
Abstract
Glioblastoma (GBM) is an aggressive and lethal type of brain tumor in human adults. The standard of care offers minimal clinical benefit, and most GBM patients experience tumor recurrence after treatment. In recent years, significant advancements have been made in the development of novel immunotherapies or other therapeutic strategies that can overcome immunotherapy resistance in many advanced cancers. However, the benefit of immune-based treatments in GBM is limited because of the unique brain immune profiles, GBM cell heterogeneity, and immunosuppressive tumor microenvironment. In this review, we present a detailed overview of current immunotherapeutic strategies and discuss the challenges and potential molecular mechanisms underlying immunotherapy resistance in GBM. Furthermore, we provide an in-depth discussion regarding the strategies that can overcome immunotherapy resistance in GBM, which will likely require combination therapies.
Similar content being viewed by others

Introduction
Glioblastoma (GBM), representing approximately half of all primary central nervous system (CNS) malignancies, is the most common primary malignant brain tumor in adults, with an annual incidence of approximately 3 cases per 100,000 people [1]. The standard of care (SOC) therapy for GBM includes maximum safe surgical tumor resection followed by radiotherapy (RT) with concurrent and adjuvant temozolomide (TMZ) chemotherapy [2]. Despite these aggressive treatments, the median overall survival (mOS) of GBM patients remains dismally low, typically ranging from 12–18 months postdiagnosis [3]. The treatment outcomes have remained largely unchanged in recent decades, and most GBM patients experience tumor recurrence. The unique location of GBM tumors in a crucial organ characterized by an immunosuppressive tumor microenvironment (TME) and physical shielding by the blood‒brain barrier (BBB) represents major challenges for GBM treatment and drug delivery [4]. Furthermore, although combination treatment with RT and TMZ can improve the survival of GBM patients [5], this strategy may also trigger TME remodeling to foster a resistant and invasive tumor phenotype [6, 7]. Therefore, novel and effective therapeutic approaches are urgently needed to address these problems and improve GBM patient outcomes.
Cancer immunosurveillance refers to the ability of the immune system to identify and eliminate cancer cells [8]. However, some tumors can evade immunosurveillance through a variety of mechanisms, such as downregulating the expression of tumor antigens and major histocompatibility complex (MHC) molecules, expressing immune inhibitory proteins, and accumulating specific metabolites [9]. In recent years, the field of cancer treatment has undergone significant advancements, with breakthroughs in the development of innovative immunotherapies that strategically modulate the immune system to target tumors [10]. This approach aims to promote cancer eradication by overcoming tumor immunoresistance and offers sustained clinical benefits. Notably, immunotherapy has been shown to eliminate tumors in some types of solid tumors (e.g., melanoma and non-small cell lung cancer) and is recommended as part of the SOC for these tumors [11, 12]. However, the impact of immunotherapy varies across cancer types because of the differences in intrinsic tumor characteristics and the immunosuppressive TME. With respect to GBM, despite limited clinical success, immunotherapy remains a crucial area of ongoing investigation in the field. The promising results of several early clinical trials in GBM and the success in other cancer types offer hope for the development of novel and effective immunotherapies for GBM patients. In this review, we present a detailed overview of the current immunotherapeutic strategies for GBM treatment and discuss the challenges and potential resistance mechanisms of immunotherapy in GBM. Furthermore, we provide an in-depth summary of the strategies that can overcome immunoresistance and improve the antitumor efficacy of immunotherapy in GBM, with a special emphasis on combination treatment options.
Current immunotherapy state for GBM
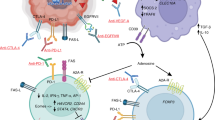
The immune system plays an important role in GBM progression, and a range of different immunotherapeutic approaches have been developed to treat GBM patients [13]. In this section, we discuss the current immunotherapeutic strategies designed for GBM, which include immune checkpoint inhibitors (ICIs), adoptive T-cell therapies, tumor vaccines, and oncolytic viral (OV) therapies (Fig. 1 and Tables 1, 2).

Immunotherapeutic strategies for GBM treatment. Four main immunotherapeutic strategies (immune checkpoint inhibitors, adoptive T-cell therapies, cancer vaccines, and oncolytic viral therapies) have been developed for GBM. A Immune checkpoint inhibitors are monoclonal antibodies that target classical (e.g., PD-1, PD-L1, and CTLA-4) and novel (e.g., LAG-3, TIM-3, TIGIT, and IDO1) immune checkpoints. B Adoptive T-cell therapy involves the infusion of activated (TILs) or engineered (CMV-specific T cells and CAR-T cells) autologous T cells to increase their antitumor activity. C Cancer vaccines use tumor antigens to activate the adaptive immune system of GBM patients, which can be delivered in the form of peptides, DCs, DNA/RNA, and viral vectors. D Oncolytic viral therapies (e.g., HSVs, poliovirus, adenovirus, and retrovirus) use replication-competent viruses to selectively infect and destroy cancer cells. Abbreviations: CAR chimeric antigen receptor, CMV cytomegalovirus, CTLA-4 cytotoxic T lymphocyte associated protein 4, DC dendritic cell, EGFRvIII epidermal growth factor receptor variant III, GBM glioblastoma, HSPPC-96 heat shock protein peptide complex-96, IDO1 indoleamine 2,3-dioxygenase 1, IL-13Rα2 interleukin-13 receptor subunit alpha 2, LAG-3 lymphocyte activation gene-3, PD-1 programmed cell death protein 1, PD-L1 programmed cell death ligand 1, TAAs tumor associated antigens, TIGIT T-cell immunoreceptor with immunoglobulin and ITIM domain, TILs tumor infiltrating lymphocytes, TIM-3 T-cell immunoglobulin and mucin domain 3, TSAs tumor specific antigens
Immune checkpoint inhibitors
Immune checkpoints are a class of regulatory molecules expressed on the surface of certain immune cells, particularly T cells, that are crucial for maintaining immune system homeostasis and preventing autoimmunity [14,15,16]. Under physiological conditions, these molecules are essential for self-tolerance and protecting tissues from immune-related damage [17,18,19,20]. However, in the context of cancer, they can be exploited by tumor cells to evade immune surveillance and suppress antitumor immunity [15, 16, 21, 22]. In the past few decades, several immune checkpoints (Fig. 1A) have been identified, including programmed cell death protein 1 (PD-1) and its ligand PD-L1, cytotoxic T-lymphocyte associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3), T-cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), T-cell immunoglobulin and mucin domain 3 (TIM-3), V-domain Ig suppressor of T-cell activation (VISTA), and indoleamine 2,3-dioxygenase 1 (IDO1) [14, 23,24,25,26]. Blocking these immune checkpoints with monoclonal antibodies, known as ICIs, has shown significant success in treating multiple advanced cancers, such as melanoma, non-small cell lung cancer, colorectal cancer, gastric cancer, and hepatocellular carcinoma [10, 27]. However, a large number of clinical trials have shown minimal clinical benefit for GBM patients [28].
Three phase III trials have been designed to test the antitumor efficacy of the anti-PD-1 antibody nivolumab in GBM patients [29,30,31]. In the CheckMate 498 study, 560 newly diagnosed GBM patients with an unmethylated O6-methylguanine-DNA methyltransferase (MGMT) promoter received RT in combination with nivolumab or TMZ. However, the study did not meet its primary endpoint of OS, with mOS values of 13.4 and 14.9 months for the nivolumab and TMZ groups, respectively [29]. Similarly, the CheckMate 548 study involving 716 newly diagnosed GBM patients with a methylated MGMT promoter also failed to improve the mOS (28.9 vs. 32.1 months) or median progression-free survival (mPFS, 10.6 vs. 10.3 months) upon the addition of nivolumab concurrent with the SOC [30]. In another setting, the CheckMate 143 study compared the antitumor efficacy of nivolumab combined with the antiangiogenic drug bevacizumab in recurrent GBM patients. This study enrolled 369 GBM patients at their first recurrence following RT and TMZ therapy and revealed that nivolumab treatment did not improve patients’ mOS (9.8 vs. 10.0 months), mPFS (1.5 vs. 3.5 months), or objective response rate (ORR, 7.8 vs. 23.1%) [31]. Similarly, another anti-PD-1 antibody, pembrolizumab, has shown limited clinical benefit both as monotherapy and in combination with bevacizumab for recurrent GBM patients in phase I and II trials [32,33,34]. Notably, neoadjuvant anti-PD-1 immunotherapy has demonstrated encouraging outcomes in selected recurrent GBM patients in window-of-opportunity trials [35, 36]. For example, a notable study with 35 recurrent GBM patients demonstrated that neoadjuvant pembrolizumab therapy combined with adjuvant therapy prolonged patient survival compared with patients who received only postsurgical pembrolizumab treatment (mOS, 13.7 vs. 7.5 months; mPFS, 3.3 vs. 2.4 months) [36]. Mechanistic studies have demonstrated that this neoadjuvant pembrolizumab therapy increases T-cell infiltration and interferon-γ (IFN-γ)-related gene expression [36] and enhances chemokine expression and T-cell receptor (TCR) clonal diversity [37]. In addition to these investigations using anti-PD-1 antibodies, the efficacy and safety of PD-L1 antibodies (e.g., durvalumab, atezolizumab, and avelumab) have also been evaluated in GBM patients, with results showing potential antitumor efficacy and favorable tolerability [38,39,40,41].
For the anti-CTLA-4 antibody ipilimumab, a phase II clinical trial has been performed, and the results showed that ipilimumab combined with TMZ treatment did not lead to clinical benefit over TMZ treatment alone (mOS, 22.7 vs. 26.4 months; mPFS, 10.9 vs. 12.5 months) [42]. Another ongoing randomized open-label phase II/III trial (NCT04396860) aims to test the antitumor effect of combining ipilimumab, nivolumab, and RT in newly diagnosed and unmethylated MGMT GBM patients. For recurrent GBM patients, the phase I CheckMate 143 trial revealed that intravenous administration of ipilimumab and nivolumab has no clinical benefit but increases immune toxicity compared with nivolumab treatment alone (mOS, 9.2 vs. 10.4 months; mPFS, 1.5 vs. 1.9 months) [43]. However, one phase I clinical trial in recurrent GBM patients involving the intracerebral administration of ipilimumab and nivolumab showed a promising survival benefit (mOS, 8.9 months; mPFS, 2.7 months), encouraging further investigations [44].
In addition to “classic” immune checkpoints, novel targets such as LAG-3, TIM-3, TIGIT, and IDO1 are actively under investigation for GBM treatment. Given that LAG-3 is an early marker of exhausted T cells, treatment with anti-LAG-3 drugs might offer therapeutic benefits in cancer patients [45]. In the context of GBM, two phase I clinical trials (NCT02658981 and NCT03493932) are underway to explore the antitumor effect of the LAG-3 inhibitor relatlimab (BMS-986016) alone or in combination with nivolumab in recurrent or newly diagnosed GBM patients. TIM-3 is a coinhibitory molecule expressed on immune cells, and TIM-3-targeted therapies are currently under investigation in various cancer types, including GBM [46]. For example, treatment with the TIM-3 inhibitor sabatolimab combined with the PD-1 inhibitor spartalizumab and stereotactic radiosurgery is underway for recurrent GBM patients (NCT03961971). TIGIT is a T-cell coinhibitory receptor that is expressed mainly by T cells and natural killer (NK) cells [47]. Preclinical studies have shown that inhibition of TIGIT improves the antitumor immune response [47]. A phase 0/I study (NCT04656535) is recruiting recurrent GBM patients to evaluate the safety and tolerability of the anti-TIGIT antibody domvanalimab (AB154) combined with the anti-PD-1 antibody zimberelimab (AB122). In addition, the antitumor efficiency of IDO1 inhibitors (e.g., BMS-986205 and indoximod) combined with RT and TMZ is also currently under investigation in different clinical trials (NCT04047706 and NCT02052648) for newly diagnosed GBM patients.
T-cell therapies
Adoptive T-cell therapy is a personalized cancer immunotherapy technique in which a patient’s T cells are isolated, expanded ex vivo and then reinfused back into the patient to target tumors [48]. Before reinfusion, patients undergo a lymphodepleting regimen with lymphocyte-directed chemotherapy to increase treatment effectiveness by creating a favorable environment for infused cells [48]. An increasing number of clinical trials have demonstrated a transient antitumor response with manageable side effects, providing hope for the development of effective adoptive T-cell therapies for GBM. Here, we discuss the current status of adoptive T-cell therapies, including tumor-infiltrating lymphocyte (TIL) therapy, cytomegalovirus (CMV)-specific T-cell therapy, and chimeric antigen receptor (CAR)-T-cell therapy, in GBM (Fig. 1B).
Tumor-infiltrating lymphocyte therapy
The autologous TILs for adoptive T-cell therapy are prepared by culturing a resected tumor specimen in a high concentration of recombinant interleukin-2 (IL-2), along with IL-15 and IL-21 if necessary, then selecting and expanding them in vitro and transferring them back to the patient [49]. This is a time-consuming process with a low success rate, and its application is limited. A pilot study revealed that autologous TILs combined with IL-2 had limited antitumor effects on malignant gliomas, potentially due to the heterogeneity of TILs in terms of TCR diversity and exhaustion levels [50]. Two other phase I clinical trials (NCT05333588 and NCT04943913) are currently recruiting GBM patients to evaluate the safety of TIL therapy. In addition, engineered autologous TILs that secrete antibodies targeting PD-1 (PD-1-TILs, aiming to prevent infused T-cell exhaustion induced by PD-1-mediated immunosuppression) are well tolerated, with no unexpected high-grade adverse events in GBM patients, and show improved antitumor efficacy compared with that of normal TILs, representing a new TIL treatment strategy for GBM [51].
Cytomegalovirus-specific T-cell therapy
Autologous CMV-specific T-cell therapy is another feasible adoptive T-cell therapy since the majority of GBM tumors, but not the surrounding normal brain tissues, express CMV antigens, which contribute to GBM progression [52]. To this end, peripheral blood mononuclear cells are isolated from the blood and expanded in vitro with synthetic CMV peptides to generate CMV-specific T cells, which are then reinfused back into the patient [52]. A phase I study revealed that autologous CMV-specific T cells are safe for primary GBM patients and provide encouraging clinical evidence for improving patient survival if therapy is offered before tumor recurrence (mOS, 23.0 vs. 14.0 months) [53]. However, larger controlled trials are needed to reproduce these observations.
Chimeric antigen receptor-T-cell therapy
A significant advancement in adoptive cell therapy is the development of CAR-T-cell therapy [54]. The autologous T cells can be genetically engineered to express CARs, which combine the antigen recognition domains of antibodies with T-cell activation domains derived from the CD3ζ chain and costimulatory receptors, such as CD28 and 4-1BB [55]. This enables CAR-T cells to specifically target tumor antigens in an MHC-independent manner, thereby bypassing antigen presentation and enhancing their ability to recognize and kill cancer cells [55]. Despite the remarkable clinical response to CAR-T-cell therapy in patients with hematologic malignancies, such as acute lymphoblastic leukemia [56] and diffuse large B-cell lymphoma [57], extending this strategy to solid tumors (e.g., GBM) is still challenging. However, significant work has aimed to develop CAR-T-cell therapies for GBM. Here, we discuss several tumor antigens, such as interleukin-13 receptor subunit alpha 2 (IL-13Rα2) and epidermal growth factor receptor variant III (EGFRvIII), that have been targeted to develop CAR-T-cell therapies for GBM treatment.
IL-13Rα2
Initial efforts in CAR-T-cell therapy focused on IL-13Rα2 since it is expressed in more than 75% of GBM cases [58]. A pilot study evaluated the safety and feasibility of CD8+ CAR-T cells targeting IL-13Rα2 in three recurrent GBM patients, and the results revealed a good safety profile and transient antitumor activity [59]. Another investigation of a recurrent GBM patient used dual-route CAR-T-cell infusions, including intracranial infusion into the resection cavity followed by intrathecal delivery into the ventricular system, which led to a favorable clinical response with the regression of all intracranial and spinal tumors for a duration of 7.5 months without severe toxicity [60]. Following these two breakthrough studies, Brown et al. conducted a phase I clinical trial in 65 patients with recurrent high-grade glioma (the majority of whom had recurrent GBM) by administering IL-13Rα2-targeted CAR-T cells through three different routes, including intratumoral (ICT), intraventricular (ICV), and dual ICT/ICV. This study confirmed the feasibility and safety of locoregional CAR-T-cell administration. Stable disease or better clinical outcomes were observed in 50% of patients. The mOS for recurrent GBM patients increased to 10.2 months from 7.7 months after dual ICT/ICV IL-13Rα2-targeted CAR-T-cell treatment [61]. Moreover, another phase I trial explored the feasibility and safety of off-the-shelf allogeneic IL-13Rα2-targeted CAR-T cells that were genetically engineered to resist treatment with glucocorticoids, which are commonly prescribed to control cerebral edema and inflammation in GBM, in six patients with unresectable recurrent GBM [62]. The results revealed that local intracranial administration of IL-13Rα2-targeted CAR-T-cell therapy in combination with recombinant human IL-2 was well tolerated and resulted in transient tumor reduction or necrosis at the infusion site in four patients [63]. Although all of these patients experienced GBM recurrence during the study, these findings provide a new direction for adoptive therapy using off-the-shelf, zinc finger-modified, and/or glucocorticoid-resistant CAR-T cells. Together, these findings highlight the potential of IL-13Rα2-targeted CAR-T-cell therapy for recurrent GBM, and further investigations are merited.
EGFRvIII
EGFRvIII is a mutated variant of the EGFR with deletion of exons 2-7 of the EGFR gene [64]. EGFRvIII is a tumor-specific protein in GBM that represents the predominant form of EGFR in tumors, with approximately half of the EGFR-amplified GBM cases expressing this variant [65]. Despite the accumulation of promising preclinical data, the clinical efficacy of CAR-T cells targeting EGFRvIII in GBM patients remains limited. In a first-in-human phase I clinical trial, ten patients with recurrent EGFRvIII+ GBM received a single intravenous infusion of autologous EGFRvIII-directed CAR-T cells. The treatment was proven to be safe and feasible (without cross-reactivity to wild-type EGFR, cytokine-release syndrome, or neurotoxicity) and was shown to downregulate tumoral EGFRvIII and upregulate immunosuppressive factors (e.g., IDO1 and PD-L1) and regulatory T cells (Tregs). However, the mOS of these patients was not affected by this therapy [66]. The treatment selective pressure induced by EGFRvIII antigen escape might contribute to the low efficacy of EGFRvIII-directed CAR-T-cell therapy in GBM patients. To address this issue, an engineered T-cell product (named CARv3-TEAM-E) has been developed to target EGFRvIII through a second-generation CAR and secrete a T-cell-engaging antibody molecule (TEAM, a bispecific antibody) that can recognize wild-type EGFR in GBM cells and CD3 in T cells. The intraventricular administration of CARv3-TEAM-E resulted in a promising safety profile and dramatic radiographic tumor regression. However, it should be noted that the antitumor responses were transient in two out of three patients [67]. Given the heterogeneous expression of target antigens in GBM cells, targeting multiple antigens could be a possible direction for developing new CAR-T-cell therapies. Indeed, a recent phase I clinical trial in six patients with multifocal recurrent GBM showed that the intrathecal injection of bivalent CAR-T cells targeting both IL-13Rα2 and EGFR led to tumor reduction in all patients, although none met the criteria for ORR [68]. Overall, these first-in-human data prove the efficacy and safety of EGFR-IL13Rα2-targeted CAR-T cells in recurrent GBM.
Cancer vaccines
Cancer vaccines aim to activate the patient’s adaptive immune system against specific endogenous and exogenous tumor antigens and are being explored in GBM [69]. Endogenous antigens, including tumor-specific antigens (TSAs, also known as neoantigens) and tumor-associated antigens (TAAs), are intracellular proteins from tumor cells. Exogenous antigens originate from the infection of exogenous pathogens, such as CMV [70, 71]. Cancer vaccines can be created using “predefined” antigens (known antigens, which include shared antigens expressed in the majority of patient tumors or personalized antigens specific to individual patients) or “anonymous” antigens (unknown antigens, which can be colocalized with antigen-presenting cells (APCs) ex vivo or engineered for reinjection into patients to activate endogenous APCs in situ) [72, 73]. After administration, antigens are presented by APCs to naïve or memory T cells, and these primed T cells then migrate into tumor sites, initiating tumor regression and establishing long-lasting memory responses against tumor recurrence [72, 73]. Various vaccination strategies are under investigation for GBM treatment [69]. In this section, we discuss four different vaccine platforms, including peptide vaccines, dendritic cell (DC) vaccines, DNA/RNA vaccines, and viral vector vaccines, and their use for GBM treatment (Fig. 1C).
Peptide vaccines
Peptide vaccines utilize synthetic peptides to mimic TSA or TAA epitopes, producing new or enhanced tumor-specific T-cell responses [74]. These peptides can be engulfed and presented by APCs to autologous CD4+ and CD8+ T cells after in vivo administration and then circulate to the tumor site, where they exhibit cytotoxicity [74]. Given the inability of these peptides to activate the innate immune system, additional immune adjuvants are usually combined with peptide vaccines, ensuring sufficient costimulatory signals from APCs to elicit a robust T-cell response [75]. Here, we discuss the common peptide vaccines used in GBM, which include rindopepimut (CDX-110), survivin vaccine (SurVaxM), IMA950, heat shock protein peptide complex-96 (HSPPC-96)-specific vaccine, and personalized neoantigen vaccines.
CDX-110
CDX-110 is a 13-amino acid peptide vaccine targeting the EGFRvIII mutation, and it consists of an EGFRvIII-specific peptide coupled with keyhole limpet hemocyanin to increase immunogenicity. Despite the promising results from phase II clinical trials [76,77,78,79], the phase III trial (NCT01480479) with CDX-110 treatment combined with chemotherapy failed to show clinical benefits in newly diagnosed GBM patients compared with chemotherapy alone (mOS, 20.1 vs. 20.0 months; mPFS, 8.0 vs. 7.4 months) [80]. Notably, approximately 60% of the patients (regardless of receiving CDX-110 treatment) experienced EGFRvIII loss, underscoring the necessity for biopsy-confirmed EGFRvIII expression before enrollment for treatment [80]. In addition, the dynamic nature of EGFRvIII raises concerns about its reliability as a stable target for vaccine development [64], suggesting the necessity of developing novel therapeutic strategies with multitarget potential.
SurVaxM
The SurVaxM vaccine targets survivin, an antiapoptotic protein that is highly expressed in GBM tumors but not in normal brain tissues [81]. An early phase I clinical trial (NCT01250470) evaluated its safety in 9 survivin+ recurrent glioma patients [82]. Moreover, a recent phase II study (NCT02455557) in newly diagnosed GBM patients demonstrated that SurVaxM treatment combined with TMZ resulted in improved mPFS (11.4 months) and mOS (25.9 months) compared with those of historical controls [83]. A multicenter and randomized controlled phase IIb trial (SURVIVE) is underway to investigate the efficacy and safety of SurVaxM combined with adjuvant TMZ in newly diagnosed GBM patients [84].
IMA950
IMA950 is a multipeptide vaccine consisting of 11 TAAs (9 MHC class I-restricted peptides and 2 MHC class II-restricted peptides) that are overexpressed in GBM cells. The first human phase I trial with IMA950 in newly diagnosed GBM (NCT01222221) demonstrated that treatment with IMA950 in combination with granulocyte‒macrophage colony‒stimulating factor (GM-CSF) was well tolerated and generated potent T-cell immune responses in at least 30% of patients [85]. Similarly, a phase I/II trial (NCT01920191) in 16 newly diagnosed GBM patients revealed good tolerability and immunogenicity when IMA950 was combined with the adjuvant immunostimulant poly-ICLC, a synthetic analog of dsRNA and toll-like receptor 3 ligand [86, 87]. Notably, the mOS of these patients reached 19.0 months, although 4 patients experienced short-term cerebral edema [88]. Although the IMA950/poly-ICLC peptide vaccine has shown no clinical benefit for recurrent GBM [89], a phase II clinical trial (NCT03665545) was designed to evaluate the antitumor efficacy of the IMA950/poly-ICLC vaccine combined with the anti-PD-1 antibody pembrolizumab in recurrent GBM patients [90].
HSPPC-96
Heat shock proteins (HSPs) are overexpressed in many cancers, where they contribute to cancer cell proliferation, differentiation, infiltration, and metastasis [91]. HSPs and autologous tumor antigen polypeptides can form complexes, named HSP-peptide complexes (HSPPCs), to mediate cell endocytosis and antigen presentation by binding to APC membrane receptors, activating CD4+ and CD8+ T cells and triggering immune responses against tumor antigen peptides [92]. The HSPPC-96 vaccine consists of the HSP glycoprotein 96 and its associated cellular neopeptides, which target multiple TSAs [93]. A phase II study evaluated the safety and antitumor efficacy of the HSPPC-96 vaccine in recurrent GBM patients and reported an mOS of 9.9 months [92]. Further studies demonstrated that the HSPPC-96 vaccine can improve the survival of GBM patients who undergo SOC and revealed that peripheral myeloid cell expression of PD-L1 might impact the antitumor efficacy of the vaccine [94].
Personalized neoantigen vaccines
Personalized polypeptide vaccines have been developed by utilizing whole-exome sequencing data to identify patient-specific neoantigens. These immune targets are designed individually according to the specific mutation site, type, and expression of the tumor neoantigen to reduce off-target effects and adverse effects. The European GAPVAC trial [95] and the American NEOVAX trial [96] have shown that these vaccines can stimulate circulating robust immune responses involving CD8+ and CD4+ T cells with a memory phenotype. The GAPVAC trial reported mPFS and mOS values of 14.2 and 29.0 months, respectively, in newly diagnosed GBM patients [95], whereas the NEOVAX trial reported mPFS and mOS values of 7.6 and 16.8 months, respectively, in newly diagnosed and unmethylated GBM patients [96]. However, another phase III trial for recurrent GBM did not meet its primary or secondary endpoints (personalized vaccine vs. placebo: mOS, 8.4 vs. 8.0 months) [97]. Thus, further studies are needed to validate and confirm the clinical benefit of personalized vaccines in GBM patients.
Dendritic cell vaccines
The intrinsic antigen presentation ability of DCs has stimulated researchers to create DC vaccine-based immunotherapies to activate adaptive immune responses [98, 99]. To make the vaccine, autologous DCs are harvested from the peripheral blood directly or differentiated from monocytes or CD34+ hematopoietic stem cells upon stimulation with cytokines, such as IL-4 and GM-CSF [98, 99]. These DCs are subsequently exposed to several forms of antigens, including DNA/RNA, peptides and tumor lysates. These tumor antigen-loaded DCs possess high antigen-presenting efficiency with sufficient exogenous costimulatory signals and can prime CD4+ T cells via peptide-MHC II complexes and CD8+ T cells via peptide-MHC I molecules [98, 99]. To date, several DC vaccines (e.g., DCVax-L, ICT-107, and CMV-DCs) have been tested in GBM patients.
DCVax-L
DCVax-L, an autologous DC vaccine pulsed with autologous tumor lysate ex vivo, is the most studied DC vaccine. The efficacy of DCVax-L was tested in a phase III clinical trial (NCT00045968) for newly diagnosed GBM patients after they received SOC. Patients were randomized at a 2:1 ratio to receive DCVax-L or placebo, with the option of crossover to the DCVax-L group in cases of disease progression or relapse during treatment [100]. Owing to the crossover design, approximately 90% of all GBM patients with recurrence received DCVax-L treatment, which resulted in depletion of the placebo group and necessitated the use of external controls for statistical analysis. Notably, compared with external controls, DCVax-L treatment led to increased mOS for both newly diagnosed GBM patients (19.3 vs. 16.5 months) and recurrent GBM patients (13.2 vs. 7.8 months) [101]. However, these survival data should be interpreted with caution considering the high crossover rate, a shift in the primary endpoint from PFS to OS, and an inappropriate selection of external controls.
ICT-107
ICT-107 is a DC vaccine consisting of autologous, monocyte-derived DCs pulsed with 6 well-known GBM TAAs, including melanoma-associated antigen 1, absent in melanoma 2, human epidermal growth factor receptor 2, tyrosinase-related protein 2, glycoprotein 100, and IL-13Rα2. A phase II study (NCT01280552) demonstrated the safety and immunogenicity of ICT-107 in newly diagnosed GBM patients. Although a modest improvement in PFS was observed in the ICT-107 group compared with the control group (11.2 vs. 9.0 months), the mOS was not significantly affected (17.0 vs. 15.0 months) [102].
CMV-DC vaccines
The CMV-DC vaccine (known as the CMV-pp65 RNA-pulsed DC vaccine) consists of autologous DCs pulsed with mRNA encoding the human CMV matrix protein pp65, which aims to kill GBM cells by stimulating CMV-specific T-cell immunity. Three separate clinical trials using CMV-DC vaccines have been conducted in newly diagnosed and SOC-treated GBM patients [103,104,105]. Notably, nearly one-third of GBM patients receiving CMV-DC vaccines exhibit no tumor recurrence at 5 years after diagnosis [105]. Given that CMV-DCs can trigger a CMV-specific CD8+ T-cell response [103, 104], they further conducted a pilot trial in which newly diagnosed GBM patients received both CMV pp65-specific T cells and a CMV-DC vaccine [106]. This combination treatment resulted in an increase in activated CMV-specific CD8+ T cells, which was correlated with better patient OS. However, further clinical trials should be conducted to confirm the antitumor effect of this treatment strategy in GBM patients.
DNA/RNA vaccines
Nucleic acid-based vaccines, including DNA (as plasmids) and RNA (as mRNA) vaccines, represent a new area of vaccine development for cancer treatment [107, 108]. DNA vaccines are generated by incorporating genes that encode TAAs into a bacteria-derived plasmid, which can induce robust CD4+ and CD8+ T-cell responses by presenting antigens on MHC class I/II molecules and producing humoral responses [107].
VXM01 is a DNA plasmid vaccine containing an attenuated strain of Salmonella typhimurium that encodes murine vascular endothelial growth factor receptor-2 (VEGFR-2), a protein that contributes to tumor angiogenesis [109]. A phase I clinical trial (NCT02718443) evaluated the antitumor efficiency of VXM01 in recurrent GBM patients who do not respond to SOC and revealed that VXM01 was well tolerated and elicited a VEGFR-2-specific T-cell immune response. Notably, a subset of patients with prolonged survival expresses lower intratumoral PD-L1, suggesting that the combination of VXM01 with anti-PD-L1 treatment is beneficial [110]. Another two DNA vaccines, INO-5401 (synthetic DNA plasmids encoding human telomerase reverse transcriptase, Wilms’ tumor gene 1, and prostate-specific membrane antigen), and INO-9012 (synthetic DNA plasmid encoding IL-12), in combination with the PD-1 inhibitor cemiplimab, are being investigated in an ongoing phase I/II trial (NCT03491683) for newly diagnosed GBM patients [111]. The safety, immunological effectiveness, and potential survival advantages of these treatments have been supported by interim results [112]. However, further trials are needed to validate the clinical benefits of these DNA plasmid vaccines in GBM patients.
mRNA vaccines transduce mRNA into cells, especially APCs, to generate translated peptides. This type of vaccine has significant safety advantages because it has the properties of rapid degradation and minimal risk of infection or insertional mutations [108]. To improve their preservation and membrane permeability, they are usually delivered by various packaging nanoparticles, such as lipid nanoparticles. One preclinical study demonstrated that mRNA-loaded lipid nanoparticles can induce a favorable antitumor response and sensitize poorly immunogenic murine GBM tumors to ICIs [113]. However, no clinical trials using mRNA vaccines to treat GBM have yet been reported.
Viral vector vaccines
Viral vector vaccines are novel vaccines that have been utilized for antigen delivery in clinical settings [114]. Genetically modified viral vectors can lose their toxicity and contain genes encoding targeted antigens, which, in turn, generate both innate and adaptive immune responses [114]. VBI-1901 is a viral vector vaccine that targets two highly immunogenic CMV antigens (glycoprotein B and phosphoprotein 65). The safety and antitumor efficacy of VBI-1901 in recurrent GBM patients have been evaluated in a randomized controlled phase IIa clinical trial (NCT03382977). Early data from this trial demonstrated that the mOS was 12.9 months, and the 12-month OS rate was 62.5% in 16 patients treated with the highest dose of VBI-1901 [115].
Oncolytic viral therapies
OV therapy represents another class of immunotherapies being intensively investigated for GBM [116] (Fig. 1D). OVs are replication-competent viruses that selectively infect and replicate in cancer cells, causing lysis of GBM cells followed by the release of viral pathogen-associated molecular patterns, damage-associated molecular patterns, TAAs, and proinflammatory cytokines into the TME, which, in turn, induce antitumor immune responses and transform the TME from “cold” to “hot” [117]. Specifically, OVs can facilitate the activation and migration of APCs to activate cytotoxic CD8+ T cells, enhancing the overall immune response against GBM.
Herpes simplex virus (HSV) is the first engineered viral strain for GBM treatment in a murine model [118]. HSV-1, a neurotropic virus from the Herpesviridae family, is a double-stranded DNA virus. Several genetically engineered versions (e.g., HSV-1716, G207, and G47∆) of HSV-1 have been evaluated in GBM. Notably, both copies of the RL1 gene encoding the virus protein ICP34.5 were deleted in all HSV recombinants, resulting in increased tumor selectivity [119, 120]. HSV-1716 is the first-generation HSV, and its safety has been proven in several phase I clinical trials [121,122,123]. The second generation of the HSV construct, G207, featuring the deletion of both copies of the RL1 gene and an inactivating insertion of the UL39 gene encoding infected cell protein 6 (ICP6), can generate preferential replication in dividing cells [124, 125]. The results from several clinical trials demonstrated that G207 is safe for GBM patients [126,127,128]. G47∆ (Teserpaturev) represents the third generation of oncolytic HSV, which is constructed on the basis of G207 with an additional deletion of the α47 gene, thus increasing viral replication and triggering antitumor immune responses via the upregulation of MHC I molecules [129]. A phase I/II trial at the University of Tokyo confirmed the safety of G47∆ in recurrent GBM patients [129, 130], followed by a phase II trial revealing its antitumor efficacy following intratumoral injections in residual or recurrent GBM [131]. As a result, G47∆ has been granted conditional time-limited approval by Japan’s Pharmaceuticals and Medical Devices Agency for brain tumor treatment. Moreover, a recent study developed a new version of G47∆ expressing IL-2 (G47Δ-mIL2), which can significantly prolong the survival of different GBM mouse models associated with increased intratumoral CD8+ T cells, suggesting that G47Δ-mIL2 may represent a new direction of HSV treatment for GBM patients [132], but further clinical trials are needed.
Polioviruses are positive- and single-stranded RNA viruses, and PVSRIPO is a nonpathogenic poliovirus/rhinovirus chimeric virus that targets tumor cells via the poliovirus receptor CD155, which is overexpressed in GBM tumors [133]. A phase I clinical trial (NCT01491893) demonstrated that intratumoral infusion of PVSRIPO in recurrent GBM patients was tolerated and produced increased mOS (12.5 vs. 11.3 months) and 24-month (21% vs. 14%) and 36-month (21% vs. 4%) survival rates compared with those of historical controls [134]. Currently, several clinical trials, including a phase II trial (NCT02986178) evaluating PVSRIPO as a monotherapy and phase I/II (NCT03973879) and phase II (NCT04479241) trials investigating the combination of PVSRIPO with the anti-PD-L1 antibody atezolizumab or the anti-PD-1 antibody pembrolizumab, are underway in GBM patients.
Adenoviruses are nonenveloped, double-stranded DNA viruses with icosahedral structures that have been extensively studied for cancer treatment [135]. DNX-2401 (known as tasadenoturev or Delta-24-RGD) is an engineered oncolytic adenovirus designed to selectively target and replicate in cancer cells with aberrant retinoblastoma (Rb) pathways. Specifically, DNX-2401 contains a genetic modification in the E1A gene, which is a 24 bp deletion responsible for Rb binding, enabling this adenovirus to replicate selectively in cancer cells with defects in the Rb pathway and decreasing virus replication in normal cells. In addition, it involves the insertion of an Arg-Gly-Asp (RGD) peptide motif, which can improve its attachment to GBM cells by binding to αV integrins. Phase I clinical trials in recurrent GBM patients demonstrated that DNX-2401 was safe and had significant antitumor effects on long-term survival in some patients [136, 137]. Furthermore, DNX-2401 in combination with TMZ was also well tolerated [138], whereas the addition of IFN-γ to DNX-2401 resulted in poor tolerability without an additional clinical benefit [139]. CRAd-S-pk7 is another glioma-tropic oncolytic adenovirus that contains the tumor-specific survivin promoter (S) and a fiber protein polylysine modification (pk7), with potential antineoplastic activity [140, 141]. Interestingly, this oncolytic adenovirus can be delivered by neural stem cells (NSCs), which can enhance the glioblastoma stem cell (GSC)-targeting ability of CRAd-S-pk7 by combining the unique tumor tropism of NSCs [142]. The safety and therapeutic potential (mOS, 18.4 months; mPFS, 9.1 months) of NSC-CRAd-S-pk7 in newly diagnosed GBM has been proven by a phase I study (NCT03072134) [143], and further clinical trials are needed to confirm its antitumor efficacy in GBM.
Moreover, adenoviruses can be modified to act as delivery vectors for exogenous genes, such as IL-12, the Fas-chimera transgene, and the HSV thymidine kinase (HSV-TK) gene. Ad-RTS-hIL-12 is an engineered adenoviral vector that delivers IL-12 controlled by the RTS promoter upon the oral administration of veledimex (VDX) [144, 145]. The tolerability and antitumor effect of Ad-RTS-hIL-12 in patients with resected recurrent GBM were evaluated in a phase I clinical trial (NCT02026271), with the results showing that peritumoral injection of Ad-RTS-hIL-12 was tolerated and resulted in an mOS of 12.7 months [146]. VB-111 is a nonreplicating adenovirus-engineered vector that can carry a Fas-chimera transgene, leading to Fas-mediated tumor apoptosis and vascular disruption [147]. In a phase I/II study (NCT01260506), combined treatment with VB-111 and bevacizumab significantly improved survival in recurrent GBM patients compared with treatment with VB-111 alone (mOS, 13.8 vs. 7.4 months; mPFS, 3.0 vs. 3.0 months) [148]. However, the subsequent phase III trial (GLOBE, NCT02511405) using combination therapy did not show a clinical benefit in recurrent GBM patients (mOS, 6.8 vs. 7.9 months; mPFS, 3.4 vs. 3.7 months) [149]. Another adenoviral vector that delivers tumoricidal gene is the HSV-TK gene, which converts prodrugs (e.g., valacyclovir, acyclovir, and ganciclovir) into toxic nucleotide analogs to kill tumor cells. The aglatimagene besadenovec (AdV-tk), a nonreplicating adenovirus expressing the HSV-TK gene, was found to be tolerated in phase I trials [150,151,152]. Two phase II trials demonstrated favorable PFS and OS associated with AdV-tk-based therapy in GBM patients [153, 154]. Moreover, a randomized and controlled phase III study also revealed that, compared with SOC treatment, AdV-tk gene treatment extended the survival of GBM patients (mOS, 12.9 vs. 8.6 months) [155]. However, another randomized phase III trial (ASPECT) demonstrated that sitimagene ceradenovec, another adenoviral vector used to deliver the HSV-TK gene, failed to improve survival in newly diagnosed GBM patients (mOS, 16.6 vs. 15.1 months; mPFS, 10.3 vs. 8.9 months), although it increased the time to death or reintervention [156]. Therefore, adenovirus-based delivery of HSV-TK needs to be further investigated for GBM treatment. However, a phase I study (NCT01811992) in newly diagnosed GBM patients revealed an mOS of 21.3 months when AdV-tk was combined with another adenovirus expressing FMS-like tyrosine kinase 3 ligand (Flt3L) [157].
In addition to adenovirus, the HSV-TK gene can be delivered by retroviruses. However, a phase III study revealed that retrovirus-based HSV-TK gene therapy did not generate significant clinical benefit for GBM patients compared with SOC treatment (mOS, 12.0 vs. 11.8 months; mPFS, 6.0 vs. 6.1 months) [158]. Vocimagene Amiretrorepvec (Toca 511) is another retrovirus-based treatment iteration that encodes a yeast cytosine deaminase gene and can convert the prodrug 5-fluorocytosine (5-FC) to toxic 5-fluorouracil (5-FU) [159]. Although the phase I trial resulted in encouraging efficacy data and a good safety profile [160, 161], the subsequent phase II/III trial failed to confirm the survival benefit of Toca 511/FC in GBM patients compared with SOC treatment (mOS, 11.1 vs. 12.2 months; mPFS, 6.0 vs. 6.1 months) [162]. Therefore, certain approaches (e.g., selecting the most effective virus type, optimizing the delivery method, refining genetic engineering, and combining other immunotherapy strategies) should be taken into consideration for enhancing the efficacy of virus-based therapies in GBM.
Challenges and resistance mechanisms of immunotherapies in GBM
Despite the rapid development of immunotherapy for GBM treatment, only a few strategies provide clinical benefits. Multiple processes, including the unique anatomical brain location protected by the BBB, the heterogeneity of GBM cells between and within patients, and the immunosuppressive TME, restrain immune system activity in GBM patients [163]. In this section, we summarize the challenges of immunotherapy in GBM and discuss the multiple mechanisms underlying immunotherapy resistance in GBM in detail (Fig. 2).

Challenges and molecular mechanisms of immunotherapy in GBM. Multiple processes restrain the response of GBM to immunotherapies, including the immunologically distinct brain (e.g., unique lympatic draining pathway, anatomical location protected by the blood‒brain barrier, and resident microglia), the immunosuppressive TME (e.g., T-cell dysfunction and exhaustion, and immunosuppressive myeloid cells, such as TAMs and MDSCs), and the characteristics of GBM cells (e.g., intertumoral and intratumoral heterogeneity, highly invasive nature, low TMB, and context-dependent GBM cells and their associated TAM biology). GBM glioblastoma, LAG-3 lymphocyte-activation gene 3, MDSCs myeloid-derived suppressor cells, PD-1 programmed cell death protein 1, TAM tumor-associated macrophage and microglia, TILs tumor-infiltrating lymphocytes, TIM-3 T-cell immunoglobulin and mucin domain 3, TMB tumor mutation burden, TME tumor microenvironment, Tregs regulatory T cells
“Immunologically distinct” brain
The brain has historically been considered an “immune privileged” organ, given that many studies have shown that heterotopic tissues are not rejected by animal brains, whereas the same tissues are eradicated by the host immune system when they are introduced into peripheral tissues [164, 165]. This is attributed to the lack of lymphatic drainage in the brain [166,167,168,169,170]. However, more studies have demonstrated that the brain engages with the immune system. Routes for antigenic egress from the brain to deep cervical lymph nodes were identified in the 1980s [167, 168, 170]. In 2015, the discovery of the presence of a dural lymphatic system within the CNS revealed a mechanism for draining CNS antigens from cerebrospinal fluid into cervical lymph nodes, facilitating immune surveillance [166, 169]. Moreover, the glial-lymphatic (glymphatic) pathway has been recently identified, providing insight into how fluids and solutes are cleared from the brain [171, 172]. Specifically, cerebrospinal fluid enters the brain along arterial perivascular spaces, engages in the interstitium through aquaporin 4 water channels, and then exits through venous perivascular spaces, draining into deep cervical and lumbar lymph nodes [172]. Therefore, the brain is an immunologically dynamic organ and should be described as “immunologically distinct” rather than “immune privileged” (Fig. 2).
The BBB, which consists of capillary endothelial cells, the basement membrane, the perivascular space, and glia limitans, is one of the inherent obstacles that impedes the entry of immune cells and immunotherapeutic drugs into the brain [173]. The BBB serves as a structural barrier that restricts the diffusion of large and hydrophilic molecules into the CNS. It also protects the brain from most blood-borne pathogens and exogenous compounds (i.e., drugs and neurotoxins) that might damage the CNS. Although the BBB is relatively impenetrable to circulating immune cells and antibodies in a quiescent state, peripheral immune cells can cross the BBB and execute vigorous inflammatory responses when danger signals are detected, providing the foundation for immunotherapy against brain tumors [174, 175]. In GBM, the BBB is partially disrupted with increased permeability due to increased angiogenesis and the release of cytokines and chemical mediators [176]. Although BBB disruption in GBM primarily occurs within the tumor core, which may enhance therapeutic delivery, the intact BBB at tumor edges can still impede the distribution of immune-related drugs [177]. To address this problem, an increasing number of studies have aimed to improve drug penetration ability by increasing drug liposolubility with liposomes or by remodeling the BBB properties via the regulation of efflux pumps and tight junctions. In addition, local administration of immunotherapeutic drugs (e.g., intrathecal drug administration, convection-enhanced delivery, and the use of implantable pharmaceutical formulations) might be another effective method for enhancing drug delivery [178]. However, randomized clinical trials are needed to evaluate the safety and effectiveness of these approaches.
Another unique immune characteristic of the brain is its resident microglia, which originate from yolk sac myeloid progenitors during development [179]. Microglia may undergo phenotypic changes in response to inflammatory stimuli [179]. Although the role of resident microglia in adaptive immunity is not fully understood, they perform a variety of critical functions, including phagocytosis, cytotoxicity, and immune regulation [179]. In GBM, resident microglia can be polarized from proinflammatory to anti-inflammatory phenotypes, which exert multiple protumorigenic activities (e.g., promoting GBM cell proliferation and invasion and stimulating angiogenesis) and inducing immunotherapy resistance. These immunosuppressive microglia inhibit effector T-cell infiltration, proliferation, and immune reactivity via distinct mechanisms, thus contributing to tumor immune evasion [180].
Immunosuppressive tumor microenvironment
GBM is considered an immunologically ‘cold’ tumor largely due to the low number of TILs and other immune effector cell types, which are related to poor responses to immunotherapies, such as ICIs [181]. Additionally, GBM can also induce systemic immunosuppression by sequestering naïve T cells within the bone marrow, primarily through the downregulation of sphingosine-1-phosphate receptor 1, which is crucial for T-cell egress from the thymus and secondary lymphoid organs into the bloodstream [182].
Within the GBM TME, many immunosuppressive cells, such as tumor-associated macrophages and microglia (TAMs), myeloid-derived suppressor cells (MDSCs), and Tregs, together contribute to immunosuppression and GBM cell immune evasion [179, 183, 184] (Fig. 2). Among them, TAMs, which include yolk sac-derived microglia and bone marrow-derived macrophages (BMDMs), constitute the predominant cell population in the GBM TME, representing as many as 50% of the total number of cells in the entire tumor mass [179]. Recent studies demonstrated that TAMs can be educated by tumor cells, which, in turn, promote tumor progression, inhibit antitumor immunity, and induce immunotherapy resistance [34]. Tregs are a subset of T cells that contribute to tumor progression and immunotherapy resistance [185]. Specifically, cancer and microenvironmental cell-secreted IDO1, IL-10, C-C motif chemokine ligand 2 (CCL2) and transforming growth factor beta (TGF-β) promote the expansion of immunosuppressive Tregs within the GBM TME [186]. As a result, these Tregs express immune checkpoint molecules (e.g., PD-1 and CTLA-4) to suppress the effector functions of T cells [184]. MDSCs are a highly heterogeneous population of myeloid cells that contribute to tumor immunosuppression [187]. GBM cell-derived macrophage migration inhibitory factor recruits MDSCs from the bone marrow into the TME [188], and tumor-infiltrating MDSCs can suppress cytotoxic T-cell activity by expressing arginase to reduce TCR expression and induce oxidative stress to secrete reactive oxygen species [189]. Additionally, MDSCs can promote T-cell exhaustion by expressing PD-L1 [190]. Together, these findings highlight that the presence of an immunosuppressive TME represents one of the key challenges for immunotherapy in GBM.
Moreover, recent reports suggest that environmental factors, including age and sex, are associated with GBM onset, severity, and the immune response. The onset of GBM generally occurs later in life, and aging remains a major variable that can impact outcomes [191]. In relation to immunotherapies, recent work has demonstrated changes in the immune microenvironment as a result of age, leading to older individuals being more refractory to ICIs [192]. In addition to age, sex is a major biological variable that impacts GBM growth and therapeutic resistance. From an epidemiological standpoint, males develop GBM more frequently [193] and have poorer outcomes [194]. A recent clinical report also revealed sex differences in MGMT methylation, where a survival advantage was observed only in females [195]. While these differences can be attributed to different signaling networks [196], recent work in preclinical models and human samples has shown that male T cells are more prone to exhaustion and more responsive to ICIs [197], whereas MDSC subset localization is sex biased, with males having an enrichment of monocytic MDSCs in the TME, whereas females having increased granulocytic MDSCs in their circulation [198]. Moreover, these differences were also leveraged for preclinical validation of sex-specific immunotherapies [198]. These observations further support earlier work in which sex differences in microglia also impacted GBM growth [199, 200]. Taken together, age and sex are emerging as key biological variables in GBM and have implications for the GBM immune response and immunotherapy strategies.
Unique GBM cell characteristics
In addition to the immunosuppressive TME, the biological activities of cancer cells also contribute to immunosuppression and immunotherapy resistance in GBM (Fig. 2). First, the complexity and heterogeneity of GBM cells pose significant challenges for immunotherapies. GBM tumors exhibit both intertumoral and intratumoral heterogeneity, impacting their different responses to immunotherapies on the basis of molecular profiles, such as dysregulated p53, Rb, and phosphoinositide 3-kinase pathways [201]. In addition to interpatient heterogeneity, intratumoral variability further increases the difficulty of immunotherapy in GBM. For example, the individual tumor mass harbors a complex and dynamic architecture of tumor cells, exhibiting variability at the epigenetic, transcriptomic, protein, and metabolic levels [202, 203]. Moreover, treatment may induce GBM tumor phenotype switching, and relapsed GBM tumors might have different tumor subtypes and accessible targets than newly diagnosed tumors do [204]. Therefore, developing immunotherapy approaches that specifically target tumor cell subpopulations might be a new direction for GBM treatment.
Second, the highly invasive capacity of GBM cells might contribute to tumor recurrence and treatment resistance [205]. The different invasive potentials of GBM cells increase their intratumoral heterogeneity. Specifically, cells in the tumor core exhibit heightened proliferative capacity, whereas those at the tumor periphery are more prone to infiltration, enabling them to penetrate surrounding normal brain tissues [205]. After infiltrating brain tissues, GBM cells remodel the extracellular matrix, cytoskeleton, and metabolism [206]. Although GBM cells rarely metastasize to distant organs [207], this invasion and infiltration decrease the possibility of complete surgical resection and lead to a high chance of tumor recurrence even after multiple postoperative adjuvant therapies, including immunotherapy.
Third, GBM cells exhibit a low tumor mutation burden (TMB) and consequently have limited neoantigen presentation for effective T-cell recognition [208, 209]. This is primarily because the presentation of neoantigens depends on the abundance of mutations capable of generating neoepitopes [210]. Although some mutations can be immunogenic and presented by APCs, the majority of mutations do not become MHC-presented neoepitopes that can be recognized and targeted by T cells [211]. Moreover, tumor subclones without highly antigenic peptides might evade immune surveillance [211]. The low TMB-induced smaller antigen pool allows fewer immunogenic neoantigens to be exposed following immunotherapy, which increases the challenges of immunotherapy in GBM.
Fourth, GBM cells can express immunosuppressive molecules, such as PD-L1, IDO1, IL-10 and TGF-β, to cause the dysfunction and exhaustion of TILs [212]. For example, TGF-β secreted by GBM cells decreases the expression of the activated receptor natural killer group 2D on CD8+ T cells and NK cells, thereby reducing their cytotoxic effects on GBM cells [213]. Moreover, tumor cells also decrease their MHC expression levels to reduce the likelihood of tumor antigen presentation [214]. On the other hand, context-dependent signaling from GBM cells can affect myeloid cell biology, such as TAM migration, polarization, and activation (Fig. 3), generating context-dependent tumor-TAM symbiotic interactions to promote GBM progression and immunosuppression [212, 215]. For example, PTEN deletion/mutation in GBM cells increases the expression and secretion of lysyl oxidase to promote macrophage infiltration via activation of the β1 integrin-protein-tyrosine kinase 2 signaling axis in macrophages [216]. TP53 gain-of-function mutation leads to the upregulation of CCL2 and tumor necrosis factor α (TNF-α) via nuclear factor-kappa B (NF-κB) signaling, which, in turn, increases the infiltration of microglia and monocyte-derived immune cells [217]. In addition to genetic alterations, metabolic changes resulting from lactate dehydrogenase A (LDHA) overexpression in GBM cells increase CCL2 and CCL7 expression to attract macrophages into the TME [218]. Eventually, these GBM-associated TAMs are polarized toward an immunosuppressive phenotype, thus inhibiting T-cell-mediated antitumor immunity and inducing immunotherapy resistance [216,217,218].

The immunosuppressive TME and tumor-TAM crosstalk in GBM. The immunosuppressive TME of GBM is a highly heterogeneous dynamic system that includes tumor cells (GBM cells and GSCs), low numbers of TILs, high infiltration of immunosuppressive cells (e.g., TAMs, MDSCs, and Tregs), normal brain cells (e.g., astrocytes and neurons), and soluble molecules. Crosstalk between tumor cells and TAMs is an important mechanism within the GBM TME that promotes tumor growth and induces immunosuppression. Under different conditions (e.g., PTEN, P53 and NF1 deletion/mutation, CLOCK and TFPI2 overexpression/amplification, and metabolic changes), tumor cells can secrete various cytokines and chemokines, such as LOX, TNF-α, OLFML3, LGMN, TFPI2, CCL2, CCL5, CCL7, and CX3CL1, to promote the migration and immunosuppressive polarization of TAMs, which in turn promotes tumor progression and immunosuppression. APCs antigen-presenting cells, CCL2 C-C motif chemokine ligand 2, CD162 cluster of differentiation 162, CLOCK circadian locomotor output cycles kaput, CX3CL1 C-X3-C motif ligand 1, CX3CR1 C-X3-C motif chemokine receptor 1, GBM glioblastoma, GSCs glioblastoma stem cells, LDHA lactate dehydrogenase A, LOX lysyl oxidase, LGMN legumain, MDSCs myeloid-derived suppressor cells, NF1 neurofibromin 1, OLFML3 olfactomedin like 3, TAMs tumor-associated macrophages and microglia, TFPI2 tissue factor pathway inhibitor 2, TILs tumor infiltrating lymphocytes, TME tumor microenvironment, TNF-α tumor necrosis factor α, TNFR, CCR2 C-C motif chemokine receptor 2, TNFR TNF-α receptor, Tregs regulatory T cells
Finally, GSCs constitute a subpopulation of GBM cells that are characterized by stem cell-like capabilities, which drive tumor self-renewal and clonal tumor initiation, eventually promoting intratumoral heterogeneity and therapeutic resistance [219]. While GSCs account for only a small fraction (~10%) of the total GBM cells within tumor tissues, a large population of proliferative cancer cells are composed of GSCs [219]. GSCs can escape immune surveillance by recruiting immunosuppressive PD-L1+ macrophages [220]. Crosstalk between GSCs and TAMs is an important mechanism within the GBM TME that promotes the immune escape of GBM tumors [212, 215]. For example, a gain-of-function screen of epigenetic regulators identified circadian locomotor output cycles kaput (CLOCK, a gene amplified in approximately 5% of GBM cases) as a key hit in GSCs that not only promotes stemness but also triggers microglial infiltration and immunosuppressive polarization by transcriptionally upregulating olfactomedin-like 3 and legumain (LGMN). As a result, these microglia promote tumor growth and suppress antitumor immunity [221, 222]. The Kunitz-type protease inhibitor TFPI2 is another example that is amplified in approximately 4% of GBM cases and can regulate GSC–microglia interactions. Specifically, TFPI2 is highly expressed in GSCs, where it not only promotes GSC self-renewal and tumor growth but also triggers microglial infiltration and immunosuppressive polarization to induce immunosuppression [223]. Moreover, neurofibromin 1 (Nf1)-deficient GSCs isolated from tumors of the Nf1 genetically engineered mouse model can produce the chemokines C-X3-C motif ligand 1 and CCL5 to recruit microglia into the TME [224], which ultimately exhibit an immunosuppressive function.
Combinational strategies to overcome immunotherapy resistance
Current observations suggest that a single immunotherapeutic approach is ineffective for GBM, which is highly complex and heterogeneous, as described above. GBM may develop various resistance mechanisms for monoimmunotherapy. For example, tumor vaccines and CAR-T cells can lead to the continuous loss of tumor antigens and increased numbers of inhibitory cells and factors in the TME [66, 77]. The efficacy of ICIs is also limited by the immunosuppressive TME and low immunogenicity of GBM cells [28]. Given the multiple mechanisms that mediate monoimmunotherapy resistance, combining immune-based approaches with SOC or other immune-remodeling strategies represents a new direction for GBM treatment and has the potential to overcome immunotherapy resistance. In this section, we discuss distinct combinational strategies that have the potential to overcome immunotherapy resistance in GBM (Fig. 4).

Strategies for overcoming immunotherapy resistance in GBM. Multiple combination strategies, including SOC combined with immunotherapies, ICIs combined with other types of immunotherapies (e.g., CAR-T-cell therapies, cancer vaccines, and OV therapies), and myeloid cell-targeted therapies combined with ICIs, have been developed to overcome immunotherapy resistance in GBM. APC antigen-presenting cell, CAR chimeric antigen receptor, GBM glioblastoma, ICIs immune checkpoint inhibitors, OV oncolytic virus, RT radiotherapy, SOC standard of care, TMZ temozolomide
Standard of care in combination with immunotherapies
The combination of immunotherapies with chemotherapy, particularly TMZ, is one of the most investigated strategies for GBM treatment. A phase II study in newly diagnosed GBM patients demonstrated that combining the anti-PD-1 antibody pembrolizumab with TMZ and tumor treatment resulted in an mPFS of 12.0 months compared with 5.8 months in case-matched controls [225]. However, the adjuvant anti-CTLA4 antibody ipilimumab combined with TMZ after surgery and chemoradiotherapy did not result in a survival benefit for newly diagnosed GBM patients (mOS, 22.7 vs. 26.4 months; mPFS, 10.9 vs. 12.5 months) [42]. IDO1-targeted ICI therapy is being investigated in combination with TMZ and RT in newly diagnosed GBM patients (NCT04047706 and NCT02052648). Moreover, other immunotherapeutic strategies, including the peptide vaccine SurVaxM [83, 84], the DC vaccine DCVax-L [100], and the oncolytic adenovirus DNX-2401 [138], in combination with TMZ are being evaluated in clinical trials, and some of them have already shown encouraging results in GBM patients.
RT can cause molecular damage (e.g., DNA breaks and base modifications) and trigger immunogenic cell death in GBM cells [226]. With respect to the immune response, RT can promote the release of tumor neoantigens and inflammatory cytokines, recruit effector T cells to the tumor site, and activate the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, laying the foundation for RT in combination with immunotherapies [226]. Recently, several clinical trials have been designed to explore the safety and efficacy of such combination therapy. For example, a phase I study demonstrated that combination treatment with hypofractionated RT (5 × 6 Gy), the anti-PD-1 antibody pembrolizumab, and the anti-VEGF antibody bevacizumab was well tolerated in GBM patients and induced promising survival results (mOS, 13.4 months; mPFS, 7.9 months) [227]. Further clinical studies are needed to explore the optimal dose and fractionation scheme of this combination strategy for GBM patients.
Although the development of a therapeutic approach that combines immunotherapy with SOC is promising, some concerns and associated solutions should be taken into consideration. First, systemic TMZ chemotherapy could cause a reduction in lymphocytes, and for this reason, local TMZ treatment might be a better administration approach when combined with ICIs [228]. Second, patients whose MGMT promoter is not methylated are resistant to TMZ treatment [229], and MGMT promoter methylation may be considered a biomarker for patient selection. Third, immunotherapy efficiency is affected by the dose and treatment timing, which should be determined before clinical trials. Together, the combination of immunotherapy with SOC shows great therapeutic potential, and further clinical trials are needed to validate the efficiency of such a combination treatment strategy in GBM patients.
Immune checkpoint inhibitors in combination with other immunotherapies
Although ICIs are widely used to treat a variety of cancers, such as melanoma and non-small cell lung cancer, the efficacy of ICIs in GBM patients is still disappointing [163, 230]. One of the key reasons for their ineffectiveness is the low immunogenicity of GBM with a relatively low number of TILs. Indeed, the efficacy of ICIs in GBM is highly dependent on the abundance and activation status of TILs, especially T cells [28]. Therefore, other immunotherapeutic approaches aimed at increasing T-cell infiltration and activation, such as CAR-T-cell therapy, cancer vaccines, and OV therapy, might be potential candidates for overcoming ICI resistance in GBM. Here, we discuss the therapeutic potential of combining ICIs with other types of immunotherapies for GBM treatment (Fig. 4).
Combination of CAR-T-cell therapies with immune checkpoint inhibitors
CAR-T-cell therapy, a breakthrough in cancer treatment, has broadened the immunotherapy landscape [55]. However, antigen loss, immunoediting, and adaptive resistance are common phenomena that typically occur after CAR-T-cell therapy [66, 231]. In addition, increased immunosuppression (e.g., upregulation of PD-1 expression) is always accompanied by CAR-T-cell therapy [232, 233]. Therefore, combining CAR-T-cell therapy with other treatment modalities (e.g., ICIs) might be an efficient way to overcome the immunoresistance associated with a single treatment [234]. The clinical exploration of combining CAR-T-cell therapy with ICIs for GBM treatment is still in the early stage. A recent phase I clinical trial (NCT03726515) revealed that the combination of EGFRvIII-CAR-T-cell therapy with anti-PD-1 therapy, pembrolizumab, did not provide clinical benefit for newly diagnosed and EGFRvIII+ GBM patients (mOS, 11.8 months; mPFS, 5.2 months), although the combination strategy was well tolerated in patients [235]. Another phase I clinical trial (NCT04003649) combining IL-13Rα2 CAR-T-cell therapy with or without anti-PD-1 therapy, nivolumab, and anti-CTLA-4 therapy, ipilimumab, is underway, highlighting the potential for GBM treatment via this new combination treatment strategy. Furthermore, engineered CAR-T cells expressing immune checkpoint antibodies (e.g., CAR-T cells expressing anti-PD-1 and anti-CTLA-4 mAbs) may represent a new direction for GBM treatment, and several clinical trials (e.g., NCT02873390, NCT02937844, and NCT03182816) are ongoing.
Combination of cancer vaccines with immune checkpoint inhibitors
Cancer vaccines can increase antitumor immunity by increasing immunogenicity and activating peripheral T cells, which is important for increasing the antitumor efficacy of ICIs [236]. Moreover, the increased expression of immune checkpoints (e.g., PD-L1) after cancer vaccine therapy provides a further theoretical basis for the combination of cancer vaccines with ICIs [77], which has been found to benefit GBM patients [237]. DC vaccines have shown promising results when combined with ICIs in GBM patients. A two-arm randomized trial in recurrent GBM patients (NCT02529072) demonstrated that treatment with a DC vaccine in combination with the anti-PD-1 agent nivolumab resulted in prolonged survival compared with single nivolumab treatment (mOS, 15.3 vs. 8.0 months; mPFS, 6.3 vs. 4.3 months). Moreover, a recent case report revealed that cotreatment with DC vaccines, anti-PD-1, and poly I:C after SOC exhibited profound antitumor efficacy, and the patient remained disease free for 69 months [238]. Another clinical trial combining the DC vaccine autologous tumor lysate-pulsed DC with the anti-PD-1 agent pembrolizumab in progressive and recurrent GBM patients is ongoing (NCT04201873). In addition to DC vaccines, other cancer vaccines have also been used in combination with ICIs. A recent phase II clinical trial (NCT03018288) evaluated the safety and antitumor efficacy of combining the peptide vaccine HSPPC-96 with anti-PD-1, pembrolizumab, and chemoradiotherapy in newly diagnosed GBM patients, but no clinical benefits were observed. The other exciting progress in the field includes current efforts aimed at testing the clinical benefits of a treatment strategy that combines novel tumor vaccines (e.g., personalized vaccines and DNA/RNA vaccines) with ICIs in GBM patients (NCT02287428, NCT05743595, NCT03665545 and NCT03491683).
Combination of oncolytic viral therapies with immune checkpoint inhibitors
OVs are designed to induce immunogenic cell death through direct virus-mediated cytotoxicity in cancer cells, thus releasing tumor antigens and proinflammatory cytokines [117]. OV therapies can not only “heat up” the GBM TME by recruiting and activating TILs but also increase PD-L1 expression in tumors to sensitize them to ICIs [239]. Therefore, combining OV therapies with ICIs is a promising strategy for GBM treatment. Among OVs, adenoviruses have been mostly used in combination with ICIs. A phase I/II clinical trial (NCT02798406) evaluated the antitumor efficiency of the oncolytic adenovirus DNX-2401 combined with the anti-PD-1 antibody pembrolizumab in recurrent GBM patients [240]. Compared with the prespecified control, the combination treatment regimen led to a significantly greater 12-month OS rate (52.7% vs. 20%). Ad-RTS-hIL-12 is an adenovirus engineered with an IL-12 transgene controlled by the RTS promoter, and the results from a phase I trial (NCT03636477) revealed that the combination of Ad-RTS-hIL-12 and the anti-PD-1 agent nivolumab was well tolerated in recurrent GBM patients and resulted in an mOS of 16.9 months [241]. These encouraging results lead to an ongoing phase II clinical trial (NCT04006119) evaluating the efficacy of this combination strategy in recurrent or progressive GBM patients. AdV-tk is another excellent example of an adenovirus suitable for combination with ICIs to treat GBM. A preclinical study demonstrated that combination treatment with AdV-tk and an anti-PD-1 antibody increased intratumoral T-cell infiltration and prolonged the survival of GBM-bearing mice [242]. Encouraged by these preclinical findings, a phase I clinical trial (NCT03576612) evaluating the combination strategy (AdV-tk combined with nivolumab) in GBM patients is underway. In addition to adenoviruses, the antitumor effects of the poliovirus PVSRIPO combined with or without the anti-PD-1 agent pembrolizumab (NCT04479241) or the anti-PD-L1 agent atezolizumab (NCT03973879) in GBM patients are currently being evaluated in two ongoing phase I/II trials.
Combinations of myeloid cell-targeted therapies with immune checkpoint inhibitors
Myeloid cells play crucial roles in regulating GBM progression and immunosuppression and inducing immunotherapy resistance [179, 243]. GBM is considered a “cold” tumor infiltrating high levels of immunosuppressive myeloid cells [212]. TAMs constitute the largest population of myeloid cells in the GBM TME, where their infiltration and immunosuppressive polarization are usually triggered by factors secreted by GBM cells [179]. As a result, these polarized TAMs contribute to several GBM tumor hallmarks, including immunosuppression [215]. Blockade of TAM infiltration and immunosuppression markedly suppresses tumor progression and activates antitumor immunity in GBM mouse models [218, 221,222,223]. These findings suggest that targeting TAMs is a promising antitumor strategy that has the potential to affect ICI therapy efficacy in GBM. Here, we discuss advances in therapeutic strategies that combine myeloid cell-targeted therapies with ICIs in GBM.
The cGAS-STING pathway serves as a sensor of cellular stress that can activate innate immunity and antigen presentation by myeloid cells and is recognized as the most promising therapeutic target for GBM treatment [244,245,246]. Indeed, STING agonists function as potential therapeutic drugs to influence the TME, including the infiltration of inflammatory macrophages and neutrophils and the upregulation of PD-L1 [247]. As a result, the combination of the clinical-grade STING agonist 8803 with an anti-PD-1 antibody markedly enhances the survival of GBM tumor-bearing mice [248]. Histone 3 lysine 27 demethylase (KDM6B) is an epigenetic enzyme that is highly expressed in immunosuppressive myeloid cells (e.g., immunosuppressive macrophages). Inhibition of myeloid cell KDM6B genetically (using myeloid cell Kdm6b-specific knockout mice) or pharmacologically (using the KDM6B inhibitor GSK-J4) sensitizes GBM-bearing mice to anti-PD-1 therapy [249]. LGMN is a protease that is highly enriched in TAMs. Inhibition of LGMN genetically and pharmacologically enhances T-cell-mediated antitumor immunity and synergizes with anti-PD-1 therapy in GBM tumor-bearing mice [250]. In addition to LGMN, sialic acid binding Ig like lectin 9 (SIGLEC9), which is expressed in a unique immunosuppressive macrophage subpopulation from GBM patients who do not respond to anti-PD-1 treatment, has been identified. Deletion of SIGLEC9 in GBM mouse models synergizes with anti-PD-1 or anti-PD-L1 therapy [251]. Similarly, MDSC-targeted therapies have the potential to improve the antitumor efficacy of ICIs. For example, C-X-C motif chemokine receptor 4 (CXCR4) is commonly overexpressed in tumor-associated MDSCs, and anti-CXCR4 therapy can synergize with anti-PD-1 therapy to increase the survival of GBM tumor-bearing mice [252]. Together, these findings suggest that strategies that target myeloid cells can increase the antitumor efficacy of ICIs in GBM mouse models.
Emerging evidence indicates that the symbiotic interaction between GBM cells and myeloid cells is critical for immunotherapy resistance [212, 253]. On the basis of the understanding and progress in the field, here, we summarize recent findings highlighting the therapeutic potential of targeting tumor–TAM symbiosis to increase the effectiveness of ICIs in GBM. For example, the inhibition of GSC–microglia symbiosis via the targeting of CLOCK and its downstream CD162 can activate antitumor immunity and synergize with anti-PD-1 therapy in GBM mouse models [221]. TFPI2 is another key mediator that can modulate GSC–microglia interactions. Blockade of this symbiosis by genetic depletion of TFPI2 in GSCs or pharmacologic inhibition of the TFPI2 receptor CD51 and its downstream STAT6 in microglia activates antitumor immunity and enhances the therapeutic efficiency of anti-PD-1 therapy in animal models of GBM tumors [223]. In addition to these newly identified strategies, we recently reviewed the accumulating approaches that can target tumor-TAM symbiosis to improve the effectiveness of ICIs in GBM [212, 253]. Although these findings highlight the importance and therapeutic potential of targeting myeloid cells and/or tumor-myeloid cell symbiosis to improve the effectiveness of ICIs in GBM, the current knowledge in this field is still at an early stage, and clinical trials are needed to validate this combination therapeutic strategy in GBM patients.
Conclusions and perspectives
GBM is the most aggressive and malignant form of glioma, with an mOS of only 12–18 months [3]. With the revolutionary success of immunotherapy in multiple cancer types, such as FDA-approved pembrolizumab and nivolumab for treating non-small cell lung cancer and melanoma, an increasing number of immunotherapies, including ICIs, adoptive T-cell therapies, tumor vaccines, and OV therapies, have been tested in preclinical and clinical studies for GBM [254]. However, the area of immunotherapy in GBM is facing enormous challenges, and the majority of phase III clinical trials have failed to yield clinically beneficial outcomes. The high failure rate of clinical trials highlights the urgent need to better understand GBM biology, including the unique features of the brain, GBM cell biology, the immunosuppressive TME, and tumor-TME symbiosis [181, 255].
The unique immune features of the brain, including the immunologically quiescent state, the function of the BBB, and the population of resident myeloid cells, contribute to the distinct immunological nature of GBM tumors, raising challenges for immunotherapies [4]. Tumor heterogeneity, including both intertumoral and intratumoral heterogeneity, is another important factor in the failure of immunotherapies. Novel approaches, such as single-cell RNA sequencing and spatial transcriptomics, may help to identify potential powerful immunotherapeutic targets for GBM [256, 257]. Furthermore, the highly immunosuppressive TME accounts for the ineffectiveness of current immunotherapies for GBM patients. Together, the low numbers of TILs and high infiltration of immunosuppressive cells (e.g., TAMs, MDSCs and Tregs) contribute to the immune evasion of GBM cells. Importantly, the interactions between tumor cells and myeloid cells (e.g., tumor-TAM symbiosis) further promote immune escape and immunotherapy resistance in GBM. Many preclinical studies have demonstrated that inhibition of tumor-TAM symbiosis can significantly prolong the survival of GBM tumor-bearing mice and synergize with ICIs, especially anti-PD-1 antibodies [212, 221, 223, 253, 258]. Finally, recent findings of the impact of age and sex may also influence the immune response and outcome and should be integrated into both preclinical studies and powered appropriately in clinical trials. Taken together, additional studies are needed to explore immunotherapy resistance mechanisms in depth and translate these preclinical findings into clinical settings.
Current immunotherapies focus mainly on the effector arm of the immune system, such as reinvigorating the T-cell response by blocking immune checkpoints, using tumor vaccines to activate adaptative immune responses, or directly transferring engineered T cells into the tumor. However, such treatment may not provide optimal results, as certain tumors, including GBM, have a relatively low number of TILs in the TME. Moreover, these lymphocytes can develop into an exhausted state after therapy [259]. Therefore, more attention should be given to enhancing the infiltration of TILs and blocking the function of immunosuppressive factors in the TME. Furthermore, given the inefficiency of a single immunotherapeutic agent, combination treatment, such as combination with SOC or other different immune-based therapies, might be a potential strategy to overcome the immunotherapy resistance observed in GBM patients. Efforts focused on developing combinational therapies and other novel strategies to turn the ‘cold’ GBM TME into a ‘hot’ TME have increasingly been recognized in both preclinical and clinical studies. Novel strategies that simultaneously target innate and adaptive immunity might be an efficient way to achieve better tumor clearance. In addition, identifying novel biomarkers that can predict which population of GBM patients can respond better to specific types of immunotherapies is critical. Given tumor heterogeneity, context-dependent biomarkers and treatment strategies might lead to maximum clinical benefit for GBM patients. Overall, despite many obstacles, immunotherapy is still one of the most promising therapeutic approaches for GBM, and developing novel combinational therapeutic strategies aimed at “heating up” GBM tumors might overcome immunotherapy resistance.
References
Miller KD, Ostrom QT, Kruchko C, Patil N, Tihan T, Cioffi G, et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J Clin. 2021;71:381–406.
Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J Clin. 2020;70:299–312.
Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and european society of neuro-oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22:1073–113.
DeCordova S, Shastri A, Tsolaki AG, Yasmin H, Klein L, Singh SK, et al. Molecular heterogeneity and immunosuppressive microenvironment in glioblastoma. Front Immunol. 2020;11:1402.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Chien CH, Hsueh WT, Chuang JY, Chang KY. Dissecting the mechanism of temozolomide resistance and its association with the regulatory roles of intracellular reactive oxygen species in glioblastoma. J Biomed Sci. 2021;28:18.
Becherini C, Lancia A, Detti B, Lucidi S, Scartoni D, Ingrosso G, et al. Modulation of tumor-associated macrophage activity with radiation therapy: a systematic review. Strahlenther Onkol. 2023;199:1173–90.
Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50.
Kim SK, Cho SW. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front Pharm. 2022;13:868695.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5.
Mamdani H, Matosevic S, Khalid AB, Durm G, Jalal SI. Immunotherapy in lung cancer: current landscape and future directions. Front Immunol. 2022;13:823618.
Knight A, Karapetyan L, Kirkwood JM. Immunotherapy in melanoma: recent advances and future directions. Cancers. 2023;15.
Majc B, Novak M, Kopitar-Jerala N, Jewett A, Breznik B. Immunotherapy of glioblastoma: current strategies and challenges in tumor model development. Cells. 2021;10:265.
Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48.
Gaikwad S, Agrawal MY, Kaushik I, Ramachandran S, Srivastava SK. Immune checkpoint proteins: signaling mechanisms and molecular interactions in cancer immunotherapy. Semin Cancer Biol. 2022;86:137–50.
He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30:660–9.
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51.
Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T-cell tolerance. J Exp Med. 2006;203:883–95.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Zou W, Chen L. Inhibitory B7-family molecules in the tumor microenvironment. Nat Rev Immunol. 2008;8:467–77.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T-cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57.
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–41.
Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T-cell responses. J Exp Med. 2011;208:577–92.
Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. 2002;169:5392–5.
Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal. 2022;20:44.
Arrieta VA, Dmello C, McGrail DJ, Brat DJ, Lee-Chang C, Heimberger AB, et al. Immune checkpoint blockade in glioblastoma: from tumor heterogeneity to personalized treatment. J Clin Investig. 2023;133:e163447.
Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro Oncol. 2023;25:123–34.
Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24:1935–49.
Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1003–10.
Reardon DA, Kim TM, Frenel JS, Simonelli M, Lopez J, Subramaniam DS, et al. Treatment with pembrolizumab in programmed death ligand 1-positive recurrent glioblastoma: results from the multicohort phase 1 KEYNOTE-028 trial. Cancer. 2021;127:1620–9.
Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27:1048–57.
de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020;22:539–49.
Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, et al. Neoadjuvant PD-1 blockade induces T-cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun. 2021;12:6938.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–86.
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470–6.
Awada G, Ben Salama L, De Cremer J, Schwarze JK, Fischbuch L, Seynaeve L, et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: a stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J Immunother Cancer. 2020;8.
Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn G, Lim M, et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J Clin Oncol. 2019;37:2032.
Nayak L, Standifer N, Dietrich J, Clarke JL, Dunn GP, Lim M, et al. Circulating immune cell and outcome analysis from the phase II study of PD-L1 blockade with durvalumab for newly diagnosed and recurrent glioblastoma. Clin Cancer Res. 2022;28:2567–78.
Lukas RV, Rodon J, Becker K, Wong ET, Shih K, Touat M, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol. 2018;140:317–28.
Brown NF, Ng SM, Brooks C, Coutts T, Holmes J, Roberts C, et al. A phase II open label, randomized study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol. BMC Cancer. 2020;20:198.
Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20:674–86.
Duerinck J, Schwarze JK, Awada G, Tijtgat J, Vaeyens F, Bertels C, et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial. J Immunother Cancer. 2021;9:e002296.
Mariuzza RA, Shahid S, Karade SS. The immune checkpoint receptor LAG3: structure, function, and target for cancer immunotherapy. J Biol Chem. 2024;300:107241.
Guo Q, Shen S, Guan G, Zhu C, Zou C, Cao J, et al. Cancer cell intrinsic TIM-3 induces glioblastoma progression. iScience. 2022;25:105329.
Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8:e000957.
Wang J, Shen F, Yao Y, Wang LL, Zhu Y, Hu J. Adoptive cell therapy: a novel and potential immunotherapy for glioblastoma. Front Oncol. 2020;10:59.
Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–75.
Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J Neurooncol. 1999;45:141–57.
Yao Y, Chen D, Tang C, Ji C, Li Z, Qian Q. Safety, efficacy, and biomarker analysis of response to engineered tumor-infiltrating lymphocytes secreting anti-PD-1 antibody in recurrent glioblastoma: an open-label, two-arms, phase 1 study. J Clin Oncol. 2023;41:2042.
Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014;74:3466–76.
Smith C, Lineburg KE, Martins JP, Ambalathingal GR, Neller MA, Morrison B, et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J Clin Investig. 2020;130:6041–53.
Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94:S3–S9.
Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. 2020;38:473–88.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18.
Westin J, Sehn LH. CAR T cells as a second-line therapy for large B-cell lymphoma: a paradigm shift? Blood. 2022;139:2737–46.
Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B, et al. Expression of nine tumor antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. 2007;81:139–48.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–72.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–9.
Brown CE, Hibbard JC, Alizadeh D, Blanchard MS, Natri HM, Wang D, et al. Locoregional delivery of IL-13Ralpha2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. 2024;30:1001–12.
Afshari AR, Sanati M, Aminyavari S, Shakeri F, Bibak B, Keshavarzi Z, et al. Advantages and drawbacks of dexamethasone in glioblastoma multiforme. Crit Rev Oncol Hematol. 2022;172:103625.
Brown CE, Rodriguez A, Palmer J, Ostberg JR, Naranjo A, Wagner JR, et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 2022;24:1318–30.
An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37:1561–75.
Felsberg J, Hentschel B, Kaulich K, Gramatzki D, Zacher A, Malzkorn B, et al. Epidermal growth factor receptor variant III (EGFRvIII) positivity in EGFR-amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res. 2017;23:6846–55.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9:eaaa0984.
Choi BD, Gerstner ER, Frigault MJ, Leick MB, Mount CW, Balaj L, et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N Engl J Med. 2024;390:1290–8.
Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Ralpha2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. 2024;30:1320–9.
Xiong Z, Raphael I, Olin M, Okada H, Li X, Kohanbash G. Glioblastoma vaccines: past, present, and opportunities. EBioMedicine. 2024;100:104963.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Li J, Xiao Z, Wang D, Jia L, Nie S, Zeng X, et al. The screening, identification, design and clinical application of tumor-specific neoantigens for TCR-T cells. Mol Cancer. 2023;22:141.
Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. 2022;3:911–26.
Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21:360–78.
Zhang L, Huang Y, Lindstrom AR, Lin TY, Lam KS, Li Y. Peptide-based materials for cancer immunotherapy. Theranostics. 2019;9:7807–25.
Zhao T, Cai Y, Jiang Y, He X, Wei Y, Yu Y, et al. Vaccine adjuvants: mechanisms and platforms. Signal Transduct Target Ther. 2023;8:283.
Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–33.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9.
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 2015;17:854–61.
Reardon DA, Desjardins A, Vredenburgh JJ, O’Rourke DM, Tran DD, Fink KL, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): results of a double-blind randomized phase II trial. Clin Cancer Res. 2020;26:1586–94.
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomized, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–85.
Tong X, Yang P, Wang K, Liu Y, Liu X, Shan X, et al. Survivin is a prognostic indicator in glioblastoma and may be a target of microRNA-218. Oncol Lett. 2019;18:359–67.
Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65:1339–52.
Ahluwalia MS, Reardon DA, Abad AP, Curry WT, Wong ET, Figel SA, et al. Phase IIa study of SurVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma. J Clin Oncol. 2023;41:1453–65.
Ahluwalia MS, Ciesielski MJ, Reardon DA, Butowski NA, Aiken R, Venur V, et al. A multicenter, randomized controlled phase 2b trial of survivin vaccine SurVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma (SURVIVE). J Clin Oncol. 2024;42:TPS2099–TPS.
Rampling R, Peoples S, Mulholland PJ, James A, Al-Salihi O, Twelves CJ, et al. A cancer research UK first time in human phase I trial of ima950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res. 2016;22:4776–85.
Zhu X, Fallert-Junecko BA, Fujita M, Ueda R, Kohanbash G, Kastenhuber ER, et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol Immunother. 2010;59:1401–9.
Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10.
Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol. 2019;21:923–33.
Boydell E, Marinari E, Migliorini D, Dietrich PY, Patrikidou A, Dutoit V. Exploratory study of the effect of IMA950/Poly-ICLC vaccination on response to bevacizumab in relapsing high-grade glioma patients. Cancers. 2019;11:464.
Dutoit V, Marinari E, Dietrich PY, Migliorini D. Ctim-08. Combination of the ima950/poly-iclc multipeptide vaccine with pembrolizumab in relapsing glioblastoma patients. Neuro Oncol. 2020;22:ii34.
Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat shock proteins and cancer. Trends Pharm Sci. 2017;38:226–56.
Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. 2014;16:274–9.
Caudill MM, Li Z. HSPPC-96: a personalized cancer vaccine. Expert Opin Biol Ther. 2001;1:539–47.
Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O’Rourke DM, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. 2017;23:3575–84.
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565:240–5.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T-cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–9.
Narita Y, Arakawa Y, Yamasaki F, Nishikawa R, Aoki T, Kanamori M, et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. 2019;21:348–59.
Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95.
Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun. 2019;10:5408.
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16:142.
Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. 2023;9:112–21.
Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. 2019;25:5799–807.
Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–9.
Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017;23:1898–909.
Batich KA, Mitchell DA, Healy P, Herndon JE 2nd, Sampson JH. Once, twice, three times a finding: reproducibility of dendritic cell vaccine trials targeting cytomegalovirus in glioblastoma. Clin Cancer Res. 2020;26:5297–303.
Reap EA, Suryadevara CM, Batich KA, Sanchez-Perez L, Archer GE, Schmittling RJ, et al. Dendritic cells enhance polyfunctionality of adoptively transferred T cells that target cytomegalovirus in glioblastoma. Cancer Res. 2018;78:256–64.
Lopes A, Vandermeulen G, Preat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38:146.
Barbier AJ, Jiang AY, Zhang P, Wooster R, Anderson DG. The clinical progress of mRNA vaccines and immunotherapies. Nat Biotechnol. 2022;40:840–54.
Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153:13–9.
Wick W, Wick A, Sahm F, Riehl D, von Deimling A, Bendszus M, et al. VXM01 phase I study in patients with progressive glioblastoma: final results. J Clin Oncol. 2018;36:2017.
Reardon DA, Brem S, Desai AS, Bagley SJ, Kurz SC, De La Fuente MI, et al. Ltbk-01. Ino-5401 and ino-9012 delivered intramuscularly (im) with electroporation (ep) in combination with cemiplimab (regn2810) in newly diagnosed glioblastoma. Neuro Oncol. 2020;22:ii237.
Reardon DA, Brem S, Desai AS, Bagley SJ, Kurz SC, De La Fuente MI, et al. INO-5401 and INO-9012 delivered intramuscularly (IM) with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma (GBM): interim results. J Clin Oncol. 2020;38:2514.
Sayour EJ, Grippin A, De Leon G, Stover B, Rahman M, Karachi A, et al. Personalized tumor RNA loaded lipid-nanoparticles prime the systemic and intratumoral milieu for response to cancer immunotherapy. Nano Lett. 2018;18:6195–206.
Wang S, Liang B, Wang W, Li L, Feng N, Zhao Y, et al. Viral vectored vaccines: design, development, preventive and therapeutic applications in human diseases. Signal Transduct Target Ther. 2023;8:149.
Iwamoto F, Nissen N, Reardon D, Forst D, Lee E, Berthoud T, et al. Ctim-27. peripheral biomarker analysis of T-cell-mediated collagen remodeling correlates with tumor responses in a phase iia trial of vaccine immunotherapeutic candidate (Vbi-1901). Neuro Oncol. 2023;25:v68.
Webb MJ, Sener U, Vile RG. Current status and challenges of oncolytic virotherapy for the treatment of glioblastoma. Pharmaceuticals. 2023;16:793.
Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2:295–300.
Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci USA. 2000;97:2208–13.
Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35.
Holman HA, MacLean AR. Neurovirulent factor ICP34.5 uniquely expressed in the herpes simplex virus type 1 delta gamma 1 34.5 mutant 1716. J Neurovirol. 2008;14:28–40.
Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumor following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11:1648–58.
Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9:398–406.
Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–66.
Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multimutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–43.
Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol. 1988;62:196–205.
Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74.
Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22:1048–55.
Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated preand posttumor resection for recurrent GBM. Mol Ther. 2009;17:199–207.
Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA. 2001;98:6396–401.
Todo T, Ino Y, Ohtsu H, Shibahara J, Tanaka M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat Commun. 2022;13:4119.
Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. 2022;28:1630–9.
Bommareddy PK, Wakimoto H, Martuza RL, Kaufman HL, Rabkin SD, Saha D. Oncolytic herpes simplex virus expressing IL-2 controls glioblastoma growth and improves survival. J Immunother Cancer. 2024;12:e008880.
Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci USA. 2000;97:6803–8.
Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl J Med. 2018;379:150–61.
Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000;18:723–7.
van Putten EHP, Kleijn A, van Beusechem VW, Noske D, Lamers CHJ, de Goede AL, et al. Convection enhanced delivery of the oncolytic adenovirus delta24-RGD in patients with recurrent GBM: a phase I clinical trial including correlative studies. Clin Cancer Res. 2022;28:1572–85.
Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36:1419–27.
Alonso MM, García-Moure M, Gonzalez-Huarriz M, Marigil M, Hernandez-Alcoceba R, Buñales M, et al. Abstract CT027: oncolytic virus DNX-2401 with a short course of temozolomide for glioblastoma at first recurrence: clinical data and prognostic biomarkers. Cancer Res. 2017;77:CT027–CT.
Lang FF, Tran ND, Puduvalli VK, Elder JB, Fink KL, Conrad CA, et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. J Clin Oncol. 2017;35:2002.
Kim JW, Auffinger B, Spencer DA, Miska J, Chang AL, Kane JR, et al. Single dose GLP toxicity and biodistribution study of a conditionally replicative adenovirus vector, CRAd-S-pk7, administered by intracerebral injection to Syrian hamsters. J Transl Med. 2016;14:134.
Ulasov IV, Zhu ZB, Tyler MA, Han Y, Rivera AA, Khramtsov A, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther. 2007;18:589–602.
Ahmed AU, Thaci B, Tobias AL, Auffinger B, Zhang L, Cheng Y, et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J Natl Cancer Inst. 2013;105:968–77.
Fares J, Ahmed AU, Ulasov IV, Sonabend AM, Miska J, Lee-Chang C, et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021;22:1103–14.
Barrett JA, Cai H, Miao J, Khare PD, Gonzalez P, Dalsing-Hernandez J, et al. Regulated intratumoral expression of IL-12 using a rheoswitch therapeutic system((R)) (RTS((R))) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018;25:106–16.
Cai H, Sun L, Miao J, Krishman S, Lebel F, Barrett JA. Plasma pharmacokinetics of veledimex, a small-molecule activator ligand for a proprietary gene therapy promoter system, in healthy subjects. Clin Pharm Drug Dev. 2017;6:246–57.
Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: results of a phase 1 trial. Sci Transl Med. 2019;11:eaaw5680.
Gruslova A, Cavazos DA, Miller JR, Breitbart E, Cohen YC, Bangio L, et al. VB-111: a novel anti-vascular therapeutic for glioblastoma multiforme. J Neurooncol. 2015;124:365–72.
Brenner AJ, Peters KB, Vredenburgh J, Bokstein F, Blumenthal DT, Yust-Katz S, et al. Safety and efficacy of VB-111, an anticancer gene therapy, in patients with recurrent glioblastoma: results of a phase I/II study. Neuro Oncol. 2020;22:694–704.
Cloughesy TF, Brenner A, de Groot JF, Butowski NA, Zach L, Campian JL, et al. A randomized controlled phase III study of VB-111 combined with bevacizumab vs bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro Oncol. 2020;22:705–17.
Germano IM, Fable J, Gultekin SH, Silvers A. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. J Neurooncol. 2003;65:279–89.
Trask TW, Trask RP, Aguilar-Cordova E, Shine HD, Wyde PR, Goodman JC, et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1:195–203.
Chiocca EA, Aguilar LK, Bell SD, Kaur B, Hardcastle J, Cavaliere R, et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to upfront surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29:3611–9.
Ji N, Weng D, Liu C, Gu Z, Chen S, Guo Y, et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget. 2016;7:4369–78.
Wheeler LA, Manzanera AG, Bell SD, Cavaliere R, McGregor JM, Grecula JC, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016;18:1137–45.
Immonen A, Vapalahti M, Tyynela K, Hurskainen H, Sandmair A, Vanninen R, et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomized, controlled study. Mol Ther. 2004;10:967–72.
Westphal M, Yla-Herttuala S, Martin J, Warnke P, Menei P, Eckland D, et al. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomized, open-label, phase 3 trial. Lancet Oncol. 2013;14:823–33.
Umemura Y, Orringer D, Junck L, Varela ML, West MEJ, Faisal SM, et al. Combined cytotoxic and immune-stimulatory gene therapy for primary adult high-grade glioma: a phase 1, first-in-human trial. Lancet Oncol. 2023;24:1042–52.
Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389–401.
Perez OD, Logg CR, Hiraoka K, Diago O, Burnett R, Inagaki A, et al. Design and selection of Toca 511 for clinical use: modified retroviral replicating vector with improved stability and gene expression. Mol Ther. 2012;20:1689–98.
Cloughesy TF, Landolfi J, Vogelbaum MA, Ostertag D, Elder JB, Bloomfield S, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018;20:1383–92.
Cloughesy TF, Landolfi J, Hogan DJ, Bloomfield S, Carter B, Chen CC, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8:341ra75.
Cloughesy TF, Petrecca K, Walbert T, Butowski N, Salacz M, Perry J, et al. Effect of vocimagene amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high-grade glioma: a randomized clinical trial. JAMA Oncol. 2020;6:1939–46.
Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–42.
Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69.
Billingham RE, Brent L, Medawar PB, Sparrow EM. Quantitative studies on tissue transplantation immunity. I. the survival times of skin homografts exchanged between members of different inbred strains of mice. Proc R Soc Lond B Biol Sci. 1954;143:43–58.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–9.
Bradbury MW, Westrop RJ. Factors influencing exit of substances from cerebrospinal fluid into deep cervical lymph of the rabbit. J Physiol. 1983;339:519–34.
Widner H, Jonsson BA, Hallstadius L, Wingardh K, Strand SE, Johansson BB. Scintigraphic method to quantify the passage from brain parenchyma to the deep cervical lymph nodes in rats. Eur J Nucl Med. 1987;13:456–61.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41.
Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J Leukoc Biol. 2006;80:797–801.
Eide PK, Vatnehol SAS, Emblem KE, Ringstad G. Magnetic resonance imaging provides evidence of glymphatic drainage from human brain to cervical lymph nodes. Sci Rep. 2018;8:7194.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra11.
Bechmann I, Galea I, Perry VH. What is the blood‒brain barrier (not)? Trends Immunol. 2007;28:5–11.
Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. 2004;172:550–9.
Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113–21.
Mo F, Pellerino A, Soffietti R, Ruda R. Blood‒brain barrier in brain tumors: biology and clinical relevance. Int J Mol Sci. 2021;22:12654.
Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood‒brain barrier truly disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018;20:184–91.
Ahmed MH, Canney M, Carpentier A, Thanou M, Idbaih A. Unveiling the enigma of the blood‒brain barrier in glioblastoma: current advances from preclinical and clinical studies. Curr Opin Oncol. 2023;35:522–8.
Khan F, Pang L, Dunterman M, Lesniak MS, Heimberger AB, Chen P, Macrophages and microglia in glioblastoma: heterogeneity, plasticity, and therapy. J Clin Investig. 2023;133:e163446.
Dumas AA, Pomella N, Rosser G, Guglielmi L, Vinel C, Millner TO, et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumor microenvironment. EMBO J. 2020;39:e103790.
Bikfalvi A, da Costa CA, Avril T, Barnier JV, Bauchet L, Brisson L, et al. Challenges in glioblastoma research: focus on the tumor microenvironment. Trends Cancer. 2023;9:9–27.
Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24:1459–68.
Mi Y, Guo N, Luan J, Cheng J, Hu Z, Jiang P, et al. The emerging role of myeloid-derived suppressor cells in the glioma immune suppressive microenvironment. Front Immunol. 2020;11:737.
Humphries W, Wei J, Sampson JH, Heimberger AB. The role of tregs in glioma-mediated immunosuppression: potential target for intervention. Neurosurg Clin N Am. 2010;21:125–37.
Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19:116.
Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol. 2012;14:584–95.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485–98.
Alban TJ, Bayik D, Otvos B, Rabljenovic A, Leng L, Jia-Shiun L, et al. Glioblastoma myeloid-derived suppressor cell subsets express differential macrophage migration inhibitory factor receptor profiles that can be targeted to reduce immune suppression. Front Immunol. 2020;11:1191.
Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumor progression. Br J Cancer. 2019;120:16–25.
Dubinski D, Wolfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 2016;18:807–18.
Kim M, Ladomersky E, Mozny A, Kocherginsky M, O’Shea K, Reinstein ZZ, et al. Glioblastoma as an age-related neurological disorder in adults. Neurooncol Adv. 2021;3:vdab125.
Ladomersky E, Zhai L, Lauing KL, Bell A, Xu J, Kocherginsky M, et al. Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin Cancer Res. 2020;26:5232–45.
Ostrom QT, Kinnersley B, Wrensch MR, Eckel-Passow JE, Armstrong G, Rice T, et al. Sex-specific glioma genome-wide association study identifies new risk locus at 3p21.31 in females, and finds sex-differences in risk at 8q24.21. Sci Rep. 2018;8:7352.
Ostrom QT, Rubin JB, Lathia JD, Berens ME, Barnholtz-Sloan JS. Females have the survival advantage in glioblastoma. Neuro Oncol. 2018;20:576–7.
Barnett AE, Ozair A, Bamashmos AS, Li H, Bosler DS, Yeaney G, et al. MGMT methylation and differential survival impact by sex in glioblastoma. Cancers. 2024;16:1374.
Yang W, Warrington NM, Taylor SJ, Whitmire P, Carrasco E, Singleton KW, et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci Transl Med. 2019;11:eaao5253.
Lee J, Nicosia M, Hong ES, Silver DJ, Li C, Bayik D, et al. Sex-biased T-cell exhaustion drives differential immune responses in glioblastoma. Cancer Discov. 2023;13:2090–105.
Bayik D, Zhou Y, Park C, Hong C, Vail D, Silver DJ, et al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov. 2020;10:1210–25.
Toonen JA, Solga AC, Ma Y, Gutmann DH. Estrogen activation of microglia underlies the sexually dimorphic differences in Nf1 optic glioma-induced retinal pathology. J Exp Med. 2017;214:17–25.
Turaga SM, Silver DJ, Bayik D, Paouri E, Peng S, Lauko A, et al. JAM-A functions as a female microglial tumor suppressor in glioblastoma. Neuro Oncol. 2020;22:1591–601.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Yabo YA, Niclou SP, Golebiewska A. Cancer cell heterogeneity and plasticity: a paradigm shift in glioblastoma. Neuro Oncol. 2022;24:669–82.
Eisenbarth D, Wang YA. Glioblastoma heterogeneity at single cell resolution. Oncogene. 2023;42:2155–65.
Larsson I, Dalmo E, Elgendy R, Niklasson M, Doroszko M, Segerman A, et al. Modeling glioblastoma heterogeneity as a dynamic network of cell states. Mol Syst Biol. 2021;17:e10105.
Hatoum A, Mohammed R, Zakieh O. The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag Res. 2019;11:1843–55.
Velasquez C, Mansouri S, Mora C, Nassiri F, Suppiah S, Martino J, et al. Molecular and clinical insights into the invasive capacity of glioblastoma cells. J Oncol. 2019;2019:1740763.
Lun M, Lok E, Gautam S, Wu E, Wong ET. The natural history of extracranial metastasis from glioblastoma multiforme. J Neurooncol. 2011;105:261–73.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Merchant M, Ranjan A, Pang Y, Yu G, Kim O, Khan J, et al. Tumor mutational burden and immunotherapy in gliomas. Trends Cancer. 2021;7:1054–8.
Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. 2023;8:9.
Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumor mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–6.
Pang L, Khan F, Heimberger AB, Chen P. Mechanism and therapeutic potential of tumor-immune symbiosis in glioblastoma. Trends Cancer. 2022;8:839–54.
Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12:7–13.
Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Investig. 2005;85:328–41.
Xuan W, Lesniak MS, James CD, Heimberger AB, Chen P. Context-dependent glioblastoma-macrophage/microglia symbiosis and associated mechanisms. Trends Immunol. 2021;42:280–92.
Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell. 2019;35:868–84.e6.
Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK, Shin YJ, et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019;26:409–25.
Khan F, Lin Y, Ali H, Pang L, Dunterman M, Hsu WH, et al. Lactate dehydrogenase A regulates tumor-macrophage symbiosis to promote glioblastoma progression. Nat Commun. 2024;15:1987.
Prager BC, Bhargava S, Mahadev V, Hubert CG, Rich JN. Glioblastoma stem cells: driving resilience through chaos. Trends Cancer. 2020;6:223–35.
Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184:2454–70.e26.
Xuan W, Hsu WH, Khan F, Dunterman M, Pang L, Wainwright DA, et al. Circadian regulator CLOCK drives immunosuppression in glioblastoma. Cancer Immunol Res. 2022;10:770–84.
Chen P, Hsu WH, Chang A, Tan Z, Lan Z, Zhou A, et al. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discov. 2020;10:371–81.
Pang L, Dunterman M, Guo S, Khan F, Liu Y, Taefi E, et al. Kunitz-type protease inhibitor TFPI2 remodels stemness and immunosuppressive tumor microenvironment in glioblastoma. Nat Immunol. 2023;24:1654–70.
Guo X, Pan Y, Gutmann DH. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol. 2019;21:1250–62.
Tran DD, Ghiaseddin AP, Chen DD, Le SB. Final analysis of 2-THE-TOP: a phase 2 study of TTFields (Optune) plus pembrolizumab plus maintenance temozolomide (TMZ) in patients with newly diagnosed glioblastoma. J Clin Oncol. 2023;41:2024.
Awada H, Paris F, Pecqueur C. Exploiting radiation immunostimulatory effects to improve glioblastoma outcome. Neuro Oncol. 2023;25:433–46.
Sahebjam S, Forsyth PA, Tran ND, Arrington JA, Macaulay R, Etame AB, et al. Hypofractionated stereotactic reirradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: results from a phase I study. Neuro Oncol. 2021;23:677–86.
Chaichana KL, Pinheiro L, Brem H. Delivery of local therapeutics to the brain: working toward advancing treatment for malignant gliomas. Ther Deliv. 2015;6:353–69.
Shaw R, Basu M, Karmakar S, Ghosh MK. MGMT in TMZ-based glioma therapy: multifaceted insights and clinical trial perspectives. Biochim Biophys Acta Mol Cell Res. 2024;1871:119673.
Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16.
Sterner RC, Sterner RM. CAR-T-cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69.
Sengupta S, Katz SC, Sengupta S, Sampath P. Glycogen synthase kinase 3 inhibition lowers PD-1 expression, promotes long-term survival and memory generation in antigen-specific CAR-T cells. Cancer Lett. 2018;433:131–9.
Pant A, Lim M. CAR-T therapy in gbm: current challenges and avenues for improvement. Cancers. 2023;15:1249.
Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471–82.
Bagley SJ, Binder ZA, Lamrani L, Marinari E, Desai AS, Nasrallah MP, et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. 2024;5:517–31.
Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7.
Segura-Collar B, Hiller-Vallina S, de Dios O, Caamano-Moreno M, Mondejar-Ruescas L, Sepulveda-Sanchez JM, et al. Advanced immunotherapies for glioblastoma: tumor neoantigen vaccines in combination with immunomodulators. Acta Neuropathol Commun. 2023;11:79.
Zhu P, Li SY, Ding J, Fei Z, Sun SN, Zheng ZH, et al. Combination immunotherapy of glioblastoma with dendritic cell cancer vaccines, anti-PD-1 and poly I:C. J Pharm Anal. 2023;13:616–24.
Lovatt C, Parker AL. Oncolytic viruses and immune checkpoint inhibitors: the “Hot” new power couple. Cancers. 2023;15:4178.
Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. 2023;29:1370–8.
Chiocca EA, Gelb AB, Chen CC, Rao G, Reardon DA, Wen PY, et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: an open-label, multi-institutional phase I trial. Neuro Oncol. 2022;24:951–63.
Speranza MC, Passaro C, Ricklefs F, Kasai K, Klein SR, Nakashima H, et al. Preclinical Investigigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol. 2018;20:225–35.
Lin YJ, Wu CY, Wu JY, Lim M. The role of myeloid cells in GBM immunosuppression. Front Immunol. 2022;13:887781.
Samson N, Ablasser A. The cGAS-STING pathway and cancer. Nat Cancer. 2022;3:1452–63.
Tripathi S, Najem H, Mahajan AS, Zhang P, Low JT, Stegh AH, et al. cGAS-STING pathway targeted therapies and their applications in the treatment of high-grade glioma. F1000Res. 2022;11:1010.
Low JT, Brown MC, Reitman ZJ, Bernstock JD, Markert JM, Friedman GK, et al. Understanding and therapeutically exploiting cGAS/STING signaling in glioblastoma. J Clin Investig. 2024;134:e163452.
Berger G, Knelson EH, Jimenez-Macias JL, Nowicki MO, Han S, Panagioti E, et al. STING activation promotes robust immune response and NK cell-mediated tumor regression in glioblastoma models. Proc Natl Acad Sci USA. 2022;119:e2111003119.
Najem H, Lea ST, Tripathi S, Hurley L, Chen CH, William I, et al. STING agonist 8803 reprograms the immune microenvironment and increases survival in preclinical models of glioblastoma. J Clin Investig. 2024;134:e175033.
Goswami S, Raychaudhuri D, Singh P, Natarajan SM, Chen Y, Poon C, et al. Myeloid-specific KDM6B inhibition sensitizes glioblastoma to PD1 blockade. Nat Cancer. 2023;4:1455–73.
Pang L, Guo S, Khan F, Dunterman M, Ali H, Liu Y, et al. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep Med. 2023;4:101238.
Mei Y, Wang X, Zhang J, Liu D, He J, Huang C, et al. Siglec-9 acts as an immune-checkpoint molecule on macrophages in glioblastoma, restricting T-cell priming and immunotherapy response. Nat Cancer. 2023;4:1273–91.
Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J Neurooncol. 2019;143:241–9.
Pang L, Khan F, Dunterman M, Chen P. Pharmacological targeting of the tumor-immune symbiosis in glioblastoma. Trends Pharm Sci. 2022;43:686–700.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumors. Nat Rev Cancer. 2020;20:12–25.
Nelson TA, Dietrich J. Investigational treatment strategies in glioblastoma: progress made and barriers to success. Expert Opin Investig Drugs. 2023;32:921–30.
Kreatsoulas D, Bolyard C, Wu BX, Cam H, Giglio P, Li Z. Translational landscape of glioblastoma immunotherapy for physicians: guiding clinical practice with basic scientific evidence. J Hematol Oncol. 2022;15:80.
Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun. 2022;13:767.
Pang L, Dunterman M, Xuan W, Gonzalez A, Lin Y, Hsu WH, et al. Circadian regulator CLOCK promotes tumor angiogenesis in glioblastoma. Cell Rep. 2023;42:112127.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23:124–36.
Acknowledgements
This work was supported in part by National Institutes of Health (NIH) R01 NS127824 (PC), NIH R01 NS124594 (PC), NIH P01 CA245705 (JDL), R35 NS127083 (JDL), and the Department of Defense (DoD) Career Development Award W81XWH-21-1-0380 (P.C.), as well as the Lerner Research Institute and Case Comprehensive Cancer Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JDL reports being named a coinventor on pending and issued patents held by the Cleveland Clinic relating to cancer therapies, but these are not directly relevant to this review. No potential conflicts of interest are disclosed by the other authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Zhou, F., Ali, H. et al. Immunotherapy for glioblastoma: current state, challenges, and future perspectives. Cell Mol Immunol 21, 1354–1375 (2024). https://doi.org/10.1038/s41423-024-01226-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-024-01226-x
Keywords
This article is cited by
-
CSF1R inhibitor C19 for glioma immunotherapy enabled by brain-targeting liposomal delivery
Acta Pharmacologica Sinica (2026)
-
Synergy of radiotherapy, focused ultrasound, and immunotherapy in the treatment of brain metastases
Journal of Neuro-Oncology (2026)
-
Prevalence of human cytomegalovirus in glioblastoma multiforme: a systematic review and meta-analysis
Journal of Neuro-Oncology (2026)
-
A manganese metabolism-related gene signature for prognosis prediction and immune microenvironment description of glioblastoma
Discover Oncology (2026)
-
Immunotherapy and targeted therapy for high grade gliomas: current and future directions
Journal of Neuro-Oncology (2026)
