria, possibly through the AMPK and mTOR pathways (Fig. 9). This study suggests that pharmacological deletion of p16-positive senescent cells may be a novel therapeutic strategy for DKD treatment.

A schematic representation of the roles and mechanisms of p16-positive senescent cells in the progression of DKD. Upregulated p16 in DKD kidneys, leading to an increase of senescence, resulted in (i) inhitbition of CDK4 and the phosphorylation of Rb, which could inhibit E2F, resulting in a downregulation of the expression of genes in the regulation of cell proliferation; (ii) increasing the genes of the key glycolytic enzymes and decreasing the production of ATP; (iii) abnormalizing the expression of the genes associated with mitochondrial metabolism, through AMPK and mTOR pathway; (iiii) increasing the expression of SASPs. They all promote the progression of diabetic kidney disease.
Growing evidence indicates that an accumulation of senescent cells is associated with the loss of kidney function in DKD [27]. In diabetic kidneys, senescence is induced by multiple stressors, including high blood glucose, accumulation of advanced glycation end products (AGE), hypertension, oxidative stress, and inflammation, resulting in a decline of renal function and changes in renal structures [28]. One of the key features of this study is to determine the direct role of p16-positive senescent cells in DKD by using the INK-ATTAC transgenic mouse model. The INK-ATTAC mouse has been generated by researchers at the Mayo Clinic [9], which includes a 2617 bp fragment of the promoter of the p16 INK4A gene and an internal ribosome entry site (IRES), followed by an open reading frame (ORF) encoding enhanced green fluorescent protein (EGFP) to allow detection of p16 INK4A-positive senescent cells with the treatment of drug AP20187 [9]. AP20187 is a synthetic drug that induces dimerization of the membrane-bound myristylated FK506-binding-protein-caspase-8 (FKBP-Casp8) fusion protein via the p16 promoter to selectively induce p16-positive senescent cell death in INK-ATTAC mice [9]. We induced type 1 diabetes in the INK-ATTAC transgenic mice and found that clearance of p16-positive senescent cells with the treatment of AP20187 alleviated kidney injury and the progression of DKD, which supports a direct role of p16-positive senescent cells in DKD.
The second key feature of this study is to determine how p16-positive senescent cells promote the progression of DKD. Our results suggest two potential mechanisms in this process. First, the increase of p16-positive senescent cells alters the expression of key enzymes involved in glycolysis and mitochondrial metabolism. Second, p16-positive senescent cells may affect the biology and function of neighboring cells through the secretion of SASPs, such as TNF-α, IL-6, IL-1β, and MCP-1, in a paracrine manner [28]. This may explain how a relatively small number of p16-positive senescent cells promotes DKD progression. To support the hypothesis that SASPs have potent local and systemic effects on neighboring normal cells, we isolated PTECs from vehicle and AP20187-treated DM INK-ATTAC kidneys and found that after the deletion of senescent cells from the diabetic mouse kidneys, the biology and function of the neighboring PTECs were normalized in DM INK-ATTAC kidneys compared to vehicle-treated controls.
Senescent cells secreted SASPs contain complement proteins, pro-inflammatory cytokines, and pro-fibrotic or pro-clotting factors [29]. These factors act either alone or together to affect renal cell biology and function, leading to pathological changes in different types of renal cells to promote renal tubulointerstitial fibrosis, podocyte hypertrophy and loss of foot process, mesangial stroma dilation, glomerular sclerosis, and glomerular hypertension [30]. Clearance of p16-positive senescent cells not only prevented cell cycle arrest but should also reduce SASP secretion via the decrease of their expression in diabetic mice, resulting in systemic benefits, including lower blood glucose. The beneficial effect of AP20187 treatment on alleviating DKD injury may also be attributed to the reduction of blood glucose mediated by SASPs. These effects may also be related to the elimination of senescent cells in adipose tissues, thereby improving glucose-induced insulin secretion and glucose tolerance in DKD kidneys [29, 31].
The third key feature of this study is that p16-positive senescent cells can affect ATP production and energy metabolism in DKD kidneys. DKD is characterized by abnormal kidney energy metabolism, but the exact mechanism is unclear. Recent studies have demonstrated that abnormal glucose metabolism in DKD is mainly manifested by increased glycolysis and elevated lactate level, which are associated with albuminuria and kidney injury [32]. In tumor cells, even when oxygen levels are normal, glycolysis is prioritized to produce energy, known as the Warburg effect [33]. However, in DKD, the increase of renal glycolysis is mainly attributed to the obstruction of tricarboxylic acid circulation, resulting in impaired oxidative phosphorylation and ATP production [34, 35]. The enhanced glycolytic activity reveals elevated glycolysis in HK2 cells cultured in high glucose medium. Glycolysis increases the production of lactic acid by producing pyruvate, which is associated with the upregulation of lactate dehydrogenase (LDH), pyruvate kinase (PKM), and hexokinase (Hk1) [36]. PKM is responsible for the last conversion step of glycolysis to produce pyruvate [37]. PKM is also thought to indirectly increase lactic acid production [38]. Meanwhile, the upregulation of LDH results in an increase of lactate levels in senescent cells [39], leading to multiple senescence-associated events, such as tumorigenesis, wound healing, and evasion of immune responses [40]. Clearance of p16-positive senescent cells not only increases ATP levels but also normalizes the expression of key enzymes in glycolysis, including hexokinase, lactate dehydrogenase, and pyruvate kinase in DKD kidneys, suggesting that an improvement of energy and glucose metabolism is a critical benefit of clearing p16-positive senescent cell therapy in DKD.
The fourth key feature of this study is that p16-positive senescent cells can affect mitochondrial function in DKD kidneys. Increasing studies revealed the role of mitochondrial dysfunction in the pathogenesis of DKD [41]. Insulin resistance and other related metabolic disorders in DKD may damage mitochondria and limit their ability to select glucose as the preferred substrate for oxidation, known as metabolic inflexibility [18, 41]. The primary biological function of mitochondria is responsible for generating a substantial amount of ATP, thereby supplying the energy source for basal cell functions in the kidneys [39]. ATP production in mitochondria is accomplished by a continuous reaction called oxidative phosphorylation (OXPHOS), and changes in OXPHOS function have been observed in various senescent models [39, 42, 43]. During senescence, dysfunctional mitochondria produce excess oxidative stress (ROS), leading to unnecessary oxidation of the proteins involved in OXPHOS and impacting their function [44]. Changes in mitochondrial metabolism, with reduced dependence on OXPHOS but increased dependence on glycolysis, are one of the characteristic changes observed during senescence [45].
Mitochondrial glucose metabolism is regulated by the mTOR pathway and its upstream regulator, AMPK [46]. It has been reported that chronic hyperglycemia can lead to an inhibition of pyruvate dehydrogenase (PDH) through mTOR-S6 signaling, which is triggered by glycolytic metabolites upstream of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [23]. In DKD, the activity of AMPK is decreased and the activity of mTOR is increased, resulting in altered expression of genes involved in mitochondrial metabolism [47]. Clearance of p16-positive cells increases the activity of AMPK and decreases the activity of mTOR, leading to a normalization of the expression of mitochondrial metabolic genes, including the downregulation of Pdk1 (encoding pyruvate dehydrogenase kinases 1) in DKD kidneys. Pdk1 can phosphorylate PDH to inhibit its activity, in which the PDH complex is able to convert pyruvate to acetyl-CoA bin the mitochondria [48]. Thus, downregulation of Pdk1 mediated by the deletion of p16-positive senescent cells should restore pyruvate dehydrogenase activity and increase pyruvate entry into the tricarboxylic acid cycle in DKD kidney, which should be one of the mechanisms to decrease DKD progression.
Last, during normal conditions, the complex of CDK4 and CDK6, as well as cyclin D, phosphorylates Rb to promote the transition of cells from the G1 phase to S phase [49]. When p16 is elevated, it competes with cyclin D for the binding of CDK4, thereby decreasing the phosphorylation of Rb and S-phase entry [50]. We found that the expression of p16 was mainly increased in renal tubules but not in glomeruli in DKD kidneys, which should promote senescence of p16-positive tubular epithelial cells. Tubular and interstitium accounts for more than 90% of renal parenchyma with important functions. It has been reported that tubular structural abnormalities predate glomerular structural changes in DKD kidneys and play an important role in the progression of DKD [51]. However, the underlying mechanisms need to be further investigated. In addition, although treatment with high glucose could increase the expression of p16 and senescence in HK2 cells, it remains to be determined how high glucose regulates the expression of p16 in DKD kidneys.
Methods
Cell lines and primary cells
Human kidney proximal tubular epithelial cells (HK2, ATCC, Cat# CRL-2190) were cultured in DMEM/F12 (Hyclone, Logan, UT, USA) supplemented with 10% FBS (Sigma, St. Louis, MO, USA) and 1% penicillin-streptomycin (Gibco, Grand Island, NY, USA) at 37 °C in 5% CO2 incubator. The cells cultured in 5 mM glucose were named the NG group, and those cultured in 25 mM glucose were named the HG group. Primary tubular epithelial cells (PTECs) were isolated from AP20187 and vehicle-treated DM INK-ATTAC mice. The renal cortices were chopped and digested with 1 mg/ml collagenase II in PBS at 37 °C for 10 min. DMEM-F12 supplemented with 10% FBS were used to stop digestion. The mixture was passed orderly through a 100-μm and 70-μm filter (BD Falcon). The filtrate was collected at the bottom and centrifuged. The cells were seeded on a culture dish in DMEM-F12 supplemented with 10% FBS and 1% (v/v) penicillin-streptomycin.
Western blot analysis
Cells were homogenized in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM Na3VO4, 25 mM β-glycerol-phosphate, 0.1 mM PMSF, Roche complete protease inhibitor set, and Sigma-Aldrich phosphatase inhibitor set, St. Louis, MO, USA), and centrifuged at 20,000×g for 20 min. Protein concentration was measured using the BCA Pierce Protein assay kit (Thermo Fisher, Waltham, MA, USA) and normalized to the lowest concentration. Protein samples were subjected to standard SDS-PAGE gels, transferred to immuno-blot PVDF membranes (Millipore, Burlington, MA, USA), blocked with 10% nonfat dry milk, and then incubated overnight at 4 °C with primary antibodies. Primary antibodies used in this study are listed as follows: mouse monoclonal antibodies against CDKN2A/p16 (F12, Santa Cruz, Dallas, TX, USA, sc-1661, 1:500 dilution), α-SMA (Cell Signaling Technology, Boston, MA, #69313, 1:1000 dilution), Rb (CST, #9309, 1:1000 dilution), CDK4 (3F121, sc-70831, 1:1000 dilution), β-actin (AC-15, Sigma, St. Louis, MO, USA, A1978, 1:3000 dilution); rabbit polyclonal antibodies against beta Galactosidase (Invitrogen, A-11132, 1:300 dilution), Fibronectin/FN1 (CST, #36779, 1:1000 dilution), Phospho-Rb (CST, #8180, 1:1000 dilution), Phospho-AMPKα (Thr172) (CST, #2531, 1:1000 dilution), AMPKα (D5A2, CST, #5831, 1:1000 dilution), Phospho-mTOR (Ser2448) (D9C2, CST, #5536, 1:1000 dilution), mTOR (7C10, CST, #2983, 1:1000 dilution), Phospho-S6 ribosomal protein (Ser240/244) (D68F8, CST, #5364, 1:1000 dilution), S6 ribosomal protein (5G10, CST, #2217, 1:1000 dilution). rabbit monoclonal antibodies anti-KIM-1 (9E1, Abcam, Waltham, MA, USA, ab302932, 1:1000 dilution). Secondary antibodies used for Western blot include donkey anti-rabbit IgG–horseradish peroxidase (sc-2313) and goat anti-mouse IgG–horseradish peroxidase (sc-2005), purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Histology and immunohistochemistry
Kidney sections were fixed in 4% paraformaldehyde (pH 7.4), and stained with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), and Masson trichrome staining methods. Immunohistochemistry was performed in 4-mm paraffin sections. For p16 staining, a monoclonal mouse anti-CDKN2A/p16 antibody (F12, Santa Cruz, Dallas, TX, USA, sc-1661, 1:100 used for immunohistochemistry [IHC]), biotinylated secondary antibody (Santa Cruz Biotechnology Inc., 1:500), and DAB substrate system were used. Kidney sections were counterstained by haematoxylin. The exposure time of the slides to the reagents was the same for the samples of all groups in each independent experiment. Images were analyzed with a NIKON ECLIPSE 80i microscope. The fibrotic area was quantified using Image J software (https://imagej.net/ij/, accessed on 5 June 2024) by Masson trichrome staining in ten random fields (original magnification ×200) as previously described [52].
Immunofluorescence staining
After antigen retrieval, tissue sections were incubated with a mouse monoclonal antibody CDKN2A/p16 (F12, Santa Cruz, Dallas, TX, USA, sc-1661, 1:100 dilution) overnight, and then were incubated with Fluro-555 anti-mouse IgG secondary antibody or Fluro-488 anti-mouse IgG secondary antibody and mounted in Prolong Gold Antifade reagent with DAPI (Invitrogen). For beta galactosidase staining, a rabbit against beta galactosidase antibody (SA-β-gal Invitrogen, A-11132, 1:300 dilution) and Fluro-555 anti-rabbit IgG secondary antibody were used. Images were analyzed using a NIKON ECLIPSE 80i microscope.
Quantitative reverse-transcription PCR (qRT-PCR)
Total RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN, Germantown, MD, USA). Total RNA (1 μg) was reverse transcribed using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) and amplified in triplicate using iTaq SYBR Green Supermix with ROX (Bio-Rad) with a real-time PCR machine (Bio-Rad), according to the manufacturer’s instructions. Genes were amplified using the following primers: Hk1-F, 5′-TCACATTGTCTCCTGCATCTC-3′; Hk1-R, 5′-CTTTGAATCCCTTTGTCCACG-3′; Ldha-F, 5′-GCTCCCCAGAACAAGATTACAG-3′; Ldha-R, 5′-TCGCCCTTGAGTTTGTCTTC-3′; Pkm2-F, 5′-CCATTCTCTACCGTCCTGTTG-3′;
Pkm2-R, 5′-TCCATGTAAGCGTTGTCCAG-3′; Ldhb-F, 5′-TACGTCACCTGGAAACTGAG-3′; Ldhb- R, 5′-CACCATCTTATGCACCTCCT-3′
Pdk1-F, 5′-TCCCCCGATTCAGGTTCAC-3′; Pdk1-R, 5′-CCCGGTCACTCATCTTCACA-3′
Sdha-F, 5′-GAGATACGCACCTGTTGCCAAG-3′; Sdha-R, 5′-GGTAGACGTGATCTTTCTCAGGG-3′
Mdh2-F, 5′-GCAACCCCTTTCACTCCTG-3′; Mdh2-R, 5′-TCTGGTCTCAATGTGACTCAGAT-3′
NDUFA4-F, 5′-TCCCAGCTTGATTCCTCTCTT-3′; NDUFA4-R, 5′-GGGTTGTTCTTTCTGTCCCAG-3′; Ndufs8-F, 5′-TTGCCTGCAAACTCTGTGAG-3′; Ndufs8-R, 5′-CTCCACAATGGCATCAACAG-3′ ; β-actin-F, 5′-AAGAGCTATGAGCTGCCTGA-3′; β-actin-R, 5′-TACGGATGTCAACGTCACAC-3′. The complete reactions were subjected to the following program of thermal cycling: 40 cycles of 10 s at 95 °C and 20 s at 61 °C. A melting curve was run after the PCR cycles, followed by a cooling step. Each sample was run in triplicate in each experiment, and each experiment was repeated three times. Expression levels of target genes were normalized to the expression level of β-actin.
Senescence-associated beta-galactosidase (SA-β-gal) staining
Renal cell senescence was evaluated using the Senescence Cells β-Galactosidase Staining Kit (#9860-Cell Signaling Technology) according to the manufacturer’s protocol. Early passage HK2 cells or primary tubular epithelial cells were washed in 1x PBS and fixed for 15 min (1x fixative solution) and then rinsed in 1x PBS two times. Add 1 ml of theβ-Galactosidase staining solution (1 ml containing 50 ul of 20 mg/ml X-gal stock solution, 930 µl of 1x Staining solution, 10 ul of 100x solution A, 10 ul of 100x solution B). Incubate the plate at 37 °C at least overnight in a dry incubator (no CO2). The development of blue dots/areas was counted as positive staining.
Renal function measurement
Mice were placed in metabolic cages for the collection of 24-h urinary samples every 4 weeks from the age of 8 weeks. Urinary microalbumin was measured by competitive ELISA according to the manufacturer’s instructions (Exocell, Philadelphia, PA). Urinary and serum creatinine was measured by an enzymatic kit (Stanbio Laboratories, Boerne, TX). Urinary albumin excretion was expressed as total urinary albumin–to–creatinine ratio (mg/mg), as previously reported [53].
Fasting blood glucose
Blood glucose levels were measured every 4 weeks by an AccuChek glucose meter (Roche Diagnostics, Basel, Switzerland) after the mice were fasted for 6 h, as recommended by the Animal Models of Diabetic Complications Consortium.
RNA interference
The RNA oligonucleotides that specifically targeted human p16 (Cat# 4392420) were purchased from Thermo Fisher. The RNA oligonucleotides were transfected with DharmaFECT siRNA transfection reagent (GE Healthcare). Twenty-four hours after transfection, cells were harvested and analyzed by Western blotting and quantitative RT-PCR (qRT-PCR).
ATP assay
The cells were transfected with p16 siRNA for 48 h and lysed with RIPA buffer. The intracellular ATP levels of cell lysates were assessed using the ATP Determination Kit (Invitrogen), according to the manufacturer’s protocol. The intracellular ATP levels were shown by normalizing to the protein amount.
Mouse strain and treatment
All animal procedures were conducted under IACUC protocol A00003756-R24 (26 July 2024), approved by the Mayo Clinic IACUC, and in compliance with National Institutes of Health, United States Department of Agriculture, and Association for Assessment and Accreditation of Laboratory Animal Care guidelines. INK-ATTAC transgenic mice, which were used for studying renal senescence in vivo, were provided by J. van Deursen at the Mayo Clinic. These INK-ATTAC mice were bred onto a C57BL/6 genetic background as heterozygotes and genotyped. All mice were randomized to each group. Unless otherwise specified, the number of mice in each group is 8. Hyperglycemia was induced in 2-month-old INK-ATTAC male mice using low-dose streptozotocin (STZ) injection following established protocols (intraperitoneally, 60 mg/kg body weight, freshly dissolved in 0.05 mol/l sterile sodium citrate, pH 4.5 for 5 consecutive days) [54, 55]. One month later, mice which were induced to have persistent hyperglycemia were randomized to the treatment of AP (eight mice) or vehicle (eight mice). Age-matched control male mice received sodium citrate. AP (B/B Homodimerizer; Clontech, Mountain View, CA) leads to dimerization and then activates FKBP-fused caspase-8 components, leading to apoptosis of cells highly expressing p16Ink4a. AP only affects cells with the ATTAC fusion protein. AP (3.3 mg/kg) was delivered by intraperitoneal injections, three times a week, for 8 weeks (total 80 mg/kg, 24 treatments).
If an improper intraperitoneal injection operation causes the death of the mouse, exclude and supplement the same amount.
Metabolic assays
The Agilent Seahorse XF mito stress test and XF glycolysis stress test were performed according to the manufacturer’s instructions. Cells were counted, and the same number of cells were plated per well in the Seahorse experiments. In addition, cells were examined microscopically for adherence to the culture plates immediately before Seahorse quantifications. Under basal conditions and following the addition of 10 mM glucose, 1 μM oligomycin, and 50 mM 2-DG, the extracellular acidification rates (ECAR) levels were measured. Oxygen consumption rate (OCR) were measured at the basal level and the addition of 1.5 μM oligomycin, 1 μM FCCP, and 0.5 μM Rot + AA.
Statistical analysis
All of the data were expressed as mean ± SEM. Statistical analyses were performed with one-way ANOVA, followed by Newman–Keuls multiple comparison from GraphPad Prism 10.0 (GraphPad Software, La Jolla, CA). In addition, a repeated-analysis ANOVA was used for albumin excretion, body weight, and fasting blood glucose analysis.
Data availability
The data used and analyzed in this study are available from the corresponding authors upon reasonable request.
References
Jadawji C, Crasto W, Gillies C, Kar D, Davies MJ, Khunti K, et al. Prevalence and progression of diabetic nephropathy in South Asian, white European and African Caribbean people with type 2 diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2019;21:658–73.
Krairittichai U, Potisat S. Survival rates and mortality risk factors of Thai patients with type 2 diabetes mellitus. J Med Assoc Thai. 2017;100:S8–15.
Xia Z, Luo X, Wang Y, Xu T, Dong J, Jiang W, et al. Diabetic kidney disease screening status and related factors: a cross-sectional study of patients with type 2 diabetes in six provinces in China. BMC Health Serv Res. 2024;24:489.
Li S, Xie H, Shi Y, Liu H. Prevalence of diabetic nephropathy in the diabetes mellitus population: a protocol for systematic review and meta-analysis. Medicine. 2022;101:e31232.
Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, et al. Changes in diabetes-related complications in the United States, 1990-2010. N Engl J Med. 2014;370:1514–23.
Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol. 2016;12:73–81.
Yokomichi H, Mochizuki M, Hirata M, Nagai A, Kojima R, Horiuchi S, et al. All-cause and cardiovascular disease mortality in underweight patients with diabetic nephropathy: biobank Japan cohort. J Diabetes Investig. 2021;12:1425–9.
Tang C, Ouyang H, Huang J, Zhu J, Gu X. Differences between diabetic and non-diabetic nephropathy patients in cardiac structure and function at the beginning of hemodialysis and their impact on the prediction of mortality. J Int Med Res. 2021;49:1221797140.
Baker DJ, Wijshake T, Tchkonia T, Lebrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6.
Ring N, Valdivieso K, Grillari J, Redl H, Ogrodnik M. The role of senescence in cellular plasticity: lessons from regeneration and development and implications for age-related diseases. Dev Cell. 2022;57:1083–101.
Mchugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65–77.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–35.
Buj R, Leon KE, Anguelov MA, Aird KM. Suppression of p16 alleviates the senescence-associated secretory phenotype. Aging. 2021;13:3290–312.
Sherr CJ. The ink4a/arf network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–7.
Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. 2016;1:e86976.
Hiroaki K, Emi S, Naohiro N, Takayasu M, Yoji AM, Yuki Y, et al. Failure to sense energy depletion may be a novel therapeutic target in chronic kidney disease. Kidney Int. 2019;95:123–37.
Afshinnia F, Nair V, Lin J, Rajendiran TM, Soni T, Byun J, et al. Increased lipogenesis and impaired beta-oxidation predict type 2 diabetic kidney disease progression in American indians. JCI Insight 2019;4.
Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, Decleves AE, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888–99.
Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. 2022;18:611–27.
Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. 2021;9:645593.
Guo J, Zheng HJ, Zhang W, Lou W, Xia C, Han XT, et al. Accelerated kidney aging in diabetes mellitus. Oxid Med Cell Longev. 2020;2020:1234059.
Gao P, Zou X, Sun X, Zhang C. Cellular senescence in metabolic-associated kidney disease: an update. Cells 2022;11:3443.
Haythorne E, Lloyd M, Walsby-Tickle J, Tarasov AI, Sandbrink J, Portillo I, et al. Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic beta-cells. Nat Commun. 2022;13:6754.
de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305:2532–9.
Afkarian M, Sachs MC, Kestenbaum B, Hirsch IB, Tuttle KR, Himmelfarb J, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol. 2013;24:302–8.
KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007;49:S12–154.
Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Ren Physiol. 2008;295:F1563–73.
Wiley CD. Role of senescent renal cells in pathophysiology of diabetic kidney disease. Curr Diab Rep. 2020;20:33.
Hickson LJ, Langhi PL, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–56.
Wang Y, Wang Y, Yang M, Ma X. Implication of cellular senescence in the progression of chronic kidney disease and the treatment potencies. Biomed Pharmacother. 2021;135:111191.
Aguayo-Mazzucato C, Andle J, Lee TJ, Midha A, Talemal L, Chipashvili V, et al. Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 2019;30:129–42.
Azushima K, Kovalik JP, Yamaji T, Ching J, Chng TW, Guo J, et al. Abnormal lactate metabolism is linked to albuminuria and kidney injury in diabetic nephropathy. Kidney Int. 2023;104:1135–49.
Vaupel P, Multhoff G. Revisiting the warburg effect: historical dogma versus current understanding. J Physiol. 2021;599:1745–57.
Zhang Z, Liang W, Luo Q, Hu H, Yang K, Hu J, et al. Pfkp activation ameliorates foot process fusion in podocytes in diabetic kidney disease. Front Endocrinol. 2021;12:797025.
Bellomo R. Bench-to-bedside review: lactate and the kidney. Crit Care. 2002;6:322–6.
Lund J, Aas V, Tingstad RH, Van Hees A, Nikolic N. Utilization of lactic acid in human myotubes and interplay with glucose and fatty acid metabolism. Sci Rep. 2018;8:9814.
Pan Y, Zheng Q, Ni W, Wei Z, Yu S, Jia Q, et al. Breaking glucose transporter 1/pyruvatekinase m2 glycolytic loop is required for cantharidin inhibition of metastasis in highly metastatic breast cancer. Front Pharm. 2019;10:590.
Zwerschke W, Mazurek S, Stockl P, Hutter E, Eigenbrodt E, Jansen-Durr P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J. 2003;376:403–11.
Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13:629–46.
Nacarelli T, Sell C. Targeting metabolism in cellular senescence, a role for intervention. Mol Cell Endocrinol. 2017;455:83–92.
Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55:S9–15.
Yoon YS, Byun HO, Cho H, Kim BK, Yoon G. Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced senescence-associated growth arrest. J Biol Chem. 2003;278:51577–86.
Yoon G, Kim HJ, Yoon YS, Cho H, Lim IK, Lee JH. Iron chelation-induced senescence-like growth arrest in hepatocyte cell lines: association of transforming growth factor beta1 (TGF-beta1)-mediated p27kip1 expression. Biochem J. 2002;366:613–21.
Kuhlbrandt W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015;13:89.
Kang HT, Park JT, Choi K, Kim Y, Choi H, Jung CW, et al. Chemical screening identifies atm as a target for alleviating senescence. Nat Chem Biol. 2017;13:616–23.
Di Domenico F, Barone E, Perluigi M, Butterfield DA. The triangle of death in alzheimer's disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling, and protein homeostasis revealed by redox proteomics. Antioxid Redox Signal. 2017;26:364–87.
Liu Y, Kou D, Chu N, Ding G. Cathelicidin-BF attenuate kidney injury through inhibiting oxidative stress, inflammation and fibrosis in streptozotocin-induced diabetic rats. Life Sci. 2020;257:117918.
Chen X, Hao B, Li D, Reiter RJ, Bai Y, Abay B, et al. Melatonin inhibits lung cancer development by reversing the warburg effect via stimulating the SIRT3/PDH axis. J Pineal Res. 2021;71:e12755.
Datta D, Anbarasu K, Rajabather S, Priya RS, Desai P, Mahalingam S. Nucleolar GTP-binding protein-1 (NGP-1) promotes G1 to S phase transition by activating cyclin-dependent kinase inhibitor p21 Cip1/Waf1. J Biol Chem. 2015;290:21536–52.
Hendrayani SF, Al-Khalaf HH, Aboussekhra A. Curcumin triggers p16-dependent senescence in active breast cancer-associated fibroblasts and suppresses their paracrine procarcinogenic effects. Neoplasia. 2013;15:631–40.
Di Vincenzo A, Bettini S, Russo L, Mazzocut S, Mauer M, Fioretto P. Renal structure in type 2 diabetes: facts and misconceptions. J Nephrol. 2020;33:901–7.
Feng M, Tang PM, Huang XR, Sun SF, You YK, Xiao J, et al. TGF-beta mediates renal fibrosis via the Smad3-Erbb4-IR long noncoding RNA axis. Mol Ther. 2018;26:148–61.
You YK, Huang XR, Chen HY, Lyu XF, Liu HF, Lan HY. C-reactive protein promotes diabetic kidney disease in db/db mice via the CD32b-Smad3-mtor signaling pathway. Sci Rep. 2016;6:26740.
Bock F, Shahzad K, Wang H, Stoyanov S, Wolter J, Dong W, et al. Activated protein cameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci USA. 2013;110:648–53.
Madhusudhan T, Wang H, Straub BK, Grone E, Zhou Q, Shahzad K, et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood. 2012;119:874–83.
Acknowledgements
XL acknowledges support from NIH R01 DK129241 and NIH R01 DK126662, and support from DOD Focused Program Award, PR221810.
Author information
Authors and Affiliations
Contributions
X.Lu. performed most experiments and data analysis. J.W. and E.A. performed some of the experiments and data analysis. X.Li. supervised the whole project, data analysis, and manuscript writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
In this study, all methods were performed in accordance with the relevant guidelines and regulations. All experimental procedures complied with the Declaration of Helsinki. The study protocol was reviewed and approved by the Mayo Clinic IACUC (Approval No. A00003756-R24), and written informed consent was obtained from all participants.
Consent to participate
We have obtained written informed consent for publication of the images from human research participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, X., Wu, J., Agborbesong, E. et al. P16-positive senescent cells promote DKD by the dysregulation of glycolysis and mitochondrial metabolism. Cell Death Discov. 11, 355 (2025). https://doi.org/10.1038/s41420-025-02650-2
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02650-2
