
Abstract
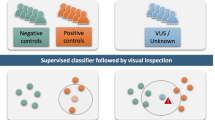
Disease-causing variants in chromatin regulator genes cause many developmental disorders. DNA methylation (DNAm) signatures are emerging as a diagnostic tool to identify disease causes and classify variants of uncertain significance (VUS). This study evaluates their diagnostic utility in a routine clinical setting. We retrospectively analyzed 298 patients from the Erasmus MC who underwent DNAm signature testing using the commercial EpisignTM platform between February 2019 and June 2023. The cohort included 75 targeted analyses for follow-up on prior genetic findings and 223 complete analyses for cases unsolved after prior diagnostic testing. In the 75 targeted analyses, DNAm signatures were positive in 18% (10/55) for (VUS), 91% (10/11) for likely pathogenic variants, and 89% (8/9) for pathogenic variants. In 223 complete analyses, a disease-linked DNAm signature was observed in 9.0% (20/223), with a (partial) phenotypic match in 55% of those (11/20) but no match in 45% (9/20). In 81.8% (9/11) of those DNAm signature positive cases with a phenotypic match, retrospective analysis identified a causative DNA variant or confirmed independently an imprinting disorder that was unidentified previously, providing valuable diagnostic insights with an overall diagnostic yield of 4.0% (9/223) for these molecular confirmed cases. In conclusion, this study supports the clinical utility of DNAm signatures to assist in interpreting and classifying VUS, but also as a complementary tool when prior genetic testing, including exome sequencing, failed to provide a diagnosis.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

Data availability
All relevant data are included in this paper. Underlying raw genetic and methylation data cannot be shared due to the given consent under which the individuals were recruited.
Code availability
All relevant data are within the paper or its Supporting Information files.
References
Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413–21.
Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607.
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1:397–400.
Weksberg R, Shuman C, Beckwith JB. Beckwith–Wiedemann syndrome. Eur J Hum Genet. 2009;18:8.
Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5:1–8.
Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25:1473.
Kerkhof J, Rastin C, Levy MA, Relator R, McConkey H, Demain L, et al. Diagnostic utility and reporting recommendations for clinical DNA methylation episignature testing in genetically undiagnosed rare diseases. Genet Med. 2024;26:101075.
Levy MA, McConkey H, Kerkhof J, Barat-Houari M, Bargiacchi S, Biamino E, et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. Hum Genet Genom Adv. 2022;3:100075.
Kerkhof J, Squeo GM, McConkey H, Levy MA, Piemontese MR, Castori M, et al. DNA methylation episignature testing improves molecular diagnosis of Mendelian chromatinopathies. Genet Med. 2022;24:51–60.
Magini P, Smits DJ, Vandervore L, Schot R, Columbaro M, Kasteleijn E, et al. Loss of SMPD4 Causes a Developmental Disorder Characterized by Microcephaly and Congenital Arthrogryposis. Am J Hum Genet. 2019;105:689–705.
Deng R, Medico-Salsench E, Nikoncuk A, Ramakrishnan R, Lanko K, Kühn NA, et al. AMFR dysfunction causes autosomal recessive spastic paraplegia in human that is amenable to statin treatment in a preclinical model. Acta Neuropathol. 2023;146:353.
Schubach M, Maass T, Nazaretyan L, Röner S, Kircher M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024;52:D1143.
Ng PC. Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20.
Danis D, Jacobsen JOB, Carmody LC, Gargano MA, McMurry JA, Hegde A, et al. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am J Hum Genet. 2021;108:1564.
Strauch Y, Lord J, Niranjan M, Baralle D. CI-SpliceAI—Improving machine learning predictions of disease causing splicing variants using curated alternative splice sites. PLoS ONE. 2022;17:e0269159.
Cheng J, Novati G, Pan J, Bycroft C, Žemgulyte A, Applebaum T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023;381:eadg7492.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405.
Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517.
Aref-Eshghi E, Kerkhof J, Pedro VP, Barat-Houari M, Ruiz-Pallares N, Andrau JC, et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am J Hum Genet. 2020;106:356.
Levy MA, Relator R, McConkey H, Pranckeviciene E, Kerkhof J, Barat-Houari M, et al. Functional correlation of genome-wide DNA methylation profiles in genetic neurodevelopmental disorders. Hum Mutat. 2022;43:1609–28.
LaFlamme CW, Rastin C, Sengupta S, Pennington HE, Russ-Hall SJ, Schneider AL, et al. Diagnostic utility of DNA methylation analysis in genetically unsolved pediatric epilepsies and CHD2 episignature refinement. Nat Commun. 2024;15:1–21.
Trajkova S, Kerkhof J, Rossi Sebastiano M, Pavinato L, Ferrero E, Giovenino C, et al. DNA methylation analysis in patients with neurodevelopmental disorders improves variant interpretation and reveals complexity. Hum Genet Genom Adv. 2024;5:100309.
Dekker J, Schot R, Bongaerts M, de Valk WG, van Veghel-Plandsoen MM, Monfils K, et al. Web-accessible application for identifying pathogenic transcripts with RNA-seq: increased sensitivity in diagnosis of neurodevelopmental disorders. Am J Hum Genet. 2023;110:251.
van der Sanden BPGH, Schobers G, Corominas Galbany J, Koolen DA, Sinnema M, van Reeuwijk J, et al. The performance of genome sequencing as a first-tier test for neurodevelopmental disorders. Eur J Hum Genet. 2022;31:81.
Sigurpalsdottir BD, Stefansson OA, Holley G, Beyter D, Zink F, Hardarson M, et al. A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes. Genome Biol. 2024;25:1–21.
Geysens M, Huremagic B, Souche E, Breckpot J, Devriendt K, Peeters H, et al. Clinical evaluation of long-read sequencing-based episignature detection in developmental disorders. Genome Med. 2025;17:1.
Acknowledgements
We thank all patients and families for their participation in this study. We thank Marielle Alders (Amsterdam UMC) for helpful discussions and Burcu Akman (IBG Izmir) for critical reading of the manuscript.
Funding
The Barakat lab acknowledges general support from the Netherlands Organization for Scientific Research and other ongoing support for rare disease research from Stichting 12q, EpilepsieNL, CURE Epilepsy and the Spastic Paraplegia Foundation, Inc. CD’s PhD project is supported by the Sophia Kinderziekenhuis Fonds (CAM19-09D) and acknowledges support from the Erasmus MC Centrum voor Zeldzame Aandoeningen. Funding bodies did not have any influence on study design, results, and data interpretation or final manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization DJS, TSB; data curation DJS, CD; formal analysis DJS, CD, TSB, RS, FF; funding acquisition TSB; methodology DJS, CD, TSB; supervision TSB, ASB; writing original draft, writing—review and editing CD, DJS, DR, AB, VJMV, LDK, SGK, YB, SD, SZ, MFD, SHD, LHH, MAS, MW, FS, MD, HTB, RM, AG, JAH, IMBHL, YI, AK, VS, KES, GMSM, MWW, TJH, TK, TSB.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Using genome-wide technologies for diagnostic purposes was previously approved by the Erasmus MC institutional review board (MEC-2012-387). All individuals or legal guardians provided written consent to use pseudoanonymized clinical and analysis data.
Web resources
OMIM, http://omim.org/. gnomAD, http://gnomad.broadinstitute.org. SIFT, https://sift.bii.a-star.edu.sg/. PolyPhen2, http://genetics.bwh.harvard.edu/pph2/.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Smits, D.J., Debuy, C., Brooks, A.S. et al. Clinical utility of DNA-methylation signatures in routine diagnostics for neurodevelopmental disorders. Eur J Hum Genet 33, 1281–1289 (2025). https://doi.org/10.1038/s41431-025-01919-5
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01919-5
This article is cited by
-
Uncertainty, ethics, and progress in genomic medicine
European Journal of Human Genetics (2025)
-
To sign or not to sign: Is this still the question?
European Journal of Human Genetics (2025)
