
Abstract
Mitochondrial ribosomal protein-small 2 (MRPS2) encodes a vital structural protein essential for assembling mitoribosomal small subunit and thus mitochondrial translation. Any defect in mitochondrial translation impacts OXPHOS activity and cellular respiration. Defects in MRPS2 have been implicated recently in four families with combined oxidative phosphorylation deficiency-36 (MIM# 617950). We herein describe two individuals from two unrelated families with variable phenotypes of acute onset severe metabolic decompensation and symptomatic hypoglycemia. Exome sequencing identified bi-allelic variants in MRPS2 (NM_016034.5) in the affected individuals: P1: c.490 G > A p.(Glu164Lys); and P2: c.413 G > A p.(Arg138His). Further evaluation of the variant c.490 G > A p.(Glu164Lys) in patient-derived skin fibroblasts revealed decreased expression of MRPS2 transcript and protein levels of MRPS2 along with expression of complex I and IV proteins. Proteomics analysis revealed decreased expression of small subunit proteins and increased expression of large subunit proteins. Also, reduced complex I and IV enzyme activities, mitochondrial respiration (OCR), and altered mitochondrial morphology on confocal imaging were observed. Additionally, mrps2 knockout zebrafish larvae demonstrated an abnormal developmental phenotype and reduced Complex IV activity. With these findings, we identify additional families with variants in MRPS2, illustrating the variable clinical spectrum and validate the pathogenicity of defects in MRPS2 through in-vitro and in-vivo assays.
Similar content being viewed by others


Introduction
Mitochondrial translation is executed by a dedicated translation machinery that includes many regulatory factors and mitochondrial–specific ribosome subunits or the mitoribosomes. The mitoribosome consists of two subunits: a 28S small subunit (mtSSU) composed of 30 mitoribosomal proteins (MRPs) and a 12S rRNA, while the 39S large subunit (mtLSU) is composed of 52 MRPs, 16S rRNA, mt-tRNAVal and mt-tRNAPhe [1, 2]. The mtLSU functions as a catalyst for peptide bond formation and attaching the ribosome to the inner mitochondrial membrane, whereas mtSSU aids in decoding the mRNA and further recruits a correct aminoacyl tRNA to the translating ribosome [3]. Mitochondrial translation is responsible for synthesizing 13 proteins of the OXPHOS system, encoded by mtDNA. Hence, translational defects in mitoribosomes have been implicated in combined OXPHOS complex deficiency [4].
MRPs, the structural components of the mitoribosome, are encoded by the nuclear genome and imported into mitochondria to facilitate mitoribosome assembly [5]. To date, variants in 14 MRP encoding genes including four MRPL genes - MRPL3 (MIM*607118), MRPL12 (MIM*602375), MRPL24 (MIM*611836), MRPL44 (MIM*611849) and ten MRPS genes i.e. MRPS2 (MIM*611971), MRPS7 (MIM*611974), MRPS14 (MIM*611978), MRPS16 (MIM*609204), MRPS22 (MIM*605810), MRPS23 (MIM*611985), MRPS25 (MIM*611987), MRPS28 (MIM*611990), MRPS34 (MIM*311994) and PTCD3 (MIM*614918) have been implicated in human mitochondrial disorders. Though pathogenic variants in these genes have been associated with variable phenotypes [6], most affected individuals present with severe, early-onset multi-organ dysfunction and are often associated with a reduced lifespan [7].
The mitochondrial ribosomal protein S2 encoded by MRPS2 (MIM*611971) is an evolutionarily conserved protein and an integral component of the mtSSU. Biallelic variants in MRPS2 have been recently reported to cause combined oxidative phosphorylation deficiency 36 (COXPD36, MIM# 617950), presenting with variable phenotypes of global developmental delay, ataxia, hypotonia, severe lactic acidosis, hypoglycemia, and sensorineural hearing loss in four unrelated individuals till date [8,9,10].
We herein describe the phenotypic and genotypic details of two unrelated individuals with biallelic variants in MRPS2. We show the impact of the novel variant in one of the individuals on the mitochondrial RNA and protein expression, OXPHOS activity, morphology, and function. We also demonstrate that loss of mrps2 leads to delayed development and abnormal developmental phenotypes in zebrafish.
Methodology
Participant recruitment
We recruited two probands (P1 and P2) from two unrelated families of Indian origin as part of an ongoing study on rare genetic disorders. Informed consents approved by the institutional ethics committee were obtained from the recruited families.
Genetic testing
The genomic DNA was extracted from whole blood using DNeasy Blood and Tissue Kits (QIAGEN, Valencia, California). Singleton exome sequencing (ES) was performed using TWIST Bioscience capture kit for P1 and P2 on an Illumina NextSeq Platform (Illumina, Inc. San Diego CA) and analysis was performed as described previously [11]. Validation and segregation analysis was performed by Sanger sequencing for prioritized variants in both families.
In silico protein modeling and analysis
In silico protein analysis was performed for the identified missense variants in both the families. Small mitoribosome subunit structure (PDB ID: 7PO3) was obtained from protein data bank (RCSB PDB) (https://www.rcsb.org/structure/7PO3; accessed on 27 January 2024). Visualization of protein structure was performed on Chimera 1.16 [12]. In silico mutagenesis for MRPS2 protein was performed and alterations in polar contacts in mutant protein were analyzed as compared to wild type. Additionally, any alterations in polar contacts of MRPS2 protein with other interacting small mitoribosome subunits were also analyzed. Multiple sequence alignment (MSA) was performed using Clustal Omega [13].
Cell culture
Dermal fibroblasts were obtained from skin biopsy of P1 and the normal controls (C1 and C2). Fibroblast cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA). Fibroblasts were enzymatically passaged in 0.25% Trypsin-EDTA (HiMedia, India). All cultures were maintained in 37 °C incubators at 5% CO2. The culture medium was replenished every two days until cells became 85% confluent. The cells were used up to 4 passages only. Further, total RNA was isolated from skin fibroblasts using the TRIzol method. The RNA quality was assessed by A260/280 ratios and converted to cDNA using SuperScript™ IV VILO™ Master Mix.
Proteomics analysis
P1-derived and control fibroblasts (volunteer donor; GM05659 and GM08333, Coriell Institute) were cultured in complete medium with Minimum Essential Medium. Cell lysate-derived tryptic peptides were labeled with tandem mass tags (TMT; Thermo Fisher Scientific, A44520) as per the manufacturer’s protocol. The P1-derived samples were used in triplicate. Following a label check, pooled peptides were fractionated by basic pH reversed-phase liquid chromatography (bRPLC) on a C18 column, and twenty-four fractions were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for total proteomics with modifications [14].
Intracellular ATP estimation
For ATP quantification, 1×106 fibroblast cells were seeded and lysed with passive lysis buffer. Intracellular ATP levels were determined in 5uL of cell lysate using the ATP determination kit (Invitrogen, USA) as per the manufacturer’s guidelines. The data were normalized against the total protein concentration [15].
Zebrafish F0 knockout generation
The F0 knock out of mrps2 in zebrafish was generated using CRISPR/Cas9 mediated gene editing, as described previously [16].
To generate the mrps2 crispants, chemically modified synthetic guide RNAs of the most efficient sgRNAs (sg1 and sg2, sourced from Synthego) (Supplementary Table S2) were injected at 1.2 ng/embryo sgRNA mix with 800 pg Cas9 protein, per embryo.
Statistical analysis
Statistical analysis was performed using GraphPad prism. ANOVA and Student unpaired t-test and data were represented as mean ± SD, and a p-value less than 0.05 was considered statistically significant. Cell-based assays were repeated thrice and performed in duplicates. The zebrafish experiments were performed in three to five sets of injectants, each with n > 70 per group.
*Detailed methodology for immunoblotting, mitochondrial complex activity, oxygen consumption rate, mitochondrial morphology analysis, and mitochondrial membrane potential is available in the supplementary information.
Results
Clinical findings
P1 was born out of a consanguineous union (Fig. 1A) with unremarkable antenatal and perinatal history. He presented with refusal to feed, excessive crying, hurried breathing, poor peripheral circulation, and weak respiratory efforts on day 41 of life. There was a family history of a deceased elder sibling with similar complaints at two months of age. On clinical examination, his weight was 3.2 kg (−2.51 SD), length 48 cm (−3.68 SD), and head circumference 36.5 cm (−1.22 SD). The arterial blood gas analysis on admission was suggestive of severe metabolic acidosis, serum lactate level was >140 mg/dl (<27 mg/dl), blood creatinine phosphokinase was 631 U/L (46–171 U/L), and liver enzymes (SGOT – 129 IU/L; SGPT – 84.9 IU/L) were mildly elevated. 2D echo evaluation was suggestive of severe pulmonary arterial hypertension, dilated right atrium, and right ventricle with moderate tricuspid regurgitation. Tandem mass spectrophotometry (TMS) from blood showed a mild derangement of the acyl-carnitine profile, whereas the gas chromatography-mass spectrophotometry (GCMS) of urine was suggestive of significantly elevated ethyl malonic acid. He recovered from this episode of decompensation and was discharged after ten days of hospital admission. He was re-evaluated at six months and later at 13 months of age and was noted to have normal development and no recent episode of decompensation.

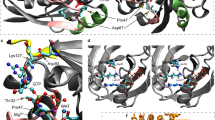
Pedigree of (A) Family 1 and (B) Family 2. Multiple sequence alignment shows (C) amino acid glutamine at 164 position and (D) arginine at 138 position is evolutionary conserved among Homo sapiens, Mus musculus, Bos taurus, Danio rerio, Drosophila melanogaster, Sus scrofa and Pan paniscus. E In silico protein modelling in MRPS2 protein structure shows (i) polar contacts (blue lines) of wild type Glu164 residue (red) with Ile142 (yellow) and Ala159 (pink) and in silico mutagenesis revealed that (ii) mutant Lys164 residue (purple) lost polar contact with Ile142 instead formed polar contact with Gly140 (dark green). F In silico protein modelling of MRPS2 and MRPS9 protein structures shows (i) wild type Glu164 residue (red) forms polar contacts (white lines) with Arg145 residue (coral) of MRPS9 and in silico mutagenesis revealed (ii) absence of polar contacts between mutant Lys164 residue (purple) of MRPS2 and any other residue of MRPS9. G In silico protein modelling of MRPS2 and MRPS23 protein structures shows (i) wild type Arg138 residue (light green) forms polar contacts (white lines) with Leu30 residue (brown) of MRPS23 and in silico mutagenesis revealed (ii) absence of polar contacts between mutant His138 (orange) of MRPS2 and any other residue of MRPS23. H Schematic representation of MRPS2 (NM_016034.5) and protein showing our variants (highlighted in red) and previously reported variants. RPS2:Ribosomal protein S2.
P2 is a one-year and five-month-old male born of a consanguineous union of family 2 (Fig. 1B). He was born of a normal vaginal delivery, and the perinatal period was uneventful. He developed generalized tonic-clonic type seizure at the age of one month. He was hospitalized at an external center where the random blood glucose was documented to be 12 mg/dl (70–150 mg/dl). The other hematological and biochemical investigations were within normal limits for his age. During admission, he had three more episodes of convulsions following a similar semiology and was further started on antiepileptics. The GCMS and TMS was performed, however, the report was not available for him. He was discharged in a stable condition and did not develop any further events of hypoglycemia-induced seizures. On evaluation at our center at one-year and five-months of age, he had been seizure-free for the past one year and was developmentally normal. His length was 78 cm (−1.46 SD), weight was 7.9 kg (−2.86 SD), and head circumference was 47 cm (−0.23 SD). Biochemical investigation revealed a random blood sugar level of <41 mg/dl whereas the other investigations were within normal limits. He had mild fullness of cheeks, mild tenting of vermilion of the upper lip, and multiple Mongolian spots over the lower lumbar region. On per abdomen examination, the liver was palpable 3 cm below the right costal margin and soft in consistency. The rest of the systemic examination was unremarkable.
Exome sequencing identifies bi-allelic variants in MRPS2
Singleton exome sequencing of P1 revealed a novel missense variant c.490 G > A p.(Glu164Lys) in exon 4 of MRPS2 (NM_016034.5) (NC_000009.12:g.135503732 G > A) in a homozygous state. This variant is observed in 26 individuals in heterozygous state in the gnomAD database (v4.1.0). Exome analysis for P2 identified a previously reported missense variant c.413 G > A p.(Arg138His) in exon 4 of MRPS2 (NM_016034.5) (NC_000009.12:g.135503655 G > A).
This variant is observed in 48 individuals in heterozygous state in the gnomAD database (v4.1.0). These variants are not observed in our in-house dataset of 3200 exomes. In silico tools such as Mutationtaster, CADD Phred and ClinPred were consistent in predicting the variants to be damaging to the protein function. Bi-allelic segregation by Sanger sequencing confirmed the carrier status of the parents of P1 and P2. The variant c.490 G > A p.(Glu164Lys) was classified as variant of uncertain significance and the variant c.413 G > A p.(Arg138His) was classified as pathogenic variant as per the American College of Medical Genetics and Genomics guidelines [17]. MSA performed using Clustal Omega showed the variant c.490 G > A and c.413 G > A being highly conserved across species (Fig. 1C, D).
In silico protein modeling and analysis
In silico mutagenesis of the p.Glu164Lys variant in MRPS2 protein structure revealed that the wildtype Glu164 forms polar contacts with Ile142 and Ala159 whereas mutant Lys164 forms polar contacts with Gly140 and Ala159 (Fig. 1E). Additionally, alterations in the interacting residues of MRPS2 with other small subunits of mitoribosomes revealed that mutant Lys164 resulted in the loss of polar interaction between MRPS2 and MRPS9 structures (Fig. 1F) On the other hand, in silico mutagenesis of the second variant p. Arg138His revealed no alterations of interactions with other residues of MRPS2. On analysis, it was observed that the wildtype residue Arg138 forms polar contacts with Leu30 residue of MRPS23 protein, however, mutant His138 does not form any polar contacts with any residue of MRPS23 (Fig. 1G).
Functional analysis of the variant, c.490G>A p.(Glu164Lys) in MRPS2
The molecular effect of the variant c.490 G > A was investigated by using primary skin fibroblast from P1 and appropriate control samples (C1 and C2). Quantitative analysis of MRPS2 expression by qRT-PCR showed reduced expression in P1 (Fig. 2A) compared with both the control samples (C1 and C2). Quantitative estimation of proteins by western blot revealed a significant reduction in MRPS2 as well as complex I (NDUFS1) and complex IV (MT-CO2, COX4) subunits in P1 (Fig. 2B (i-iv)). On proteomics analysis, a significant decrease in MRPS2, NDUFS1, and COX4 expression was noted in patient P1 when compared to controls (Supplementary Fig. 1A–C). Although the levels of MT-CO2 were also found to be decreased by proteomics analysis, this was not statistically significant (fold-change: 0.48, p = 0.2) (Supplementary Fig. 1D). All the detected small subunit components were found to be decreased, and most large subunit proteins were increased in P1 compared to controls (Fig. 2C). Analysis of cellular respiration by Seahorse XF24 Extracellular Flux Analyzer showed decreased OCR and increased extracellular acidification rate (ECAR), suggesting perturbed mitochondrial respiration capacity in fibroblast cells from P1 (Fig. 3A, B, Supplementary Fig. 2). We also observed reduced OXPHOS enzyme activity in P1, suggesting perturbed mitochondrial OXPHOS defect (Fig. 3C, D). A significant decrease in ATP levels in fibroblast cells of P1 was also observed when compared to control fibroblast cells suggesting perturbed mitochondrial ATP synthesis (Fig. 3E).

A qRT-PCR analysis showing relative MRPS2 mRNA expression in control 1 (C1), control 2 (C2) and patient (P1) cell lines. Expression of MRPS2 in P1 was significantly upregulated when compared to C1 and C2. β-Actin was used as the internal control; B Representative blot images for protein expression of MRPS2, NDUFS1, MT-CO2 and COX4; (i-iv) Western blot analysis of protein expression of MRPS2, NDUFS1, MT-CO2 and COX4 in C1, C2 and P1 fibroblast cell lines. Densitometric analysis was performed upon normalization of MRPS2, NDUFS1, MT-CO2 and COX4 protein band intensity to respective β-Actin band. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001; C Altered abundance of mitochondrial ribosomal proteins. A volcano plot showing alterations in the levels of various mitochondrial ribosomal subunit proteins. Small and large mitochondrial ribosomal subunit proteins are represented by red and green dots, respectively, as indicated. The horizontal dashed line corresponds to p = 0.05.

A Representative graph showing the OCR in control and P1 fibroblast cell lines. Injection of oligomycin, FCCP, and antimycin A and rotenone (A/R) are indicated (B) Representative graph showing the EACR in control and P1 cell lines. Injection of oligomycin, FCCP, and antimycin A and rotenone (A/R) are indicated (C) Mitochondrial complex I enzymatic activities in MRPS2 control and P1 cell lines (D) Mitochondrial complex IV enzymatic activities in control and P1 cell lines (E) Representative graph showing intracellular ATP levels. ATP determination kit was used to quantify intracellular ATP levels. *p < 0.05, **p < 0.001, ***p < 0.0001 and ****p < 0.00001.
Control and P1 mitochondria were visualized using MitoTracker Red staining and analyzed using confocal microscopy. Analysis of mitochondrial morphology and network showed reduced branch length in P1 mitochondria compared to control fibroblast mitochondria (Supplementary Fig. 3A, B). Staining for MMP with Rhodamine123 showed decreased fluorescence intensity in P1 compared to the control fibroblast cells, indicating reduced mitochondrial membrane potential (Supplementary Fig. 3C, D).
Loss of mrps2 in zebrafish impacts early development and mitoribosome subunits
To uncover the impact of a loss of mrps2 function during early development, we used the F0 knockout strategy. Guides targeting exons 2 and 3 were chosen based on editing efficiency and injected together to create F0 crispants (Fig. 4A). Extensive editing was observed at each guide target site, as shown in the representative image of the HMA PCR assay (Fig. 4B). Due to the lack of availability of an antibody that recognizes zebrafish Mrps2, we measured and observed a significant decrease in the mRNA levels of mrps2 in the crispants, presumably due to nonsense-mediated decay of the edited mRNA (Fig. 4C). The crispants and control larvae were raised to 7dpf with observation of developmental abnormalities daily. A subset of the mrps2 crispants showed abnormalities such as delayed or absent hatching, delayed yolk absorption, bent body or tail, severe oedema and dysmorphisms between 3 and 5dpf (Fig. 4D, E). As the number of injectants with biallelic genomic edits leading to a complete loss of function is likely to be low, the fraction of larvae that showed these abnormalities was also low. However, over five independent experiments, the number of larvae with abnormal phenotypes was consistently and significantly greater in the mrps2 crispants as compared to the NT (control) injectants. No significant differences in survival were observed until the age of 7dpf (Fig. 4F). In order to assess mitochondrial function, Complex IV activity was measured in mitochondria isolated from control and mrps2 crispants, and a slight reduction in activity was observed (Fig. 4G), similar to that detected in the patient fibroblasts. We also observed a decrease in the 12S rRNA levels in the mrps2 crispants, resulting in a significant reduction in the 12S/16S rRNA ratio (Fig. 4H), as observed in fibroblasts of P1, indicating a destabilization of the mitoribosome [8]. Further, transcript levels of mitoribosome complex subunits (complex I, IV, and V) were found to be reduced in the crispants (Fig. 4I).

A A schematic of the zebrafish mrps2 with the location of the selected sgRNA target sites indicated. The sequences of the two sgRNAs used in the study are provided in the table, B Assessment of editing efficiency for each sgRNA at 24 h post injection, by a heteroduplex mobility assay (HMA), C Relative expression levels of the mrps2 mRNA in the controls and injectants (crispants), D Representative bright field images of wild-type and mrps2 crispants (at 3dpf) illustrating abnormal developmental phenotypes observed in the crispants, as labeled, E, F Quantification of the number of larvae showing abnormal development at 5dpf (E) and survival at 7dpf (F) (5 experiments, NT n = 560, KO n = 820), G Complex IV activity measurement in mitochondria isolated from control and mrps2 crispants, H 12S to 16S rRNA ratio in the crispants compared to controls. Results from 3 to 5 independent experiments are quantified, and error bars represent SEM, (I) Relative mRNA levels of OXPHOS subunits genes including nd1, ndufs1 (Complex I), sdha (Complex II), cytb (Complex III), mt-co1 and mt-co2 (Complex IV), in the mrps2 crispants compared to controls.
Discussion
Combined oxidative phosphorylation deficiency is an autosomal recessive multisystem disorder with variable manifestations resulting from a defect in the mitochondrial oxidative phosphorylation (OXPHOS) system. Previously, four variants in MRPS2 have been reported in four unrelated individuals (Fig. 1H) [8,9,10]. The current report expands the phenotypic and genetic spectrum of MRPS2-related COXPD36 (MIM# 617950). The clinical presentations of P1 and P2, though variable, overlap with those of COXPD36 as well as several other OXPHOS protein defects. P1, who presented with early onset of metabolic decompensation and multisystem dysfunction, did not suffer any further episodes till the end of infancy. However, there is a history of early demise of his sibling with a similar episode in early infancy. Such acute episodes leading to early mortality have not been reported in other individuals with COXPD36. The clinical presentation of P2 with symptomatic hypoglycemia closely resembled the other reported individuals with COXPD36. Unfortunately, hearing evaluation could not be carried out for both P1 and P2 and is planned in their follow-up visit as sensorineural hearing loss has been documented in all four reported individuals [8,9,10] to date. Neuroimaging findings were unremarkable in individuals reported by Gardeitchik et al. [8] and Papadopoulos et al. (2023), however neuroimaging records were not available for individuals reported by Liu et al. [9] as well as current probands. Both P1 and P2 at present appear to have non-progressive symptoms as has been observed in other individuals with this condition. Clinical, biochemical, and genomic variant details of the present and reported individuals with MRPS2 defects have been provided in Table 1.
MRPS2 is a crucial structural component of the small subunit (mt-SSU) of the mitoribosome and plays an essential role in decoding and recruitment of mitochondrial messenger RNAs (mt-mRNAs). The intricate role of MRPS2 in mitoribosome assembly and mitochondrial translation underscores its importance in cellular bioenergetics and highlights the potential consequences of its dysfunction. It has been demonstrated that the loss of individual MRPS proteins leads to decreased 12S rRNA steady-state abundance, indicating the interdependence of these components for proper mitoribosomal function. In silico analysis suggested that the variant p.Glu164Lys would affect the intramolecular interactions within the MRPS2 protein as well as intermolecular interactions with other MRPS proteins which might lead to an unstable protein and overall, the mt-SSU. Concurrent to this, in our study, fibroblasts from P1 showed reduced mRNA and protein expression of MRPS2 when compared to control cells. Reduced MRPS2 expression level impacted mitochondrial protein translation with decreased mitochondrial OXPHOS proteins NDUFS1 (Complex I component), MT-CO2, and COX4 (Complex IV components), suggesting combined mitochondrial OXPHOS defect in P1 cell lines. In silico analysis for previously reported variant p.Arg138His, did not reveal any alterations within the MRPS2 protein structure. However, an absence of the interaction between mutant MRPS2 protein and MRPS23 was observed which is likely to impact mitoribosome assembly and thus mitochondrial protein synthesis as observed in patient fibroblasts in the previous study [8]. Additionally, to investigate the impact of the variant observed in P1 on individual subunits of the complexes, we performed proteomics analysis using LC-MS/MS. The findings are in keeping with a previous report where MRPS2-deficient fibroblasts were shown to have normal levels of the large subunit proteins on a background of impaired small subunit assembly [8] and likely indicate overall changes in mitoribosomal component synthesis and assembly in association with decreased availability of MRPS2.
The decreased assembly of mt-SSU and the resultant lack of functional mitoribosomes impairing mitochondrial translation due to defects in MRPS2 are likely to cause multiple OXPHOS deficiencies. Previously, Papadopoulos, T. et al. (2023) reported normal activities for complexes I, II, and III, but a marked decrease in the amount of complex IV and its enzymatic activity in MRPS2 mutant fibroblasts. We also found reduced enzymatic activities of mitochondrial complex I and IV in MRPS2 mutant cell lines along with depleted intracellular ATP levels. The functional consequences of the MRPS2 variant on mitochondrial function were further supported by altered bioenergetics in cell lines of P1. A decreased OCR and an increase in EACR indicated impaired mitochondrial respiration in mutant cells. Conversely, the increased ECAR reflected a shift towards glycolysis, suggesting metabolic adaptations in response to mitochondrial dysfunction caused by the MRPS2 variant. Perturbation in mitochondrial OXPHOS is known to reduce mitochondrial membrane potential [18]. Concurrently, Rhodamine123 staining showed decreased fluorescence intensity in cell lines of P1 as compared to control cells, indicating reduced membrane potential due to mitochondrial OXPHOS defect. Mitochondrial OXPHOS defect is also linked to altered mitochondrial structure. Imaging analysis showed reduced mitochondrial branch length, suggesting fragmented mitochondria in fibroblasts of P1.
Furthermore, we used the zebrafish model to evaluate the impact of mrps2 loss on the mitoribosome and early organ development. The crispants (F0 knockouts) are genetically mosaic but contain a stable genetic edit facilitating phenotypic studies from the larval to the adult stages. The F0 strategy is effective in measuring the direct consequences of the loss of a critical protein without the added layer of genetic adaptation or compensation that arises during the generation of a stable knockout line [19,20,21]. Previously, few human mitoribosomal proteins with orthologues in zebrafish have been investigated for their impact on developmental phenotypes and mitochondrial dysfunction. A morpholino mediated knockdown of Mrpl24 resulted in locomotor impairment and structural defects, arising from the impaired mitoribosome assembly [22]. A role for Mrpl4 in notch signaling was reported using the zebrafish model, however the authors did not comment on any mitochondrial or developmental phenotypes in the knockout [23]. In our studies in the mrps2 crispants, we found a significant decrease in the 12S rRNA levels, indicating the destabilization of the mitoribosome in these larvae, accompanied by changes in the expression of OXPHOS subunit genes and a reduction in Complex IV activity. This is likely the basis of the developmental delay in the crispants, seen in the form of delayed hatching and yolk absorption, shorter body length, oedema and generalized dysmorphisms. Also, similar to the observations in the case of mrpl24 morphants, we did not observe any impact on survival or a major locomotion deficit in the crispants (Supplementary Fig. 4), possibly due to the remaining mitoribosome activity and function.
In conclusion, we report additional individuals with variants in MRPS2, thus expanding the phenotypic and genotypic spectrum of COXPD36. We also demonstrate the pathogenicity of the novel variant added to this study through extensive in-vitro and omics assays performed on patient fibroblasts. We also demonstrate the impact of loss of mrps2 in zebrafish leading to severe developmental abnormalities.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD062317. The additional data supporting this study’s findings are available from the corresponding authors upon reasonable request.
References
Koc EC, Cimen H, Kumcuoglu B, Abu N, Akpinar G, Haque ME, et al. Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front Physiol. 2013;4:183.
Rorbach J, Gao F, Powell CA, D’Souza A, Lightowlers RN, Minczuk M, et al. Human mitochondrial ribosomes can switch their structural RNA composition. Proc Natl Acad Sci USA. 2016;113:12198–201.
Bieri P, Greber BJ, Ban N. High-resolution structures of mitochondrial ribosomes and their functional implications. Curr Opin Struct Biol. 2018;49:44–53.
Ferrari A, Del’Olio S, Barrientos A. The diseased mitoribosome. FEBS Lett. 2021;595:1025–61.
Amunts A, Brown A, Toots J, Scheres SHW, Ramakrishnan V. Ribosome. The structure of the human mitochondrial ribosome. Science. 2015;348:95–8.
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Prim. 2016;2:16080.
Bursle C, Narendra A, Chuk R, Cardinal J, Justo R, Lewis B, et al. COXPD9 an evolving multisystem disease; congenital lactic acidosis, sensorineural hearing loss, hypertrophic cardiomyopathy, cirrhosis and interstitial nephritis. JIMD Rep. 2017;34:105–9.
Gardeitchik T, Mohamed M, Ruzzenente B, Karall D, Guerrero-Castillo S, Dalloyaux D, et al. Bi-allelic mutations in the mitochondrial ribosomal protein MRPS2 cause sensorineural hearing loss, hypoglycemia, and multiple OXPHOS complex deficiencies. Am J Hum Genet. 2018;102:685–95.
Liu C, Zhou W, Liu Q, Peng Z. Hypoglycemia with lactic acidosis caused by a new MRPS2 gene mutation in a Chinese girl: a case report. BMC Endocr Disord. 2022;22:15.
Papadopoulos T, Gaignard P, Schiff M, Rio M, Karall D, Legendre A, et al. New description of an MRPS2 homozygous patient: Further features to help expend the phenotype. Eur J Med Genet. 2024;67:104889.
Kausthubham N, Shukla A, Gupta N, Bhavani GS, Kulshrestha S, Das Bhowmik A, et al. A data set of variants derived from 1455 clinical and research exomes is efficient in variant prioritization for early-onset monogenic disorders in Indians. Hum Mutat. 2021;42:e15–61.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Ligezka AN, Radenkovic S, Saraswat M, Garapati K, Ranatunga W, Krzysciak W, et al. Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications. Ann Neurol. 2021;90:887–900.
Adiga D, Bhat S, Shukla V, Shah HV, Kuthethur R, Chakrabarty S, et al. Double C-2 like domain beta (DOC2B) induces calcium dependent oxidative stress to promote lipotoxicity and mitochondrial dysfunction for its tumor suppressive function. Free Radic Biol Med. 2023;201:1–13.
Singh S, Danda S, Sharma N, Shah H, Madhuri V, Mir TA, et al. Biallelic variants in CCN2 underlie an autosomal recessive kyphomelic dysplasia. Eur J Hum Genet EJHG. 2025;33:30–7.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet. 2015;17:405–24.
Liu S, Liu S, He B, Li L, Li L, Wang J, et al. OXPHOS deficiency activates global adaptation pathways to maintain mitochondrial membrane potential. EMBO Rep. 2021;22:e51606.
Quick RE, Buck LD, Parab S, Tolbert ZR, Matsuoka RL. Highly efficient synthetic CRISPR RNA/Cas9-based mutagenesis for rapid cardiovascular phenotypic screening in F0 Zebrafish. Front Cell Dev Biol. 2021;9:735598.
Kroll F, Powell GT, Ghosh M, Gestri G, Antinucci P, Hearn TJ, et al. A simple and effective F0 knockout method for rapid screening of behaviour and other complex phenotypes. eLife. 2021;10:e59683.
Bek JW, Shochat C, De Clercq A, De Saffel H, Boel A, Metz J, et al. Lrp5 mutant and crispant zebrafish faithfully model human osteoporosis, establishing the zebrafish as a platform for CRISPR-based functional screening of osteoporosis candidate genes. J Bone Min Res J Am Soc Bone Min Res. 2021;36:1749–64.
Di Nottia M, Marchese M, Verrigni D, Mutti CD, Torraco A, Oliva R, et al. A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly. Neurobiol Dis. 2020;141:104880.
Mo D, Liu C, Chen Y, Cheng X, Shen J, Zhao L, et al. The mitochondrial ribosomal protein mRpL4 regulates Notch signaling. EMBO Rep. 2023;24:e55764.
Acknowledgements
We sincerely acknowledge and thank the families for their consent and participation in this study. We are grateful to all the referring physicians who made this work possible. We would like to acknowledge and thank “DBT/Wellcome Trust India Alliance” (IA/CRC/20/1/600002) and the Department of Health Research (R.11014/33/2023-GIA/HR) for funding the study. The authors gratefully acknowledge infrastructure funding from DST-FIST, K-FIST, TIFAC-CORE, DBT-BUILDER, Department of Medical Genetics (KMC), Manipal School of Life Sciences, and Manipal Academy of Higher Education. We also thank Vivek Ghose, Deepti Tomar, and Anu Jain for their support.
Funding
DBT/Wellcome Trust India Alliance for the study, “Centre for Rare Disease Diagnosis, Research and Training” (IA/CRC/20/1/600002). Department of Health Research for the study, “Delineating the genomic basis of neurodegeneration and mitochondrial disorders associated with defective DNA break repair” (R.11014/33/2023-GIA/HR). Open access funding provided by Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Contributions
AK, PH, HS and RM performed the experiments. MY, VB, SM, SH, and A.Shukla took clinical care of the patients and provided advice. MY, NM, AA, VB, and RP analyzed and interpreted the clinical data. SC and AK have validated and analyzed the molecular data. HS and A.Sevilimedu analyzed and interpreted the zebrafish data. KG and AP contributed to proteomics and data analysis. AK and MY wrote the manuscript. SC, A. Sevilimedu, A. Shukla led and supervised the work and designed the experiments. All authors edited the paper with input from the other authors, approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study is approved by the institutional ethics committee, Kasturba Medical College, and Kasturba Hospital (IEC: 363/2020).
Animal ethics procedures and approval
All experiments with zebrafish were done in a CCSEA (previously CPCSEA)-approved zebrafish facility at Dr. Reddy’s Institute of Life Sciences (1100/po/Re/s/07/CPCSEA) in Hyderabad, India. The facility also has US-NIH OLAW assurance (F22-00539). All procedures and protocols were reviewed and approved by the Institutional Animal Ethics Committee (Protocol approval DRILS/IAEC/AS/2021-1). The “Guidelines for Experimentation on Fishes, 2021” published by CPCSEA was used as a reference.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kandettu, A., Yeole, M., Sekar, H. et al. Further delineation of defects in MRPS2 causing human OXPHOS deficiency and early developmental abnormalities in zebrafish. Eur J Hum Genet 33, 1451–1461 (2025). https://doi.org/10.1038/s41431-025-01858-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01858-1
This article is cited by
-
Insights in genetics: from molecular mechanisms to patient perspectives
European Journal of Human Genetics (2025)
