
Abstract
DDX3X-related neurodevelopmental disorder is one of the most common monogenic causes of intellectual disability in females, with currently >1000 females diagnosed worldwide. In contrast, reports on affected males with DDX3X variants are scarce. The limited knowledge on this X-linked disorder in males hinders the interpretation of hemizygous DDX3X variants in clinical practice. In this study, we present a new cohort of 19 affected males (from 17 unrelated families) with (possibly) disease-causing DDX3X variants, for whom we collected clinical and molecular data. Additionally, we reviewed the existing literature on 13 males with DDX3X variants. The phenotype in males is diverse, including intellectual disability, speech/language delays, behavioural challenges and structural brain abnormalities. The vast majority of males have missense variants, including two recurrent variants (p.(Arg351Gln) and p.(Arg488Cys)). No truncating variants have been reported, consistent with the presumed embryonic lethality of complete loss-of-function of DDX3X in males. In our novel cohort, 6/17 variants are de novo in the affected male and 3/17 variants are de novo in the mother. This study provides significant insights in the genetic and phenotypic spectrum of males with DDX3X variants, by presenting the data of a combined cohort (n = 32) of novel and published individuals. Our data show that variants in DDX3X can cause an X-linked neurodevelopmental disorder in males, with unaffected or mildly affected carrier females. These findings will aid the interpretation of hemizygous missense variants in DDX3X and can guide clinical management and counselling, in particular with regard to recurrence risks in the respective families.
Similar content being viewed by others

Introduction
DDX3X is an essential gene located on the X-chromosome (Xp11.4), that partially escapes X-inactivation [1, 2]. It encodes a DEAD-box RNA helicase with numerous important functions in cellular processes, including transcription regulation, splicing, RNA metabolism, translation, DNA repair, stress granule formation, inflammation and regulation of the Wnt/β-catenin pathway [3,4,5,6]. Research on DDX3X is extensive and spans across diverse disciplines and topics (including fundamental cell biology, oncology, immunology and neurodevelopmental disorders), indicating its broad functions and associations. DDX3X-related neurodevelopmental disorder (NDD) was first described in 2015 and has been identified as one of the most frequent monogenic causes of intellectual disability (ID) in females [7,8,9,10,11]. It is associated with a broad phenotypic spectrum in affected females, including ID with speech/language problems, behavioural issues, hypotonia, structural brain anomalies, epilepsy and movement disorders [8, 9, 12,13,14,15,16,17].
To date, several cohorts of females with DDX3X-related NDD have been described in literature [3, 7,8,9, 13, 14]. Multiple types of de novo variants have been identified in affected females, including nonsense, frameshift, missense and splice site variants. In contrast to the numerous reported females with DDX3X-related NDD, only a small number of reports on males with rare hemizygous DDX3X variants have been published [7, 18, 19]. All of these males have missense or splice site variants, either maternally inherited or de novo. So far, no germline truncating variants in DDX3X have been identified in males, indicating the presumed embryonic lethality of complete loss-of-function of DDX3X in the hemizygous state.
Currently, the limited knowledge on DDX3X-related NDD in males hampers the interpretation of rare hemizygous DDX3X variants. This is particularly unfavourable given the current increase in the discovery of variants of uncertain significance, due to the expanding use of next generation sequencing in patients with developmental disorders [20]. Furthermore, in two recent studies assessing the pathogenicity of DDX3X variants, no additional evidence was found for pathogenicity of a subset of DDX3X missense variants reported in males [21, 22].
With this study we aim to gain more knowledge on the genetic and phenotypic spectrum of DDX3X-related NDD in males. We here present the largest male cohort to date (n = 19) and a literature review of previously published individuals (n = 13). This comprehensive overview of 32 individuals will facilitate variant interpretation in males with rare hemizygous DDX3X variants and guide clinical management. Our study has important consequences for counselling of the respective families, in particular with regard to the recurrence risks of these X-linked variants.
Materials and methods
Study participants and data collection
The new cohort presented in this study consists of male individuals with ID/NDD and rare variants in DDX3X identified in a diagnostic setting (except for individual 10 - identified in a research setting) and reported as possibly contributing to the phenotype. Information on these individuals was assembled from clinicians through (inter)national collaborations, including the international and Dutch DDX3X foundations, between February 2022 and October 2024. De-identified clinical and variant data were collected from the involved clinicians through a standardized questionnaire. Informed consent for the sharing and publication of medical data was provided by patients or their legal representatives. Consent for publication of photographs was obtained separately. Genetic testing and research were performed in accordance with protocols approved by the local Institutional Review Boards.
Literature review
A PubMed literature search was performed to identify published reports on male individuals with DDX3X-related neurodevelopmental disorder. Articles published until October 1st 2024 were identified using the following search strategy: ‘(DDX3X[Title/Abstract]) AND ((male* [Title/Abstract]) OR (boy [Title/Abstract]) OR (hemizygous [Title/Abstract]) OR (XY [Title/Abstract]))’. By screening the titles and abstracts, we selected relevant articles which included at least one male with a possibly disease-causing variant in DDX3X and included clinical information about the individual. We extracted genetic and phenotypic data on the male individuals from these papers and their supplementary data. For three males we received additional or updated information on clinical features after approaching the corresponding authors of the selected manuscripts (Supplementary Table S1).
Next-generation sequencing and in silico variant analyses
The DDX3X variants in the probands in our cohort were identified by exome sequencing (individuals 1,3-14 and 17) or genome sequencing (individual 2, 15, 16) in a clinical setting. The variants were classified as pathogenic, likely pathogenic or variant of unknown significance (VUS) by the local genetic laboratory. The individuals did not have clear alternative genetic diagnoses explaining the NDD (except for individual 4, see Results). Inheritance of the variants was examined either as part of trio exome/genome sequencing or by Sanger sequencing of the specific variant in other family members. In this study, variants are annotated in DDX3X transcript NM_001356.5 (MANE transcript). In silico variant analyses to assess pathogenicity were performed using Alamut Visual Plus (Version 1.7.1), CADD (v1.7), PolyPhen-2, PhyloP, SIFT scores, MetaDome (Version 1.0.1) tolerance landscapes, AlphaMissense, SpliceAI [23] and DDGun (BioFold). Population allele frequencies of the variants were extracted from gnomAD (v4.1.0, accession date 16-10-2024). We reclassified all variants based on a combination of ACMG/AMP [24] and additional criteria (Supplementary Table S2).
Three-dimensional protein structure analysis
The possible effects of the variants on DDX3X protein 3D structure were analysed using solved crystal structures of the (1) unbound DDX3X (bound to AMP) (PDB:5E7J [25]); (2) DDX3X dimer bound to dsRNA in pre-unwound state (PDB:6O5F [26]); and (3) DDX3X in post-unwound state (PDB:7LIU [not published] and 2DB3 [27]). All available structures lack the N-terminal part of the protein, so it was evaluated using the available AlphaFold2 structure [28]. The analysis was performed using the YASARA Structure v20 [29].
Results
Cohorts
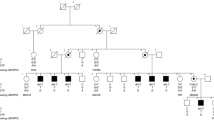
In this study, we present a novel cohort consisting of 19 males (from 17 unrelated families) with rare hemizygous (possibly) disease-causing variants in DDX3X. We also reviewed the existing literature and found seven articles which in total described 16 male probands with DDX3X variants [3, 7,8,9, 14, 18, 19]. To enable a reliable overview of clinical features, we excluded three individuals from the previous literature carrying variants with low certainty of pathogenicity (p.(Ala9Val), p.(Arg79Lys) and p.(Arg110His)). In Table 1, we provide a consolidated clinical summary encompassing both cohorts (n = 32). Additionally, facial photos and pedigrees of males from our novel cohort are presented in Fig. 1. All variants identified in our novel cohort and previous literature are listed in Table 2 and Fig. 2. In the subsequent paragraphs, we will mainly focus on the males from our novel cohort.

A Facial photgraphs. B Pedigrees. All (relevant) tested family members are indicated by + (DDX3X variant present) or - (DDX3X variant absent) (noted as x/x in females for biallelic state).

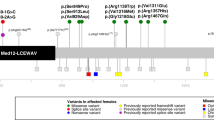
A Visual representation of DDX3X transcript (MANE transcript; NM_001356.5) with variants predicted to affect splicing. B Linear representation of the DDX3X protein including the two helicase domains (extracted from Uniprot) with locations of missense variants annotated.
Clinical characteristics
The 19 male individuals in our new cohort show overlapping neurodevelopmental symptoms with variable other features, summarized in Table 1 (details in Supplementary Table S1). The median age at detection of the DDX3X variant was 8 years (range 9 months – 47 years). In 12/17 individuals, abnormalities were present during pregnancy and/or delivery. This includes abnormalities on prenatal ultrasounds (4/17), (partial) placental abruption (3/17) and a caesarean section for various reasons (7/17). All males have developmental delays and/or an intellectual disability, which is mild in six, moderate in three and severe in two individuals (and of unknown severity in eight). IQ data are available for five individuals, demonstrating a mean total IQ of 65 (range 56–75). All males have a delayed speech and language development; the age of first words ranges from 18 months to 8 years (median 24 months). Three males have verbal dyspraxia or speech apraxia. Regarding current speech and language capacities, 4/7 individuals (aged 4 years and older) are minimally verbal, with most only speaking a few words. Motor development is delayed in 17/18 individuals, with a median age of first steps at 24 months. Behavioural difficulties are present in 14/18 individuals. Six individuals have officially been diagnosed with an autism spectrum disorder (ASD); additionally six males show features of ASD without a formal diagnosis. Three individuals have a formal diagnosis of ADHD, and two additional males show signs of ADHD.
Regarding neurological features, both hypotonia and hypertonia are reported (8/16 and 4/12 respectively). About three quarters of the individuals (14/18) experience movement problems: six show ataxia or coordination problems. Also, abnormal gait, stiff legs and frequent falls are described repeatedly. One male (individual 7) had epilepsy in the past (complex partial epilepsy), for which he received anti-epileptic medication. Three others experienced a single (febrile) seizure and one had staring spells. A cerebral MRI or CT scan shows abnormalities in ten (out of 15) males, including corpus callosum abnormalities (5/15), enlarged ventricles or hydrocephalus (4/15) and a small or thin brainstem (2/15). One individual has a retrocerebellar arachnoidal cyst, another has a Blake’s pouch cyst.
Vision problems (11/19) consist mainly of mild refractive errors, strabismus and amblyopia. Nine individuals have recurrent ear infections and/or a need for ear tubes. Four individuals have diverse congenital cardiac anomalies, encompassing a ventricular septal defect, a combination of a ventricular septal defect and atrial septal defect, a bicuspid aorta valve and an aberrant right subclavian artery. Gastrointestinal problems include gastroesophageal reflux (4/17), faecal incontinence (3/17) and constipation (3/17). Six males have one or more urogenital abnormalities, including cryptorchidism (3/18), inguinal hernia (2/18) and a hydrocele (3/18). Plagiocephaly is present in three individuals, and one male has a cleft lip-palate. One individual has a humerus cyst; another has an osteochondral defect of the left talus. No malignant neoplasms have been reported. Variable dysmorphic facial features are present in the cohort, but we did not observe a clear recognisable facial phenotype (Fig. 2A). However, a prominent/high/broad forehead, deep-set eyes and prominent ears are reported in multiple individuals.
Interestingly, five out of the twelve carrier mothers in our cohort report mild neurodevelopmental symptoms, which are notably less severe than in their affected sons. Four mothers have mild learning problems, requiring special education in two; one also has ADHD; and one has dyslexia. Two carrier mothers report a single early miscarriage in the past, whilst the mother of individual 16 had multiple early miscarriages. Also, she underwent a termination of pregnancy involving a male foetus who had cardiovascular abnormalities and Dandy-Walker syndrome; no genetic test was performed at the time.
In two families, an additional affected male was identified and confirmed to carry the DDX3X variant. This involves a cousin of the index (in family 1) and a brother (in family 8), whose clinical features are also part of this study. In family 14, the affected male has a sister with intellectual disability, autism spectrum disorder, coarse facies and a small stature. Genetic testing showed that she does not carry the DDX3X variant, and whole exome sequencing was negative.
Variants and in silico analyses
In the 17 probands in our novel cohort, thirteen unique variants were observed (Fig. 2 and Table 2). Eleven variants lead to amino acid substitutions (missense variants), while two affect a canonical splice site of the last exon. Intriguingly, one of the missense variants (p.(Arg488Cys)) is present in five unrelated individuals. Three missense variants lead to amino acid substitutions located in the helicase ATP-binding domain, six in the helicase C-terminal domain and two variants are located outside of the known functional domains of the DDX3X protein (one of which is within the Q motif just prior to the helicase ATP-binding domain).
For all variants in our new cohort, segregation analysis (or trio WES/WGS) was performed to investigate the inheritance of the variants (Fig. 1B). Six variants were found to be de novo in the affected male (four of which concerned the recurrent p.(Arg488Cys) variant), and 11 variants were found to be maternally inherited. In five of the families with maternally inherited variants, further segregation analysis was performed in the maternal grandparents. In three of these families, the DDX3X variant was found to be de novo in the mother of the proband (family 3-5); in one family the grandmother did not have the variant but the grandfather was unavailable for testing (family 7); and in one family the variant was inherited from the maternal grandmother (family 15). In family 1, the maternal grandmother was not tested, but the variant was most likely inherited from her because of the occurrence in two male cousins and their mothers, although germline mosaicism in the grandmother cannot be excluded. X-inactivation tests were performed in four carrier mothers, and showed random X-inactivation in two mothers (family 4 and 14) and extreme skewing in two (family 8 and 16).
A diverse range of in silico prediction tools was used to aid variant interpretation for all variants in our cohort and previously reported males (Table 2 and Supplementary Table S2). All variants from our novel cohort had a CADD PHRED (v1.7) score ≥23, besides the p.(Leu461Phe) with a CADD score of 15.08 (other prediction scores pointed to pathogenicity for this variant). All variants were absent from gnomAD (v4.1.0), except for p.(Ile522Thr) which was present once in a heterozygous state. The two splice site variants (c.1910-15_1910-2del and c.1910-2 A > G) are both predicted to cause loss of the canonical acceptor site of the last exon. According to prediction tools, a new acceptor site (canonical in alternative transcript ENST00000625837.2) might get activated. However, the exact effects of these variants on transcript and protein levels are currently unknown. In individual 4 another genetic variant possibly contributing to the phenotype was found: a likely pathogenic variant in KMT2C (Supplementary Table S1). Since the phenotypes of DDX3X-related NDD and KMT2C-related NDD are rather aspecific and overlapping, it is difficult to determine the relative contributions of both variants to this individual’s phenotype [30].
In the 16 male probands with DDX3X variants from previous literature (of which we excluded three for the clinical overview), 14 unique variants were reported, including 13 missense and one splice site variant (Fig. 2). In five individuals, the variant was confirmed to be de novo. The p.(Arg351Gln) variant was recurrently identified in three unrelated males. Similar to our new cohort, we found a clustering of missense variants within the two helicase domains. Interestingly, the p.(Arg568Ala) affects the same amino acid position as the variant found in individual 15 within our cohort.
We systematically reclassified all DDX3X variants from our novel cohort and previous literature, ranging from class 3 (variant of unknown significance) to class 5 (pathogenic) (Table 2 and Supplementary Table S2).
Three-dimensional protein structure analysis
DDX3X is a dynamic protein with multiple conformations and binding partners. Therefore, the effects of the variants were predicted on several available DDX3X conformational structures (see Materials and Methods). Variant analysis showed that the identified missense variants likely exhibit different molecular mechanisms of (partial) loss of the functions of DDX3X.
Out of 24 analysed missense variants, eight variants were predicted to disrupt and/or destabilize one of the helicase domain’s structure: p.(His209Leu), p.(Ala233Thr), p.(Ser269Phe), p.(Val300Phe), p.(Leu461Phe), p.(Val496Met), p.(Pro568Ala) and p.(Pro568Ser) (see Supplementary Fig. S1 and Supplementary Table S3). In addition, four variants were predicted to affect the protein’s conformation changes and/or stability in a specific conformational state: p.(Asp164Gly), p.(Pro386Ser), p.(Leu484Val) and p.(Arg488Cys). Nine of the identified missense variants were located at or near the surface of the DDX3X protein, with four variants predicted to affect binding to the second DDX3X monomer in a homodimer or binding to the RNA: p.(Arg292Leu), p.(Arg351Gln), p.(Ser456Phe), p.(Ile522Thr). The five remaining variants located at or near the surface (p.(Arg315Ser), p.(Arg362Cys), p.(Thr369Ala), p.(Arg376His) and p.(Ala467Ser)) were not predicted to affect surfaces that are known to bind to other proteins or RNA, so their effect on the protein is currently unknown. Similarly, the N-terminal part of the DDX3X contains a disordered region, so the three variants located in this region (p.(Ala9Val), p.(Arg79Lys), p.(Arg110His)) are not expected to affect the protein’s secondary structure. However, DDX3X is known to have dozens of binding partners for which exact binding sites are largely unknown. Therefore, we cannot exclude that the surface variants, as well as the N-terminal variants could disrupt binding to other proteins.
Discussion
In this study, we provide the first overview of DDX3X-related neurodevelopmental disorder (NDD) in males, by presenting a large new cohort of affected males (n = 19) and a review of published patients in the literature (n = 13). We demonstrate the clinical and molecular features of this X-linked disorder in males.
The clinical spectrum in the males within our study is diverse. All males have a developmental delay and/or intellectual disability, ranging from mild to severe. Behavioural challenges, movement problems and brain abnormalities are frequent. Although the phenotype is rather non-specific and variable, clinical features are comparable to those described in females with DDX3X-related NDD [12]. Interestingly, 5/12 of the carrier mothers of affected males in our novel cohort have mild neurodevelopmental symptoms. In previous literature, most carrier mothers were thought to be asymptomatic.
Regarding the molecular spectrum of DDX3X variants, the vast majority of males (27/30) have missense variants and three males have splice site variants. No protein truncating variants have been reported, in contrast to affected females where these loss-of-function variants are very common [4]. In males, most missense variants are located within the helicase ATP-binding domain and helicase C-terminal domain. Two missense variants are recurrently present in males: the p.(Arg351Gln) is present in three unrelated males from the literature, and the p.(Arg488Cys) variant in five unrelated males in our novel cohort. Notably, the latter variant has been described in affected females too [31], and this amino acid position has been found to be recurrently mutated [3]. To our knowledge, this is the only DDX3X variant which has been reported in both male and female probands with NDD. Additionally, three variants predicted to affect splicing are present in our study; their precise effects on the protein are currently unknown. Ten out of the 27 unique variants presented in this study have also been reported as somatic variants in various forms of cancer [32]. No malignant neoplasms have been reported in the males in our study.
In our study, we see several indicators for pathogenicity of the DDX3X variants in males. Firstly, certain missense variants are recurrently present in affected males, whilst absent from population databases. Furthermore, the DDX3X variant was de novo in 10/30 males. Additionally, three of the maternally inherited missense variants in our novel cohort were confirmed de novo in the carrier mother.
Evidently, there are different patterns of variant types present between males and females with this X-linked disorder that partially escapes X-inactivation. In females, loss-of-function is an acknowledged mode of pathogenicity. In addition, a subset of recurrent missense variants demonstrate a dominant-negative effect causing aberrant RNP granules (generally associated with a more severe phenotype) [3]. In the hemizygous state, complete loss-of-function of DDX3X is thought to be embryonically lethal, consistent with results from animal studies [33, 34]. DDX3Y, a Y-chromosome homologue of DDX3X mainly expressed in the testis, seems unable to compensate for the loss of DDX3X [4]. We propose that the missense variants reported in affected males have a partial loss-of-function (or hypomorphic) effect, supported by previous functional studies in zebrafish [7, 19]. Assumably these variants cause a phenotype in the hemizygous state, whereas the effect on females will be milder due to the existence of a second unaffected allele and/or partial X-inactivation. Accordingly, carrier mothers are either asymptomatic or mildly symptomatic.
Several other X-linked conditions show similar differences in variant types between affected males and females, including disorders resulting from pathogenic variants in USP9X, ALG13, KDM5C, IQSEC2, OFD1 and SMC1A [35,36,37,38,39,40]. These genes all (partially) escape X-inactivation, feature de novo loss-of-function variants in affected females, and milder (often maternally inherited) missense variants in males. We suggest to refer to this group of disorders (including DDX3X-related NDD) as ‘X-linked’, instead of X-linked dominant or X-linked recessive [41, 42].
In contrast to our findings, two recent studies did not find supporting evidence for pathogenicity of DDX3X variants in males. One study, using statistical enrichment analyses to identify X-linked NDD genes, showed a female bias for de novo mutations in DDX3X [21]. It is clear that the prevalence of de novo DDX3X variants is higher in females than in males, but in our view an absence of enrichment does not exclude the possibility of pathogenicity of rare variants in males. Another study, assessing pathogenicity of DDX3X variants using saturation genome editing (SGE) analyses, did not find supporting functional evidence for pathogenicity of most variants in males [22]. They suggest further evidence is needed to support the association between damaging DDX3X variants and NDD in males. We theorize that the effects of DDX3X missense variants in males might not have been picked up by these analyses, for example because the assay may not have been sensitive enough to identify variants with hypomorphic effects. In contrast to these two studies, our study provides substantial evidence supporting the occurrence of DDX3X-related NDD in males. While we cannot classify all variants as pathogenic at the moment, our data confirm that this disorder can affect males. For future research, we recommend additional functional studies for DDX3X variants to help further understanding the different modes of pathogenicity in both males and females.
In conclusion, this study shows that specific DDX3X variants in males can cause an X-linked neurodevelopmental disorder. Our overview and insights will aid in the interpretation of hemizygous DDX3X variants in clinical practice, and guide counselling and management in the corresponding males and their families.
References
Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–6.
Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda). 2017;7:2719–27.
Lennox AL, Hoye ML, Jiang R, Johnson-Kerner BL, Suit LA, Venkataramanan S, et al. Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron. 2020;106:404–20.e8.
Gadek M, Sherr EH, Floor SN. The variant landscape and function of DDX3X in cancer and neurodevelopmental disorders. Trends Mol Med. 2023;29:726–39.
Radford EJ, Tan HK, Andersson MHL, Stephenson JD, Gardner EJ, Ironfield H, et al. Saturation genome editing of DDX3X clarifies pathogenicity of germline and somatic variation. Nat Commun. 2023;14:7702.
Rosa ESI, Smetana JHC, de Oliveira JF. A comprehensive review on DDX3X liquid phase condensation in health and neurodevelopmental disorders. Int J Biol Macromol. 2024;259:129330.
Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MR, et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on WNT signaling. Am J Hum Genet. 2015;97:343–52.
Wang X, Posey JE, Rosenfeld JA, Bacino CA, Scaglia F, Immken L, et al. Phenotypic expansion in DDX3X - a common cause of intellectual disability in females. Ann Clin Transl Neurol. 2018;5:1277–85.
Tang L, Levy T, Guillory S, Halpern D, Zweifach J, Giserman-Kiss I, et al. Prospective and detailed behavioral phenotyping in DDX3X syndrome. Mol Autism. 2021;12:36.
Gillentine MA, Wang T, Eichler EE. Estimating the prevalence of de novo monogenic neurodevelopmental disorders from large cohort studies. Biomedicines. 2022;10:2865.
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62.
Johnson-Kerner B, Snijders Blok L, Suit L, Thomas J, Kleefstra T, Sherr EH. DDX3X-Related Neurodevelopmental Disorder. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993-2023, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved; 1993.
Beal B, Hayes I, McGaughran J, Amor DJ, Miteff C, Jackson V, et al. Expansion of phenotype of DDX3X syndrome: six new cases. Clin Dysmorphol. 2019;28:169–74.
Dai Y, Yang Z, Guo J, Li H, Gong J, Xie Y, et al. Expansion of Clinical and Genetic Spectrum of DDX3X Neurodevelopmental Disorder in 23 Chinese Patients. Front Mol Neurosci. 2022;15:793001.
Levy T, Siper PM, Lerman B, Halpern D, Zweifach J, Belani P, et al. DDX3X Syndrome: Summary of Findings and Recommendations for Evaluation and Care. Pediatr Neurol. 2022;138:87–94.
Ruault V, Burger P, Gradels-Hauguel J, Ruiz N, Jamra RA, Afenjar A, et al. Lessons from two series by physicians and caregivers’ self-reported data in DDX3X-related disorders. Mol Genet Genom Med. 2024;12:e2363.
Ng-Cordell E, Kolesnik-Taylor A, O’Brien S, Astle D, Scerif G, Baker K. Social and emotional characteristics of girls and young women with DDX3X-associated intellectual disability: a descriptive and comparative study. J Autism Dev Disord. 2023;53:3208–19.
Nicola P, Blackburn PR, Rasmussen KJ, Bertsch NL, Klee EW, Hasadsri L, et al. De novo DDX3X missense variants in males appear viable and contribute to syndromic intellectual disability. Am J Med Genet A. 2019;179:570–8.
Kellaris G, Khan K, Baig SM, Tsai IC, Zamora FM, Ruggieri P, et al. A hypomorphic inherited pathogenic variant in DDX3X causes male intellectual disability with additional neurodevelopmental and neurodegenerative features. Hum Genom. 2018;12:11.
Evans JP, Powell BC, Berg JS. Finding the rare pathogenic variants in a human genome. JAMA. 2017;317:1904–5.
Martin HC, Gardner EJ, Samocha KE, Kaplanis J, Akawi N, Sifrim A, et al. The contribution of X-linked coding variation to severe developmental disorders. Nat Commun. 2021;12:627.
Radford EJ, Tan HK, Andersson MHL, Stephenson JD, Gardner EJ, Ironfield H, et al. Saturation genome editing of DDX3X clarifies pathogenicity of germline and somatic variation. medRxiv. 2022:2022.06.10.22276179.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–48.e24.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Floor SN, Condon KJ, Sharma D, Jankowsky E, Doudna JA. Autoinhibitory Interdomain Interactions and Subfamily-specific Extensions Redefine the Catalytic Core of the Human DEAD-box Protein DDX3. J Biol Chem. 2016;291:2412–21.
Song H, Ji X. The mechanism of RNA duplex recognition and unwinding by DEAD-box helicase DDX3X. Nat Commun. 2019;10:3085.
Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Krieger E, Vriend G. YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics. 2014;30:2981–2.
Rots D, Choufani S, Faundes V, Dingemans AJM, Joss S, Foulds N, et al. Pathogenic variants in KMT2C result in a neurodevelopmental disorder distinct from Kleefstra and Kabuki syndromes. Am J Hum Genet. 2024;111:1626–42.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D5.
Sondka Z, Dhir NB, Carvalho-Silva D, Jupe S, Madhumita, McLaren K, et al. COSMIC: a curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 2023;52:D1210–D7.
Chen CY, Chan CH, Chen CM, Tsai YS, Tsai TY, Wu Lee YH, et al. Targeted inactivation of murine Ddx3x: essential roles of Ddx3x in placentation and embryogenesis. Hum Mol Genet. 2016;25:2905–22.
Matsumura T, Endo T, Isotani A, Ogawa M, Ikawa M. An azoospermic factor gene, Ddx3y and its paralog, Ddx3x are dispensable in germ cells for male fertility. J Reprod Dev. 2019;65:121–8.
Johnson BV, Kumar R, Oishi S, Alexander S, Kasherman M, Vega MS, et al. Partial Loss of USP9X function leads to a male neurodevelopmental and behavioral disorder converging on transforming growth factor β signaling. Biol Psychiatry. 2020;87:100–12.
Shoubridge C, Harvey RJ, Dudding-Byth T. IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Hum Mutat. 2019;40:5–24.
Fieremans N, Van Esch H, de Ravel T, Van Driessche J, Belet S, Bauters M, et al. Microdeletion of the escape genes KDM5C and IQSEC2 in a girl with severe intellectual disability and autistic features. Eur J Med Genet. 2015;58:324–7.
Jansen S, Kleefstra T, Willemsen MH, de Vries P, Pfundt R, Hehir-Kwa JY, et al. De novo loss-of-function mutations in X-linked SMC1A cause severe ID and therapy-resistant epilepsy in females: expanding the phenotypic spectrum. Clin Genet. 2016;90:413–9.
Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21.
Sakakibara N, Morisada N, Nozu K, Nagatani K, Ohta T, Shimizu J, et al. Clinical spectrum of male patients with OFD1 mutations. J Hum Genet. 2019;64:3–9.
Basta M, Pandya AM Genetics, X-Linked Inheritance. StatPearls. Treasure Island (FL): StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC.; 2023.
Dobyns WB, Filauro A, Tomson BN, Chan AS, Ho AW, Ting NT, et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A. 2004;129a:136–43.
Parra A, Pascual P, Cazalla M, Arias P, Gallego-Zazo N, San-Martín EA, et al. Genetic and phenotypic findings in 34 novel Spanish patients with DDX3X neurodevelopmental disorder. Clin Genet. 2024;105:140–9.
Acknowledgements
We thank all families and authors involved in this study for their support and contributions. We acknowledge the DDX3X Foundation and the French family group (Xtraordinaire).
Funding
This work was supported by a grant of Netherlands Organisation for Health Research and Development (10250022110003 and 91718310, to TK) and Dutch Research Council grant (015.014.036, to TK).
Author information
Authors and Affiliations
Contributions
MK, LSB and TK designed the study and drafted the paper, with all authors contributing to the revision of the paper. DR contributed by performing the three-dimensional protein structure analysis and aiding in the interpretation of other in silico prediction tools. RP contributed by assisting the critical interpretation of all male DDX3X variants presented in our study. AB, CO, CB, CM, BdV, ME, QW, MS, EFM, DP, DD, RCR, RR, RC, BC, CL, SS, SM, RL, HS and BL contributed to acquiring and providing clinical and molecular data on the individuals presented in this study. All authors approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declaration
This paper is consistent with the journal’s guidelines on issues involved in ethical publication. According to Dutch CCMO guidelines, no formal approval by an institutional review board was necessary due to the retrospective nature of the study. Consent was obtained from parents for publication of photographs.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kennis, M.G.P., Rots, D., Bouman, A. et al. DDX3X-related neurodevelopmental disorder in males – presenting a new cohort of 19 males and a literature review. Eur J Hum Genet 33, 980–988 (2025). https://doi.org/10.1038/s41431-025-01832-x
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01832-x
This article is cited by
-
Role of DEAD/DEAH-box helicases in immunity, infection and cancers
Cell Communication and Signaling (2025)
-
Genomic medicine in full bloom: a summer farewell issue
European Journal of Human Genetics (2025)
-
Expanding the understanding of DDX3X-related neurodevelopmental disorder in males
European Journal of Human Genetics (2025)

{kind=link}