
Abstract
Craniofrontonasal syndrome (CFNS; MIM #304110) is a rare craniofacial disorder characterized by hypertelorism, a broad nasal root with a bifid nasal tip, orofacial clefting, and genital malformations caused by pathogenic variants in the X-linked gene EFNB1 (MIM *300035). CFNS exhibits sex-specific heterogeneity, with increased severity in females likely secondary to cellular interference related to random X-inactivation, resulting in mosaic EFNB1 expression. Previous studies have identified over 140 variants in EFNB1, but approximately 20% of CFNS have negative molecular testing, either due to a yet undiscovered causal gene or causal variants in regulatory regions not covered by traditional genetic testing methodologies. Here, we report a two-generation family with a clinical diagnosis of CFNS and negative clinical molecular testing. Research short-read genome testing identified a 2-Mb inversion together with two smaller deletions (13- and 7-bp), about 106-Kb downstream of EFNB1, which cosegregated with CFNS. Patient-derived fibroblasts reprogrammed into induced pluripotent stem cells (iPSCs) demonstrated two distinct iPSC populations in affected females, where one or other of the two X chromosomes was inactivated. In vitro assays further demonstrated that iPSCs with the active X chromosome bearing the inversion, exhibited a significant increase in EFNB1 expression, suggesting allelic imbalance contributes to mosaic EFNB1 expression. These findings nominate a novel causal variant type of CFNS, conclude a 43-year diagnostic odyssey for an affected family, and offer new hope for family planning for affected individuals.
Similar content being viewed by others

Introduction
Craniofrontonasal syndrome (CFNS; MIM #304110) was initially described by two groups in 1979 (reference [1, 2]) as a craniofacial syndrome characterized by hypertelorism, a broad nasal root with a bifid nasal tip, orofacial clefting, dry, wooly hair, mild skeletal abnormalities, and genital malformations. Females often have more severe disease with multisystem involvement, in contrast to males, where hypertelorism can be the only presenting feature. In 2004, genetic variants in the X-linked gene EFNB1 (MIM *300035), encoding the transmembrane signaling molecule ephrin-B1, were identified as the underlying cause of CFNS [3, 4].
This paradoxical difference in disease severity between males and females, termed cellular interference, was first characterized in 2004 and relates to random X-inactivation, which forms two distinct cell populations, each expressing either the wild-type or mutant allele [3,4,5]. The coexistence of these two cell populations subsequently gives rise to mosaic EFNB1 expression, which disrupts cell adhesion and contributes to the abnormal tissue boundary formation. This hypothesis was supported by the subsequent identification of severely affected males carrying mosaic variants in the EFNB1 gene [6] and robust cell segregation observed in mixed human neuroepithelial cells with mosaic EFNB1 expression [7].
While previous studies have identified more than 140 causal variants in the EFNB1 gene, including upstream noncoding [6, 8], nonsense, missense, frameshift, splice-altering, duplication [9] and deletion [10, 11] variants, approximately 20% of CFNS patients have negative molecular testing [10, 12, 13]. In this study, we report the discovery of a 2-Mb inversion, about 106-Kb downstream of EFNB1, cosegregating with CFNS in a two-generation pedigree revealed by short-read genome sequencing. To evaluate the pathogenicity of the inversion, we reprogrammed patient-derived fibroblasts and an X-inactivation assay was used to determine which X was active in each individual clone once isolated. We further demonstrated that cells bearing the inversion allele on the active X chromosome had a significant increase in EFNB1 expression. These findings nominate a novel variant type as causal for CFNS, conclude a 43-year diagnostic odyssey for an affected family, and offer new hope for family planning for affected individuals.
Materials and methods
Genome sequencing and analysis
Whole genome sequencing was performed on four family members (I-2, II-1, II-4 and III-2) after negative exome (see Supplementary Materials and Methods) using an Illumina NovaSeq 6000 sequencer (Illumina, Inc., San Diego, CA, USA) with paired-end 100 bp reads. Libraries were generated from genomic DNA using the Illumina TruSeq DNA PCR-Free Library Prep Kit (Illumina, San Diego, CA). All the raw reads were aligned to the reference human genome using the Burrows-Wheeler Aligner [14] (BWA-Mem v0.7.12). BAM files were then processed by two short-read structural variant callers, Manta [15] and Wham [16], to capture structural variations (SVs) with default parameters. Similarly, the split and discordant reads files were generated by SpeedSeq [17] and were provided as inputs to Lumpy [18], another SV calling program. Confirmatory genotyping of the identified inversion was performed by polymerase chain reaction (PCR) and Sanger sequencing by using primers 5’-AAGGGGAGACTGGACTTCTTG-3’ and 5’-TCGAAGTCAGATCTCCAAATGT-3’, and 5’-TACCACCATTCAGACGGGAAA-3’ and 5’-AGGAGTTCAAGGCAGCAGT-3’. Thermal cycling condition was: 95 °C:5 m - (96 °C:5 s - 58 °C:5 s - 68 °C:40 s) × 35 - 72 °C:5 m - 4 °C:hold.
Fibroblast reprograming to induced pluripotent stem cells (iPSCs) and molecular karyotyping
Fibroblasts from II-1 and II-4 were cultured following standard techniques. Fibroblasts were transduced using CytoTune-iPS 2.0 Sendai Reprogramming vectors containing polycistronic hKlf4–hOct3/4–hSox2 (hKOS), hcMyc, and hKlf4 (Thermo Fisher Scientific, Waltham, MA). Following transduction, cells were plated on tissue culture plates coated with Geltrex (Thermo Fisher Scientific, Waltham, MA) in Essential 8 media (Thermo Fisher Scientific, Waltham, MA). Once colonies began to form, they were picked using a pipet tip and media was switched to StemFlex (Thermo Fisher Scientific, Waltham, MA). To ensure no copy number variations (CNVs) occurred as a result of iPSC reprograming, DNA isolated from multiple colonies were genotyped with the Illumina Infinium Global Screening Array (llumina, San Diego, CA). The PennCNV algorithm [19] was used for CNV calling and aneuploidy was screened with GenomeStudio.
X-chromosome inactivation (XCI) assay
From each of the original iPSC inductions, 20 separate individual clones were picked. Genomic DNA was isolated from each of these 20 clones. X-chromosome inactivation (XCI) analysis was performed at the androgen receptor (AR) triplet repeat locus, where the inactive X is methylated and, therefore, selectively retained after HpaII digestion. Following DNA methylation-sensitive restriction digestion with HpaII for each sample, the predigested and undigested samples were amplified by PCR. Fragment size data for digested and undigested pairs for each sample were collected using a capillary electrophoresis instrument to determine the XCI pattern [20].
Real time quantitative reverse transcription PCR (RT-qPCR)
RT-qPCR was performed following the manufacturer’s protocol. For each sample/colony, RNA was extracted with using the RNeasy Mini Kit with on-column DNaseI digestion (Qiagen, catalog nos. 74104 and 79254), according to the manufacturer’s instructions and reverse transcription was done with the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, catalog no. 4368814) using random primers. Taqman assay primers (Thermofisher), LIN28A, NANOG, OCT4 and SOX2 were used to check for iPSC markers expression and SeV, hKOS, hKlf4 and hcMyc primers (Thermo Fisher Scientific, Waltham, MA) were used to check for virus vector detection. Quantification of EFNB1 expression was performed using the Fast SYBR Green PCR kit (Thermofisher). The data were analyzed by using the ΔΔct method, determined by RT-qPCR, and were normalized to GAPDH in same cDNA samples with values expressed as a percentage of the GAPDH control. The primers for EFNB1 and GAPDH are 5’-CAATAGGCCAGAGCAGGAAATA-3’ and 5’- CTTCCATTGGATGTTGAGGTAATG-3’, and 5’-CTTTGGTATCGTGGAAGGACTC-3’ and 5’-AGTAGAGGCAGGGATGATGT-3’, respectively. Thermal cycler condition was: 95 °C:20 s - (95 °C:3 s - 60 °C:30 s) × 40.
Western blotting of iPSC lysates
Whole cells were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (complete protease inhibitor cocktail tablets, Roche, Mannheim, Germany) and phosphatase inhibitors sodium fluoride and sodium metavanadate. Approximately 60 micrograms of protein were separated on NuPAGE 4–12% Bis–Tris gels run with MOPS SDS running buffer (Life Technologies, Carlsbad, CA). Proteins were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), and membranes were blocked with 5% bovine serum albumin (BSA; Thermofisher) in phosphate buffered saline containing 0.1% tween20 (PBST) for 30 min at room temperature. Membranes were incubated with primary antibodies diluted in 5% BSA in TBST [goat anti-Ephrin-B1(R&D Systems, Minneapolis, MN, catalog number AF473, 500 ng/ml final concentration); mouse anti-beta-actin (Santa Cruz, catalog number AC-15, 500 ng/ml final concentration)] overnight at 4 °C. Following 3 washes (5 minutes each) with PBST, membranes were incubated with horse radish peroxidase-conjugated donkey-anti-goat (R&D Systems, catalog number HAF017) or horse-anti-mouse (Cell Signaling Technology, Danvers, MA; catalog number 7076) secondary antibody diluted 1:10,000 in PBST for 30 min at room temperature. Following 3 more washes with PBST, western blots were developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher) and imaged using a KwikQuant Imager (Kindle Biosciences, Greenwich CT).
Results
Clinical report and clinical diagnostic studies
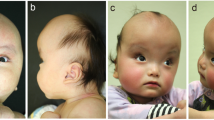
The proband (II-4; Fig. 1A), now 43 years old, was first seen in 1980 at 9 months of age by Clinical Genetics, presenting with right coronal synostosis, hypertelorism, and a bifid nose. Family history was notable for one sister (II-1; Fig. 1B) with left coronal synostosis, hypertelorism, short columella, and a flat nasal tip noted at 4 years of age and a second sister (II-2) with a nasal cleft and hypertelorism without craniosynostosis. All three siblings were diagnosed clinically with CFNS. Their father (I-2) was noted to have hypertelorism and a slight widow’s peak but was otherwise unaffected. Both of the proband’s daughters (III-2 and III-3) were diagnosed clinically with CFNS. The clinical course for individual III-2 is additionally complicated by short stature, poor weight gain, strabismus, pulmonary stenosis and vascular ring (Fig. 1A). Genetic testing, including a karyotype, microarray, MLPA study for 22q11.2 deletion, and EFNB1 gene sequencing and deletion/duplication tests, was negative.

A Pedigree and cosegregation analysis. B Facial photographs of individual II-1 at 10 months old (before surgical repairs) and 46 years old (post synostosis and hypertelorism repairs), respectively.
Additional molecular investigations with exome and genome sequencing
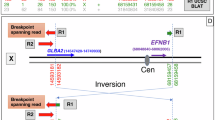
Exome sequencing followed by Sanger validation in Individual III-2 revealed a de novo pathogenic PTPN11 variant (c.236A>G, p.(Gln79Arg)), consistent with Noonan syndrome, accounting for her history of short stature, pulmonic stenosis, and failure to thrive, but not her CFNS clinical diagnosis (Fig. 1A). To identify the molecular basis of the CFNS phenotype, we performed genome sequencing, which revealed a heterozygous 2-Mb inversion at Xq13.1, about 106-Kb downstream of the EFNB1 gene, captured by Lumpy with support from Manta. Follow-up PCR and Sanger sequencing determined that the inversion spanned from chrX(GRCh37):g.68,167,790 to chrX(GRCh37):g.70,181,179 with 2-bp microhomology sequences at both breakpoints (NC_000023.10:g.68167790_70181179inv; Fig. 2A, B). Interestingly, two small deletions were also identified at both ends of the inversion: NC_000023.10:g.68167777_68167789del (13-bp) and NC_000023.10:g.70181180_70181186del (7-bp; Fig. 2A, B). Query of the ENCODE dataset [21] demonstrated that the 5’ end of the inversion and the 13-bp deletion overlap with a cis-regulatory element (ccRE; ENCODE #: EH38E2757328). This ccRE has maximum DNase, H3K4me3, H3K27ac, and CTCF Z scores of 3.50, 1.19, 1.17, 3.48, respectively, suggesting that it possesses a CTCF-bound insulator property.

A Sanger sequencing precisely mapped the breakpoints and confirmed the cosegregation pattern. B Simplified diagram showed the inversion together with two smaller deletions at both ends. C X-inactivation analysis in two affected family members (II-4 [proband) and II-1) revealed that each individual has two distinct cell populations. By analyzing AR locus fragment sizes as markers and recognizing that both individuals inherited the same X chromosome from their father, the allele with a fragment size of 279 was determined to harbor the EFNB1 inversion (also see Supplementary Fig. 1).
Characterization of the impact of the inversion downstream of EFNB1 in patient-derived iPSCs
Given the overlap, we hypothesize that the identified inversion and 13-bp deletion telomeric to EFNB1 may disrupt a three-dimensional CTCF-bound topologically associated domain (TAD), altering EFNB1 expression and contributing to allelic imbalance. To test this hypothesis, we obtained skin biopsies from two affected family members (II-1 and II-4) and reprogrammed the fibroblasts to generate stem cell lines. Genomic integrity assessment of all studied clones was assessed using the Infinium Global Screening Array, which revealed no copy number variations and no aneuploidy of any chromosomes. The iPSC lines generated in this study exhibited typical hallmarks of pluripotency, including the expression of endogenous pluripotency genes (data not shown). X-inactivation analysis was performed on 20 separate individual clones from each of the original iPSC inductions and revealed two cell populations with different inactivated X chromosomes (Fig. 2C; Supplementary Table 1). By analyzing fragment sizes and knowing that both individuals inherited the same X chromosome from their father, we were able to identify the clones bearing the inversion allele on the active X chromosome in the two affected individuals (Fig. 2C; Supplementary Fig. 1). Subsequently, RT-qPCR demonstrated significantly increased EFNB1 expression in cells bearing the inversion allele on the active X chromosome in both individuals (Fig. 3A). This increase in EFNB1 expression was further validated by Western blot analysis of iPSC whole cell lysates (Fig. 3B, C). Together, these data suggest that the inversion regulates EFNB1 expression through a long-range effect, leading to a relatively higher EFNB1 expression level.

A RT-qPCR analysis showed significantly increased EFNB1 expression in clones in which the active X chromosome bears the inversion compared to clones in which active X chromosome bears the wildtype allele in both II-4 and II-1. Normalized values are illustrated by the box-and-whisker plot with all values. **P < 0.01 by two-tailed, unpaired t test. B Representative Western blot image confirmed the reduced EFNB1 level in both individuals. C Quantification analysis of Western blot. Normalized valued are illustrated by the box-and-whisker plot with all values. **P < 0.01; ****P < 0.0001 by two-tailed, unpaired t test.
Discussion
While deletions and duplications involving the EFNB1 gene have previously been identified as causes for CFNS, the occurrence of an inversion involving only the regulatory region has not, to our knowledge, been described in CFNS. While this work was in progress, a patient with CFNS and a pericentric inversion involving the entire EFNB1 gene was reported [22]. It was proposed that the pericentric inversion disrupts interaction with a specific enhancer (GH0XJ068989) located ~150 kb downstream of EFNB1, within the inverted region identified in our study. However, the observed increase in EFNB1 expression levels suggests that the enhancer landscape may be more complex than previously thought. As multiple candidate enhancers exist [22], further investigation will be needed to pinpoint the functional enhancer(s) for EFNB1. Recently, genome sequencing has emerged as comprehensive single-test approach in molecular diagnosis, offering several advantages [23,24,25,26,27,28], including improved the variant calling of both small variants and large structural variations due to its more uniform coverage across the entire genome compared to exome sequencing. Here, we illustrate its molecular diagnostic utility in a two-generation pedigree, following a 43-year observational period and extensive genetic investigations.
CFNS represents one of the three disorders where females harboring pathogenic heterozygous variants are more severely affected than males who carry hemizygous variants. The other two examples are PCDH19-related neurodevelopmental disorder [29] (MIM #300088) and ARR3-related early-onset high myopia [30] (MIM #301010). The suspected underlying mechanism driving this disparity is that random X-inactivation in heterozygous females produces a mixture of two cell populations [29, 31], causing patchy or mosaic gene expression that disrupts cell-cell communication and adhesion, ultimately leading to the aberrant tissue boundary formation. This cellular interference model is supported for EFNB1 by animal studies, which demonstrate that female mice with heterozygous Efnb1 deficiency have a more severe phenotype than either hemizygous male or homozygous KO female mice [32].
Given the overlap between the identified rearrangement and a ccRE (E2757328; chrX(GRCh37):g.68,167,666_68,167,985), which was classified CTCF-only/CTCF-bound by the ENCODE project based on high DNase and CTCF scores, along with low H3K4me3 and H3K27ac scores, we predicted that the rearrangement might disrupt or relocate the CTCF binding site in a different orientation, potentially disrupting the formation of a TAD or creating an alternative TAD loop. Several studies have shown that inversion of CTCF-binding sites can disrupt chromosomal loops, which in turn leads to increased or decreased gene expression [33, 34]. We initially hypothesized that the X chromosome expressing the identified rearrangement would reduce EFNB1 expression levels; however, contrary to our expectation, it led to elevated EFNB1 mRNA and EFNB1 protein levels compared to the normal X chromosome. Interestingly, duplications of the EFNB1 region have been previously reported in patients with hypertelorism [9, 35], and it has been shown that the X chromosome bearing the EFNB1 duplication produces more EFNB1 transcript than the normal X chromosome [9]. Our results further support the idea that an allelic imbalance in EFNB1 expression could lead to a phenotype consistent with loss-of-function EFNB1 variants, even though the detailed mechanism remains elusive.
Our study also demonstrates the importance of broad based testing to identify a dual diagnosis. Specifically, exome sequencing for subject III-2 unexpectedly identified a de novo pathogenic PTPN11 variant. This variant explains her additional clinical presentations, including heart defects and poor weight gain. This highlights the significance of analyzing each affected family member individually when conducting genetic analysis. In this family, when four samples were analyzed collectively under the assumption of either X-linked or autosomal dominant inheritance pattern, the pathogenic PTPN11 variant would not have been identified. A clinical diagnosis of Noonan syndrome would have been difficult, given the blended dysmorphology of Noonan syndrome and CFNS. The Noonan syndrome diagnosis had significant implications for her clinical care. Notably, she experienced substantial blood loss during a previous craniosynostosis surgery (documented at ~37cc/kg) and menorrhagia. Upon receiving a Noonan syndrome diagnosis, she was evaluated by Hematology and found to have mild factor VII, IX, and XI deficiencies, underscoring the necessity for hemostasis planning prior to any procedures in the future.
Over several years, the proband (II-4) had two children who are similarly affected with CFNS. If this specific inversion downstream of EFNB1 had been identified early on, it could have provided reproductive options and enhanced decision-marking.
In summary, our study serves a compelling reminder of the necessity of comprehensive genetic analysis to uncover concealed genetic variants. Coupling with the analysis of long-range effects of the rearrangement in patient-derived iPSCs, our study brings closure to a 43-year-long molecular diagnostic journey for CFNS.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Cohen MM Jr. Craniofrontonasal dysplasia. Birth Defects Orig Artic Ser. 1979;15:85–89.
Slover R, Sujansky E. Frontonasal dysplasia with coronal craniosynostosis in three sibs. Birth Defects Orig Artic Ser. 1979;15:75–83.
Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, et al. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet. 2004;74:1209–15.
Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci USA. 2004;101:8652–7.
Wieland I, Makarov R, Reardon W, Tinschert S, Goldenberg A, Thierry P, et al. Dissecting the molecular mechanisms in craniofrontonasal syndrome: differential mRNA expression of mutant EFNB1 and the cellular mosaic. Eur J Hum Genet. 2008;16:184–91.
Twigg SR, Babbs C, van den Elzen ME, Goriely A, Taylor S, McGowan SJ, et al. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Genet. 2013;22:1654–62.
Niethamer TK, Larson AR, O’Neill AK, Bershteyn M, Hsiao EC, Klein OD, et al. EPHRIN-B1 mosaicism drives cell segregation in craniofrontonasal syndrome hiPSC-derived neuroepithelial cells. Stem Cell Rep. 2017;8:529–37.
Romanelli Tavares VL, Kague E, Musso CM, Alegria TGP, Freitas RS, Bertola DR, et al. Craniofrontonasal syndrome caused by introduction of a novel uATG in the 5’UTR of EFNB1. Mol Syndromol. 2019;10:40–47.
Babbs C, Stewart HS, Williams LJ, Connell L, Goriely A, Twigg SR, et al. Duplication of the EFNB1 gene in familial hypertelorism: imbalance in ephrin-B1 expression and abnormal phenotypes in humans and mice. Hum Mutat. 2011;32:930–8.
Twigg SR, Matsumoto K, Kidd AM, Goriely A, Taylor IB, Fisher RB, et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: frequent somatic mosaicism and explanation of the paucity of carrier males. Am J Hum Genet. 2006;78:999–1010.
Wieland I, Weidner C, Ciccone R, Lapi E, McDonald-McGinn D, Kress W, et al. Contiguous gene deletions involving EFNB1, OPHN1, PJA1 and EDA in patients with craniofrontonasal syndrome. Clin Genet. 2007;72:506–16.
Wieland I, Reardon W, Jakubiczka S, Franco B, Kress W, Vincent-Delorme C, et al. Twenty-six novel EFNB1 mutations in familial and sporadic craniofrontonasal syndrome (CFNS). Hum Mutat. 2005;26:113–8.
Wallis D, Lacbawan F, Jain M, Der Kaloustian VM, Steiner CE, Moeschler JB, et al. Additional EFNB1 mutations in craniofrontonasal syndrome. Am J Med Genet A. 2008;146A:2008–12.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2.
Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, et al. Wham: identifying structural variants of biological consequence. PLoS Comput Biol. 2015;11:e1004572.
Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12:966–8.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–74.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39.
Consortium EP, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710.
Kakadia PM, Fritz B, Bohlander SK. Craniofrontonasal syndrome in a patient with an inv(X)(p22.2q13.1), separating EFNB1 from its enhancer. Eur J Hum Genet. 2025;33:546–51.
Li D. Genome sequencing as a single comprehensive test in molecular diagnosis. Eur J Hum Genet. 2023;31:3–4.
Nomakuchi TT, Teferedegn EY, Li D, Muirhead KJ, Dubbs H, Leonard J et al. Utility of genome sequencing in exome-negative pediatric patients with neurodevelopmental phenotypes. Am J Med Genet A. 2024;194:e63817.
Schobers G, Derks R, den Ouden A, Swinkels H, van Reeuwijk J, Bosgoed E, et al. Genome sequencing as a generic diagnostic strategy for rare disease. Genome Med. 2024;16:32.
Wojcik MH, Lemire G, Berger E, Zaki MS, Wissmann M, Win W, et al. Genome sequencing for diagnosing rare diseases. N Engl J Med. 2024;390:1985–97.
Lowther C, Valkanas E, Giordano JL, Wang HZ, Currall BB, O’Keefe K, et al. Systematic evaluation of genome sequencing for the diagnostic assessment of autism spectrum disorder and fetal structural anomalies. Am J Hum Genet. 2023;110:1454–69.
Pagnamenta AT, Yu J, Walker S, Noble AJ, Lord J, Dutta P, et al. The impact of inversions across 33,924 families with rare disease from a national genome sequencing project. Am J Hum Genet. 2024;111:1140–64.
Gecz J, Thomas PQ. Disentangling the paradox of the PCDH19 clustering epilepsy, a disorder of cellular mosaics. Curr Opin Genet Dev. 2020;65:169–75.
Xiao X, Li S, Jia X, Guo X, Zhang Q. X-linked heterozygous mutations in ARR3 cause female-limited early onset high myopia. Mol Vis. 2016;22:1257–66.
Depienne C, LeGuern E. PCDH19-related infantile epileptic encephalopathy: an unusual X-linked inheritance disorder. Hum Mutat. 2012;33:627–34.
Compagni A, Logan M, Klein R, Adams RH. Control of skeletal patterning by ephrinB1-EphB interactions. Dev Cell. 2003;5:217–30.
Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162:900–10.
de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, et al. CTCF binding polarity determines chromatin looping. Mol Cell. 2015;60:676–84.
Petit F, Andrieux J, Holder-Espinasse M, Bouquillon S, Pennaforte T, Storme L, et al. Xq12q13.1 microduplication encompassing the EFNB1 gene in a boy with congenital diaphragmatic hernia. Eur J Med Genet. 2011;54:e525–527.
Funding
This study was supported in part by an Eagles Autism Foundation grant (to DL).
Author information
Authors and Affiliations
Contributions
DL conceptualized the study. DL, SD, DMM, LW, JT, EJB, HH, and EHZ conducted molecular and clinical evaluation of the affected individuals. DL, LSM, CH, and AS performed functional assessment and validation. DL wrote the original draft of the manuscript. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The Institutional Review Board of the Children’s Hospital of Philadelphia approved this study. Informed consent was obtained from all participants included in the study. The individual III-2 was consented for publication of photos.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, D., Matsuoka, L.S., Donoghue, S. et al. Modeling the long-range effect of an inversion downstream of EFNB1 concludes a 43-year molecular diagnostic odyssey for craniofrontonasal syndrome. Eur J Hum Genet 33, 1684–1689 (2025). https://doi.org/10.1038/s41431-025-01887-w
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01887-w
