
Abstract
Naïve CD8 + T cells are heterogenous, with subsets exhibiting divergent kinetics and functions post-activation. MicroRNAs, important mediators of post-transcriptional regulation, contribute to specification of different naïve T cell subsets. However, the microRNA regulatory circuits mediating functional specialization of naïve subsets are poorly understood. Here, we profiled microRNA expression in diverse subsets of naïve CD8 + T cells, revealing significant differences in their microRNA expression landscapes. We developed a novel framework, miR-Inf, to decipher microRNA regulatory programs. miR-Inf features two innovative attributes: (i) an efficient approach based on intron-exon ratios to estimate gene decay rates from a compendium of RNA-seq profiles, in order to better capture microRNA regulatory effects, and (ii) identification of cell-type-specific microRNA targets by integrating decay rate data and microRNA expression data. We applied this framework to identify consequential miRNAs in naïve CD8 + T cell subsets and predicted their subset-specific targets. Our analyses revealed that miR-29, a microRNA known to be important in CD8 + T cells, likely functions by modulating transcripts encoding epigenetic factors, thereby pre-programming different naïve T cell subsets to exhibit different immune responses post-activation. Collectively, our data and broadly applicable framework defined microRNA regulatory circuits across a variety of naïve CD8 + T cell subsets.
Similar content being viewed by others


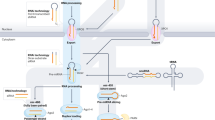
Introduction
CD8 + T cells, a major class of adaptive lymphocytes, contribute to pathogen clearance and tumor control. Upon stimulation, naïve CD8 + T cells differentiate into cytotoxic effector cells and memory cells, capable of killing infected cells and conferring prolonged protection, respectively. Although previously believed to be homogenous [1, 2], the naïve pool is now recognized as a heterogenous population comprised of subsets with distinct kinetics and functions post-activation [3,4,5,6,7,8,9]. These different fates are partly preprogrammed by gene regulatory programs established in naïve cells [6, 10, 11]. Thus, deciphering the gene regulatory programs of naïve subsets is critical to understanding T cell biology.
Subsets of naïve CD8 + T cells are distinguished by their phenotypes and developmental origins. The main phenotypic subsets are true naïve (TN) and virtual memory (VM) cells. Unlike TN cells, VM cells express markers also found on memory cells and respond rapidly to TCR stimulation [12,13,14]. Moreover, the functions of naïve cells depend on their developmental origins (fetal liver or adult bone marrow progenitors), or when they are made in the host [15]. Compared to naïve CD8 + T cells generated in adult mice, cells from neonatal mice expand more rapidly upon infection, but are limited in their ability to form immunological memory [5]. In addition, neonatal naïve cells are sensitive to stimulation by innate cytokines, enabling them to respond without antigen-specific stimulation via their TCR [16]. These differences are, in large part, specified by gene regulatory programs established in naïve cells, mediated by transcription factors and microRNAs [6, 10, 11].
MicroRNAs (miRNAs) are single-stranded non-coding RNAs that repress gene expression post-transcriptionally. With a widespread influence on the transcriptome, miRNAs play fundamental roles in mammalian biology [17]. miRNAs mediate repression by inducing mRNA decay and/or translational repression [18]; with decay the major mode of regulation accounting for ~85% of the regulatory impact [19, 20]. Within the immune system, multiple miRNAs play major roles in immune development, immune responses and signaling pathways [21]. In CD8 + T cells, the miRNAs let-7, miR-150, miR-155 and miR-181, play important roles in T cell development and function [5, 22,23,24].
Several miRNAs are differentially expressed between adult and neonatal naïve CD8 + T cells; these differences diminish following activation [10], suggesting that such miRNAs function by specifying gene regulatory differences in naïve cells. One miRNA, miR-29, is expressed at higher levels in adult naïve cells compared to neonatal counterparts [10]. Indeed, high levels of miR-29 are required to enable robust generation of immunological memory, whereas the lower levels in neonatal cells preclude memory generation [11]. However, how miR-29 directs these functional differences and what miR-29 target(s) mediate such effects have not been elucidated.
Identifying miRNA-target interactions is essential to understanding miRNA biology [25, 26]. Among target prediction methods, one class relies upon features of target sequence [27], with TargetScan [28] representing the most widely used method. Such algorithms are used extensively as they do not require experimental data as input. However, false positive predictions remain a drawback of these methods, a limitation derived from cell-type-specific gene expression and cellular contexts [29, 30]. Another class of algorithms, which account for different cellular contexts [31,32,33,34], rely upon paired expression profiles of mRNAs and miRNAs as input, and leverage the fact that miRNA expression levels can partially explain target expression patterns [32]. However, steady state mRNA levels are determined predominantly by transcriptional regulation rather than miRNAs, an inherent limitation to algorithms that rely upon transcriptome profiling. To more precisely identify cell-type-specific miRNA targets, new approaches are needed, employing measurements better reflecting miRNA regulation – mRNA stability.
In this work, we profiled miRNA expression in a compendium of different naïve CD8 + T cell subsets, and observed significant differences in their miRNA expression landscapes. We developed a novel framework, miR-Inf, for identifying consequential miRNAs and their cell-type-specific targets. Specifically, we present an efficient method based on intron-exon ratios for estimating decay rates to better capture miRNA regulation. We constructed a miRNA regulatory network and identified key miRNAs in naïve CD8 + T cell subsets and their cell-type-specific targets. Our analysis revealed that miR-29 likely specifies T cell fates through modulating chromatin remodelers in naïve cells. Collectively, our work provides a novel and efficient framework to comprehensively assess the miRNA-target interactome, which is readily applicable to diverse settings.
Methods
Mice
TCR transgenic mice specific for HSV-1 glycoprotein B498-505 peptide SSIEFARL (gBT-I) were provided by Dr. Nikolich-Zugich (University of Arizona) and crossed with Thy1.1 or C57BL/6 mice (Jackson Laboratories). Neonatal and adult gBT-I animals were used at 6–8 days old and 2–4 months old, respectively. MiR-29ab-1 fl/fl mice were provided by Dr. Costinean (Ohio State University) and crossed to B6-CyJ CD4-Cre mice. C56Bl/6 mice were purchased from the Jackson Laboratory and maintained under pathogen-free conditions at College of Veterinary Medicine in Cornell University. At the time of experimentation, mice were sex matched at 8–12 weeks of age for adult group and 7 days old for neonate group. The facilities are accredited by the American Association of Accreditation of Laboratory Animal Care.
RNA-seq
Cell sorting for RNA-seq was performed as described previously [11]. 20,000 FACS sorted cells were placed in Trizol, and RNA was isolated following manufacturer’s instructions, with an additional chloroform extraction and Glycoblue (ThermoFisher) added prior to precipitation (1 h, 4 °C), and a second wash in 70% ethanol. RNA integrity was confirmed (AATI fragment analyzer) and libraries prepared with Ultra II Directional RNA kit (NEB) using 40–50 ng total RNA, quantified (Qubit) and size distribution assessed (fragment analyzer) prior to sequencing.
Small RNA-seq
Cells and RNA were isolated as described above. Libraries were prepared with NEBNext Small RNA Library Prep Kit (New England Biolabs) or Illumina TruSeq Small RNA Prep Kit using 20–100 ng total RNA as input, size selected for insert sizes ~18–35 bp.
Small RNA-seq analysis
Mapper.pl in miRDeep2 [35] was used for preprocessing and adapter removal (-d -e -h -k TGGAATTCTCGGGTGCCAAGG -l 18 -m -v). Quantifier.pl in miRDeep2 [35] was used to map reads to miRNAs (-t mmu -d -e 0 -W). Precursors and mature miRNA sequences were downloaded from miRbase [36] v22. Reads of miRNAs from the same family were summed, with conserved miRNA families (family conservation >= 2 in TargetScan [28]) included. Families with more than 10 normalized counts in at least one profile were used for PCA. The count matrix was normalized and log2 transformed using VST function in DESeq2 [37] (blind=True). Batch effects were removed using ComBat [38], with cell type factors as covariates. PCA was conducted using prcomp in R.
RNA-seq analysis
Adapters were trimmed with trim_galore (default parameters). Reads were mapped to mm10 with HISAT2 [39]. FeatureCounts was used to count reads at genomic features using GENCODE mouse gene annotation v21 (-s 0 -Q 50). Count matrix normalization, batch effect removal and PCA as described above.
Estimating relative decay rates
Gene body read counts were generated using featureCounts [40] on gene annotations of GENCODE mouse v21 (-s 0 -Q 50), with only non-overlapping genes included. Exon read counts were generated for the same set of non-overlapping genes on exons. Intron read counts were calculated by subtracting exon counts from gene body counts, for intronic genes. Exon and intron read counts were normalized independently to transcript per million (TPM). Relative decay rates were calculated by dividing intron by exon TPMs.
miRNA regulatory network construction
Selecting target genes and miRNA families
Conserved miRNA families (family conservation >= 2, TargetScan) expressed with at least 1000 normalized counts in at least one profile were included. Genes with the following criteria were included: relative decay rates estimated; expressed at more than 10 TPM in at least one profile; and, with at least one target site of the included miRNAs predicted by TargetScan [28].
TargetScan predictions
MiRNA target predictions from TargetScanMouse [28] release 7.2 with total context scores < −0.2 were used. Scores were scaled to between 0 and 1 for the prior matrix used for network inference.
miR-Inf framework
miR-Inf framework was adapted from Inferelator, an algorithm for transcriptional regulatory network inference [41]. Several adaptations were made for miR-Inf: (1) decay rates were used instead of RNA-seq counts; (2) miRNA expression was used instead of transcription factor expression; (3) positive solves only were allowed for interaction coefficients between decay rates and miRNA expression levels; (4) only TargetScan predictions were considered as potential targets ; and, (5) a penalty for predicting a regulation negatively correlated with negative scaled total context score values. miR-Inf was used with a model size of 2 miRNAs/gene and bias 0.05 (strong prior reinforcement), and ran five times with random seeds, with the final network composed of regulatory interactions in a majority of five runs.
Analysis of miR-29 and let-7 predicted targets
Enrichment analysis and protein-protein interaction analysis on miR-29 and let-7 predicted targets were performed using STRING website version 12 [42], with genes included for network inference as background genes.
Cell culture
293 T cells were regularly cultured in the lab and obtained from American Type Culture Collection. The cell line was not recently authenticated but was authenticated at American Type Culture Collection. Mycoplasma testing was performed only if the cells were maintained in culture for more than four months. The cell lines used in this study were cultured for no longer than 8 weeks. All other cell lines maintained beyond four months tested negative for mycoplasma.
Luciferase assays
Three independently passaged 293 T cells were plated into separate wells of a 24-well plate and treated as independent replicates for the luciferase assay. 293 T cells were cotransfected (Lipofectamine 2000) with luciferase reporter plasmids containing either wild-type or mutant (2nt disruptions to predicted miRNA target sites) 3’UTRs of Zfp282, together with synthetic miR-29a mimics or a non-targeting inert control [10] (Integrated DNA Technologies). For each condition, miRNA duplexes were prepared by annealing complementary guide and passenger strands to generate either functional miR-29a or inert miRNA. Cells were harvested 24 h post-transfection, and Firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) on a Veritas Microplate Luminometer (Turner Biosystems). Firefly luciferase signals were normalized to Renilla luciferase to account for transfection efficiency. Based on the prior data and power analysis (alpha = 0.05, two-tailed t test), three biological replicates were able to provide 100% power to detect statistical differences. No samples were excluded. No randomization was used. The investigator was not blinded. A two-tailed, two-sample unequal variance t-test without adjustments for multiple comparisons was performed. The experiment was replicated three times.
The sequences used were as follows:
-
miR-29a
-
○
Guide strand: 5′-UAGCACCAUCUGAAAUCGGUUA-3′
-
○
Passenger strand: 5′-ACCGAUUUCAGAUGGUGUGAAU-3′
-
Inert control
-
○
Sense strand: 5′-UAAAAAUCGCGUGGAUUAAUG-3′
-
○
Antisense strand: 5′-UUAAUUUACGCGGUUUUUAUU-3′
Flow cytometry
For flow cytometry, cells were surface stained with pre-conjugated antibodies purchased from ThermoFisher/Life technologies, BioLegend or BD Biosciences. The following clones were used: CD4 (GK1.5, BD Biosciences #612900), CD8a (53-6.7, BioLegend #100798), CD122 (TM-β1, BD Biosciences #562960) and CD44 (IM7, Invitrogen/Life Technologies 11-0441-85). Viability staining was performed using Fixable Viability dye e780 (ThermoFisher/Life technologies). For intracellular staining, the FoxP3 buffer fixation kit (Life technologies) was used according to manufacturer’s instructions. For intracellular miR-29 targets, Dnmt3a and Hp1α, antibodies were purchased from Novus Biologicals (NB120-13888PE, NBP2-52434), clones 64B1446 and 3A11F8, respectively. Dnmt3a antibody was purchased as a PE conjugate and Hp1α staining was performed by primary antibody staining followed by secondary Alexa647 goat-anti mouse antibody (Invitrogen A21236) staining. Flow cytofluorimetric data were acquired using FACSDiva software on a BD FACSymphony A3 equipped with five lasers (BD Biosciences). Analysis was performed with FlowJo (BD Biosciences). Based on the prior data, and power analysis (alpha = 0.05, two-tailed t test) indicated that six biological replicates would provide 100% power to detect statistical differences. No samples were excluded. No randomization was used. Based on historical data and the use of unmanipulated animals, randomization was deemed unnecessary for these experiments, particularly given the limited number of mice available. The investigator was not blinded. Blinding was not necessary for the conclusion, and we reported raw fluorescence intensity. The experiment was replicated one time. An unpaired, two-tailed t-test without adjustments for multiple comparisons was performed.
Results
Naïve CD8 + T cell subsets possess distinct miRNA expression profiles
To understand the miRNA expression landscape in naïve CD8 + T cells, we generated 34 small RNA-seq profiles of naïve CD8 + T cell subsets, including cells bearing different phenotypic markers (VM, TN) and of different developmental origins (adult, neonatal; Fig. S1A). Principal component analysis (PCA) revealed that principal component 1 (PC1), accounting for 32.7% of the variance, separated adult and neonatal cells (Fig. 1A, B, S1A). Among the top miRNAs contributing to PC1, several have established roles in CD8 + T cell function, including miR-29, miR-130 and miR-150 (Fig. 1C) [10, 11, 22]. TN and VM cells were partitioned by PC2, accounting for 13.2% of the variance (Fig. 1A, B). This analysis showed that different naïve CD8 + T cell subsets have distinct miRNA expression patterns, with major differences between adult and neonatal cells.

A PCA of small RNA-seq profiles from naïve CD8 + T cell subsets. B Pearson correlation (R) between principal components from A and metadata. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. C Top 5 miRNA families contributing to positive and negative sides of PC1. D PCA of RNA-seq profiles from naïve CD8 + T cell subsets. E Pearson correlation (R) between principal components from D and metadata. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
We also investigated the mRNA expression landscape of naïve T cell subsets using RNA-seq (Fig. 1D, S1B). In contrast to our miRNA analysis, PCA of gene expression profiles revealed that TN and VM cells were partitioned by PC1 (56.5% of the variance; Fig. 1D, E), with PC2 largely coincident with adult versus neonatal cells (Fig. 1D, E). Collectively, we observed substantial changes in miRNA and mRNA expression landscapes of naïve CD8 + T cell subsets, with pronounced changes in miRNA expression between adult and neonatal cells, potentially driving their divergent functions post-activation [5, 9, 12, 13].
Estimating decay rates using RNA-seq profiles
MicroRNAs repress gene expression by inducing mRNA decay [19, 20]. Thus, we sought to estimate decay rates genome-wide to better capture miRNA regulatory effects. Previous studies approximated decay rates using measurements of transcription rate and RNA levels, assuming the cells are in steady state [43,44,45,46]. An optimal implementation of this approach is to use precision nuclear run-on sequencing (PRO-seq [47]), which measures nascent transcription, to directly assess transcription genome-wide. However, PRO-seq is a complex and labor-intensive assay requiring a significant number of cells as input. Alternatively, a practical implementation of this approach to leverage intronic read counts in RNA-seq profiles as a proxy for RNA production rate [44]. Importantly, using public data [19], we found that PRO-seq read counts and RNA-seq intronic read counts correlated well (Figs. 2A, S2A-C), validating the utility of intronic read counts as a proxy for relative RNA production. Thus, we derived estimations of relative decay rates using intronic and exonic read counts from RNA-seq data (Fig. 2B).

A Correlation between normalized intronic read counts and normalized PRO-seq counts for HEK297 cells upon miR-122 induction. B Schematic plot illustrating estimation of decay rate, assuming transcript level of a gene is stable. C Estimated decay rates of miR-122 targets and background genes in miR-122 over-expressed (OE) samples and control samples. D Cumulative distribution of log2 fold changes of the estimated decay rates between miR-122 overexpressed (OE) samples and control samples for miR-122 targets and background genes.
We sought to evaluate our decay rate estimations. Previously, we defined direct targets of multiple miRNAs in HEK293 cells using combined analysis of PRO-seq and RNA-seq (CARP), an approach to robustly partition direct miRNA targets from indirect downstream effects [19]. We estimated relative decay rates of genes in miR-122 induced cells and control cells, and observed significantly higher decay rates for CARP-identified direct miR-122 targets in miR-122 induced cells compared to those in the control cells (Fig. 2C, D). In addition, more modest increases in decay rates were observed for both TargetScan predicted miR-122 targets and for transcripts that exhibited decreased mRNA levels in response to miR-122, measured by RNA-seq (Fig. 2C, D). Similar results were observed for additional miRNAs (Fig. S2D-F). Collectively, we observed the highest estimated decay rates for direct miRNA targets, supporting the efficacy of our decay rate estimation.
Using the approach of estimating decay rates with intronic and exonic read counts from RNA-seq profiles, we derived genome-wide decay rates for different naïve CD8 + T cell subsets (Figs. S1B, S2G-I). PCA showed that the decay rate profiles of neonatal and adult cells partition by PC1, and TN and VM cells by PC2 and PC3 (Fig. S2H-I), results similar to that observed using miRNA expression profiles (Fig. 1A, B). The estimated decay rates reflect miRNA-mediated regulation, which we anticipated would enable more precise predictions of miRNA targets.
A miRNA regulatory network for naïve CD8 + T cells
To identify miRNAs with a consequential impact on the transcriptome together with their cell-type-specific targets, we developed a framework for constructing miRNA regulatory networks (Fig. 3A). Our approach was adapted from a transcriptional regulatory network construction algorithm, Inferelator [41], which has been proven in multiple contexts [41, 48, 49]. Our algorithm, miR-Inf, relies upon small RNA-seq and estimated decay rate profiles as input data. miR-Inf models differential decay rate as a sparse multivariate linear function of microRNA activities, to identify target transcripts for which differences in decay rates across subsets can be explained by corresponding changes in miRNA levels. In addition, the algorithm leverages TargetScan predictions [28] as a priori knowledge, and refines these sequence-based predictions to a subset supported by miRNA expression and mRNA decay patterns. miR-Inf performs consistently across different intron read counts thresholds (Fig. S3A-C) and identifies miRNA targets without bias toward genes with low or high intron read counts or estimated decay rates (Fig. S3D, E). Using our novel framework, we constructed a miRNA regulatory network for naïve T cell subsets.

A MiRNA regulatory network construction workflow in naïve CD8 + T cell subsets. B Top miRNA families with highest numbers of predicted targets. C Top miRNA families with highest numbers of predicted conserved targets. D Expression levels of miR-29 in adult and neonatal naïve CD8 + T cells. E Cumulative distribution of log2 fold changes of estimated decay rates and gene expression levels (F) between neonatal and adult naïve CD8 + T cells, showing miR-29 predicted targets from TargetScan (conserved: PCT > 0.75; matched: subsampled TargetScan targets with context scores matched to those predicted by miR-Inf) and miR-Inf (conserved: miR-Inf targets with PCT > 0.75 in TargetScan). Inset: P-values from Wilcoxon tests comparing miR-Inf (top three p-values) or TargetScan (bottom two p-values) to background. Also, comparing miR-Inf to TargetScan in decay rates: p = 5.8 × 10−3; comparing miR-Inf conserved to TargetScan conserved in decay rates: p = 9.1 × 10−9. G Expression levels of let-7 in TN and VM CD8 + T cells. Cumulative distribution of log2 fold changes of estimated decay rates (H) and gene expression levels (I) between VM and TN CD8 + T cells, showing let-7 predicted targets from TargetScan and miR-Inf, otherwise as described in E. Comparing miR-Inf to TargetScan in decay rates: p = 5.5 × 10−3; comparing miR-Inf conserved to TargetScan conserved in decay rates: p = 2.8 × 10−4.
The miRNA-gene network in naïve T cell subsets was comprised of 36 miRNA families and 2614 target genes. Five miRNA families had more than 145 predicted targets (Fig. 3B), suggestive of extensive roles across subsets. Several miRNA families have established roles in CD8 + T cells, such as let-7 and miR-29. Similar miRNAs were observed when we subsetted the network to retain only evolutionarily conserved miRNA targets (Fig. 3C; PCT [50] > 0.75), which are more likely be functionally relevant. let-7 modulates the effector response of CD8 + T cells, with higher let-7 levels maintaining the naïve phenotype, and lower levels enhancing effector functions [24]. miR-29 expression is significantly elevated in adult naïve CD8 + T cells compared to neonatal cells [10], and different levels of miR-29 specify rapid effector responses in neonatal cells versus long-term memory in adult cells [11]. In addition, miR-26a is indispensable for CD8 + T cell cytotoxicity [51]; miR-27 can limit cytokine production in T cells [52]. Thus, our predictions recover miRNAs with established roles in T cells, and implicate additional miRNAs.
To evaluate the predictions, we examined expression patterns of identified miRNAs and their predicted targets. miR-29 exhibited significantly higher expression in adult compared to neonatal cells (Fig. 3D), as expected [10]. Concordantly, decay rates of both miR-Inf predicted targets and sequence-based targets (TargetScan) of miR-29 were higher in adult cells, albeit to a lesser extent for TargetScan predicted targets (Fig. 3E). Similarly, expression levels of miR-29 predicted targets were reduced in adults; however, the magnitude of reduction in gene expression was much smaller than that observed in decay rates, indicating those genes were also subject to transcriptional regulation (Fig. 3F). Notably, when we subsampled the TargetScan-predicted targets to match the predicted efficacy of the target sites of the miR-Inf predicted targets, miR-Inf targets displayed substantially more significant changes in gene expression and decay rate patterns compared to the subsampled set (Fig. 3E, F), indicating that miR-Inf is capturing features of target identities not found in TargetScan predictions alone.
Our analysis also identified let-7 as a miRNA important for the naïve T cell subsets. Specifically, let-7 was more highly expressed in VM compared to TN cells (Fig. 3G), and let-7 target genes had higher decay rates and lower expression levels in VM cells, accordingly (Fig. 3H, I). This analysis demonstrated that the predicted targets exhibited increased decay rates and decreased expression in conditions with high miRNA levels. Notably, the number of targets predicted by our framework was reduced compared to TargetScan, but they appeared to be subject to stronger regulation by their cognate miRNAs (Fig. 3E, F, H-I). Thus, our method refined the TargetScan predicted miRNA targets, likely reflecting the different cellular contexts of naive subsets, providing a more confident assessment of the miRNAs’ functional targets in naïve T cells.
We compared predicted miR-29 and let-7 targets with other state-of-the-art miRNA target prediction methods (Fig. S4A–D). miRDB [53] and miRWalk [54] are machine-learning-based target predictions, which rely upon both target site sequence features and experimental data; anamiR [55] integrates RNA and miRNA profiles for cell-type-specific predictions. Among the predicted miR-29 targets, miR-Inf, with fewer targets, led to significant shifts in decay rates and gene expression levels, outperforming miRDB and miRWalk (Fig. S4A–B). While predicted targets from anamiR showed the largest expression changes, only two out of 35 contained conventional miR-29 binding sites, strongly suggesting that such predictions do not represent true miR-29 targets, at least not due to canonical miRNA-mediated regulation. We also compared let-7 targets predicted from miR-Inf, miRDB and miRWalk (anamiR did not predict any targets for let-7). miR-Inf, again, displayed more substantial changes in gene expression levels and decay rates, outperforming other methods (Fig. S4C–D). Taken together, we developed a novel framework for constructing miRNA regulatory networks and applied it to naïve CD8 + T cells. We not only identified miRNAs important for different naïve T cell subsets, but also provided cell-type-specific miRNA targets refined from sequence-based predictions.
MiR-29 targets are enriched in chromatin remodeling genes
It is well-established that miR-29 acts as a developmental switch in naïve CD8 + T cells, with low levels in neonatal cells promoting effector responses with minimal memory potential, and high levels in adult cells potentiating long-term immunological memory [11, 56]. However, the miR-29 targets that mediate such effects have not been systematically investigated. Our predicted miR-29 targets in naïve T cell subsets included Eomes and T-bet, both previously suggested as miR-29 targets [10, 11], which have established roles in CD8 + T cell development and immune response [57,58,59,60]. To obtain a comprehensive and systematic understanding of the functions of miR-29 in naïve T cells, we performed enrichment analysis on evolutionarily conserved miR-29 targets predicted by miR-Inf. Top enrichment corresponded to gene sets associated with chromatin regulation and DNA methylation (Fig. 4A). Genes driving these enrichments included DNA methyltransferases (Dnmt3a, Dnmt3b) and DNA demethylase (Tet1), as well as multiple histone modifiers and effectors, such as erasers for H3K4 (Kdm5b, Kdm5c) and H3K36 (Kdm2a) [61], readers for H3K9me3 (Cbx3, Cbx5) [62], and corepressors associated with histone deacetylase activity (Rcor1, Bcorl1) [63, 64]. To gain perspective on miR-29 targets, we searched for protein-protein interactions (PPI) within predicted targets of miR-29 using the STRING database [42]. Significant enrichment of PPI (p = 5.58 × 10−8) was observed within the predicted miR-29 targets, suggesting that these genes had interconnected functions (Fig. 4B). Together, these results suggest that miR-29 mediates epigenetic changes in naïve cells that contribute to specifying age-related differences in CD8 + T cell immune response.

A Enrichment analysis of miR-29 predicted and conserved targets. B Protein-protein interaction network of miR-29 predicted targets (enrichment p-value: 5.58 × 10−8), showing only connected nodes, with all genes included in network inference as the background set. Dnmt3a (C) and Cbx5 (D) protein expression levels in neonatal and adult CD8 + T cells (n = 6). Center represents mean; error bar represents standard error of the mean (s.e.m.). E Luciferase reporter assay assessing direct regulation of Zfp282 by miR-29 (n = 3). Constructs containing either wild-type or mutated miR-29a binding sites in 3’UTR of Zfp282 were transfected into cells with or without miR-29a induction. Center represents mean; error bar represents standard error of the mean (s.e.m.). F Enrichment analysis of let-7 predicted conserved targets. G Protein-protein interaction network of let-7 predicted targets (enrichment p-value: 2.41 × 10−7), showing only connected nodes, with all genes included in network inference as the background set.
To validate the predicted targets of miR-29, we measured protein levels of Dnmt3a and Cbx5 - both with established roles in chromatin regulation - in neonatal and adult CD8+ T cells. Both proteins were more highly expressed in neonatal T cells (Fig. 4C, D, Fig. S5A–B), as predicted, and consistent with the lower expression of miR-29 in this cell population (Fig. 3D), supporting a regulatory relationship between miR-29 and those targets. We further validated Zfp282, a transcription factor, as a direct target of miR-29 using luciferase reporter assays. Reporter constructs containing either wild-type or mutated miR-29a binding sites within the 3’ untranslated region (3’UTR) of Zfp282 were transfected into cells with or without miR-29a induction (Fig. 4E). Luciferase activity was significantly reduced in the presence of miR-29a, but only when the binding sites were intact, demonstrating that miR-29a directly represses Zfp282 through its 3’UTR. We note that multiple additional targets (Eomes, Tbx21, Dnmt3a, Dnmt3b, and Slc16a1) of miR-29 predicted by miR-Inf have been previously validated in similar luciferase assays [10, 65,66,67], providing additional validation in the predictions and efficacy of miR-Inf.
let-7 modulates CD8 + T cell immune responses, with reduced levels enhancing clonal expansion and effector functions [24]. We found that let-7 was differentially active in naïve CD8 + T cell subsets (Fig. 3G), with higher expression in VM compared to TN cells. Predicted conserved targets of let-7 include Ccr7, Rasgrp1 and Btg2, each with well-defined roles in CD8 + T cell immune responses [68,69,70]. In particular, RasGRP1 transduces TCR-mediated signals and regulates the threshold of T cell activation [70]. Rasgrp1 exhibited reduced levels in VM cells compared to TN cells (Fig. S5C), potentially due to the repression exerted by let-7 in VM cells (Fig. 3G). This suggests that let-7 may dictate the fates of TN and VM cells through its regulation of Rasgrp1.
We performed enrichment analysis on let-7 predicted conserved targets in naïve CD8 + T cells (Fig. 4F), finding enrichment in kinase activity, signal transduction and phosphorylation pathways, echoing findings on its roles in metabolism, development and immune functions [71, 72]. Finally, significant PPI enrichment (p-value = 2.41×10-7) was observed within the predicted let-7 targets (Fig. 4G).
Regulatory interactions between miR-29, transcription factors and genes
MiRNAs and transcription factors (TFs) are integral to gene regulatory networks governing T cell function, and regulatory interrelationships between miRNAs and TFs are important features of these networks [73]. To investigate such regulatory relationships, we overlaid our miRNA-gene network with a TF-gene network in naïve CD8 + T cells, constructed from a compendium of gene expression and chromatin accessibility profiles [49]. We found that certain miR-29 targets encode TFs, which themselves are predicted to regulate expression of additional miR-29 targets. For example, Zfp282 and Dnmt3a are both predicted miR-29 targets bearing conserved miR-29 target sites, with increased expression in neonatal naïve CD8 + T cells compared to adult cells (Fig. 5A–C), and Zfp282, a TF, is predicted to activate transcription of Dnmt3a, with its binding motif proximal to the Dnmt3a promoter (Fig. 5B). This analysis suggests a regulatory circuit: when miR-29 is highly expressed (in adult naïve cells), Zfp282 and Dnmt3a are both repressed; when miR-29 levels are low (in neonatal cells), Zfp282 and Dnmt3a are more highly expressed, and Dnmt3a is further activated by Zfp282. This intricate circuit would ensure high expression of Dnmt3a, encoding a DNA methyltransferase, in neonatal cells, to set up their specific chromatin environments.

A A miRNA regulatory circuit between miR-29, Zfp282 and Dnmt3a. B Coverage of gene expression at gene Zfp282 and Dnmt3a loci. C Expression levels of Zfp282 and Dnmt3a in adult and neonatal naïve CD8 + T cells. D A miRNA regulatory circuit between miR-29, Tbx21 and Slc16a1. E Coverage of gene expression at gene Tbx21 and Slc16a1 loci. F Expression levels of Tbx21 and Slc16a1 in adult and neonatal naïve CD8 + T cells.
Another exemplary gene regulatory circuit is comprised of miR-29, Tbx21 and Slc16a1 (Fig. 5D–F). Tbx21 is a master regulator controlling CD8 + T cell effector functions [58, 60]. Slc16a1 encodes a lactate transmembrane transporter, the absence of which impairs T cell proliferation [74]. Both Tbx21 and Slc16a1 were predicted as miR-29 targets by miR-Inf, with elevated expression in neonatal naïve CD8 + T cells compared to adult cells (Fig. 5D–F). Tbx21 is predicted to upregulate Slc16a1, with a Tbx21 motif at the Slc16a1 locus. This gene regulatory circuit suggests that low levels of miR-29 in neonatal cells no longer represses Tbx21 and Slc16a1, leading to increased expression of both genes. Additionally, Tbx21 activates Slc16a1, indicating that both genes may facilitate the prompt activation and elevated effector functions in neonatal cells. Collectively, our analysis revealed intricate regulatory relationships between miRNAs, TFs and other genes, exemplifying how miR-29 and specific TFs interact and mediate downstream impacts on the gene regulatory network.
Discussion
We systematically investigated miRNA regulation in functionally divergent CD8 + T cells subsets, focusing on naïve cells with varying phenotypes (VM and TN) and developmental origins (adult and neonatal). miRNAs exhibit differential activity in these naïve subsets, shaping their divergent functional traits post-activation [10, 11]; but the miRNA regulatory circuits mediating such functional differences remain unclear. Here, we profiled miRNA expression across naïve subsets and revealed notable differences. The differences were especially pronounced between neonatal and adult cells, with additional variation between VM and TN cells, indicating that miRNAs may contribute to their divergent functional properties.
We developed a novel framework, miR-Inf, to infer miRNA regulatory programs. Our framework features two innovative components: (i) estimating genome-wide decay rates, which captures miRNA regulatory effects; (ii) constructing miRNA regulatory networks leveraging the decay rate and miRNA expression data. Although prior studies have implemented cell-type-specific miRNA analysis utilizing expression data [31, 75,76,77,78,79], our framework alone employs the decay rate data derived from RNA-seq, which can more effectively capture the subtle post-transcriptional effects mediated by miRNAs. Moreover, our miRNA network construction method models differential decay rate as a sparse multivariate linear function of miRNA activities, adapted from the transcriptional network inference method Inferelator [41]. This approach improves the identification of consequential miRNAs and their functional targets. Finally, our framework integrates powerful sequence-based predictions from TargetScan [28], and generates a refined list of cell-type-specific targets supported by decay rate data and miRNA expression patterns. miR-Inf is versatile across different cell types and tissues, enabling improved identification of cell-type-specific consequential miRNAs and their functional targets.
We employed miR-Inf to study miRNA regulation in functionally divergent naïve CD8 + T cell subsets, with key miRNAs and their cell-type-specific targets identified. Top miRNAs with a significant number of predicted targets are miR-29 and let-7, both of which are involved in T cell immune response [11, 80], yet the mechanisms by which they exert their effects were not well understood. Previous studies have identified several DNA methyltransferases (DNMTs) and ten-eleven translocation enzymes (TETs) as miR-29 targets in embryo development [81], brain maturation [82] and acute myeloid leukemia [83], establishing its role in regulating DNA methylation. Histone deacetylases (HDAC4) were also targeted by miR-29 in multiple myeloma [84]. We uncovered multiple additional histone modifiers and effectors as predicted miR-29 targets, including erasers for H3K4me3 and readers for heterochromatin, further broadening our understanding in miR-29 regulation. We showed that the pre-programming of naïve T cells by miR-29 may derive from modulation of chromatin remodeling genes, thereby influencing the epigenome.
Both miRNAs and TFs are essential components of the regulatory networks governing T cell function. miRNA and TF co-regulatory network analysis has been used to identify key regulators and pathways associated with cancer [85, 86], heart failure [87, 88] and inflammation [89]. By integrating our miRNA-gene network with a TF-gene network in naïve CD8 + T cells [49], we identified interactions across miRNAs, TFs and target genes. We present two regulatory circuits involving miR-29 as examples, which deepened our understanding of the intricate gene regulatory mechanisms in T cells. In particular, miR-29 is predicted to repress both the transcription factor Zfp282 and the DNA methyltransferase Dnmt3a, which itself is positively regulated by Zfp282. Furthermore, we validated that the miRNA target sites within Zfp282 are responsive to miR-29 and that Dnmt3a protein levels are significantly lower in CD8 + T cells with high levels of miR-29. Thus, high levels of miR-29 likely repress Dnmt3a both directly and indirectly via Zfp282.
Data availability
Raw and analyzed data generated in this paper were deposited to GEO under accessions GSE280662 (RNA-seq), GSE280664 (small RNA-seq), GSE280665 (small RNA-seq), GSE280667 (small RNA-seq) and GSE280668 (small RNA-seq). Analyzed PRO-seq data was obtained from GSE140365 [19].
Code availability
Code for miR-Inf framework and data analysis for this manuscript is deposited on Github (https://github.com/hongyazhu/MicroRNA-regulation-Naive-T-Cell-Subsets).
References
Plumlee CR, Sheridan BS, Cicek BB, Lefrançois L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity. 2013;39:347–56.
Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. 2007;27:985–97.
Lee SW, Lee GW, Kim HO, Cho JH. Shaping Heterogeneity of Naive CD8+ T Cell Pools. Immune Netw. 2023;23:e2.
Richard AC. Divide and conquer: phenotypic and temporal heterogeneity within CD8+ T cell responses. Front Immunol. 2022;13:949423.
Smith NL, Wissink E, Wang J, Pinello JF, Davenport MP, Grimson A, et al. Rapid proliferation and differentiation impairs the development of memory CD8+ T cells in early life. J Immunol Balt Md 1950. 2014;193:177–84.
Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental origin governs CD8+ T cell fate decisions during infection. Cell. 2018;174:117–130.e14.
Tabilas C, Iu DS, Daly CWP, Yee Mon KJ, Reynaldi A, Wesnak SP, et al. Early microbial exposure shapes adult immunity by altering CD8+ T cell development. Proc Natl Acad Sci USA. 2022;119:e2212548119.
This S, Rogers D, Mallet Gauthier È, Mandl JN, Melichar HJ. What’s self got to do with it: Sources of heterogeneity among naive T cells. Semin Immunol. 2023;65:101702.
Wang J, Wissink EM, Watson NB, Smith NL, Grimson A, Rudd BD. Fetal and adult progenitors give rise to unique populations of CD8+ T cells. Blood. 2016;128:3073–82.
Wissink EM, Smith NL, Spektor R, Rudd BD, Grimson A. MicroRNAs and their targets are differentially regulated in adult and neonatal mouse CD8+ T cells. Genetics. 2015;201:1017–30.
Yee Mon KJ, Zhu H, Daly CWP, Vu LT, Smith NL, Patel R, et al. MicroRNA-29 specifies age-related differences in the CD8+ T cell immune response. Cell Rep. 2021;37:109969.
Haluszczak C, Akue AD, Hamilton SE, Johnson LDS, Pujanauski L, Teodorovic L, et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med. 2009;206:435–48.
Lee JY, Hamilton SE, Akue AD, Hogquist KA, Jameson SC. Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci USA. 2013;110:13498–503.
Thiele D, La Gruta NL, Nguyen A, Hussain T. Hiding in plain sight: virtually unrecognizable memory phenotype CD8+ T cells. Int J Mol Sci. 2020;21:8626.
Rudd BD. Neonatal T cells: a reinterpretation. Ann Rev Immunol [Internet]. 2020 Jan 13 [cited 2020 Aug 19]; Available from: https://www.annualreviews.org/doi/abs/10.1146/annurev-immunol-091319-083608.
Watson NB, Patel RK, Kean C, Veazey J, Oyesola OO, Laniewski N, et al. The gene regulatory basis of bystander activation in CD8+ T cells. Sci Immunol. 2024;9:eadf8776.
Bartel DP. Metazoan MicroRNAs. Cell. 2018;173:20–51.
Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37.
Patel RK, West JD, Jiang Y, Fogarty EA, Grimson A. Robust partitioning of microRNA targets from downstream regulatory changes. Nucleic Acids Res. 2020;48:9724–46.
Eichhorn SW, Guo H, McGeary SE, Rodriguez-Mias RA, Shin C, Baek D, et al. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell. 2014;56:104–15.
Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. 2016;16:279–94.
Almanza G, Fernandez A, Volinia S, Cortez-Gonzalez X, Croce CM, Zanetti M. Selected microRNAs define cell fate determination of murine central memory CD8 T cells. PloS One. 2010;5:e11243.
Amado T, Amorim A, Enguita FJ, Romero PV, Inácio D, de Miranda MP, et al. MicroRNA-181a regulates IFN-γ expression in effector CD8+ T cell differentiation. J Mol Med Berl Ger. 2020;98:309–20.
Wells AC, Daniels KA, Angelou CC, Fagerberg E, Burnside AS, Markstein M, et al. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. eLife. 2017;6:e26398.
Min H, Yoon S. Got target?: computational methods for microRNA target prediction and their extension. Exp Mol Med. 2010;42:233–44.
Riolo G, Cantara S, Marzocchi C, Ricci C. miRNA Targets: from Prediction Tools to Experimental Validation. Methods Protoc. 2020;4:1.
Riffo-Campos ÁL, Riquelme I, Brebi-Mieville P. Tools for sequence-based miRNA target prediction: what to choose? Int J Mol Sci [Internet]. 2016 Dec 9 [cited 2020 Aug 15];17. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5187787/
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005.
Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JAF, Elkon R, Agami R. A Pumilio-induced RNA structure switch in p27-3’ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–20.
Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23:1743–8.
Bradley T, Moxon S. FilTar: using RNA-Seq data to improve microRNA target prediction accuracy in animals. Bioinformatics. 2020;36:2410–6.
Li Y. Computational methods of inferring context-specific MicroRNA Regulatory Networks. University of Toronto; 2015.
Liu W, Wang X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019;20:18.
Movassagh M, Morton SU, Hehnly C, Smith J, Doan TT, Irizarry R, et al. mirTarRnaSeq: an R/bioconductor statistical package for miRNA-mRNA target identification and interaction analysis. BMC Genom. 2022;23:439.
Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52.
Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–62.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma Oxf Engl. 2014;30:923–30.
Miraldi ER, Pokrovskii M, Watters A, Castro DM, De Veaux N, Hall JA, et al. Leveraging chromatin accessibility for transcriptional regulatory network inference in T Helper 17 Cells. Genome Res. 2019;29:449–63.
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51:D638–46.
Alkallas R, Fish L, Goodarzi H, Najafabadi HS. Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nat Commun. 2017;8:909.
Gaidatzis D, Burger L, Florescu M, Stadler MB. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015;33:722–9.
Baudrimont A, Voegeli S, Viloria EC, Stritt F, Lenon M, Wada T, et al. Multiplexed gene control reveals rapid mRNA turnover. Sci Adv. 2017;3:e1700006.
Blumberg A, Zhao Y, Huang YF, Dukler N, Rice EJ, Chivu AG, et al. Characterizing RNA stability genome-wide through combined analysis of PRO-seq and RNA-seq data. BMC Biol. 2021;19:30.
Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc. 2016;11:1455–76.
Pokrovskii M, Hall JA, Ochayon DE, Yi R, Chaimowitz NS, Seelamneni H, et al. Characterization of transcriptional regulatory networks that promote and restrict identities and functions of intestinal innate lymphoid cells. Immunity. 2019;51:185–197.e6.
Zhu H, Jiang Y, McNairn AJ, Fogarty EA, Tabilas C, Patel RK, et al. Deciphering gene regulatory programs underlying functionally divergent naïve T cell subsets [Internet]. bioRxiv; 2024 [cited 2024 Nov 12]. p. 2024.11.06.621737. Available from: https://www.biorxiv.org/content/10.1101/2024.11.06.621737v1.
Friedman RC, Farh KKH, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105.
Long H, Xiang T, Luo J, Li F, Lin R, Liu S, et al. The tumor microenvironment disarms CD8+ T lymphocyte function via a miR-26a-EZH2 axis. Oncoimmunology. 2016;5:e1245267.
Pua HH, Steiner DF, Patel S, Gonzalez JR, Ortiz-Carpena JF, Kageyama R, et al. MicroRNAs 24 and 27 suppress allergic inflammation and target a network of regulators of T helper 2 cell-associated cytokine production. Immunity. 2016;44:821–32.
Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–31.
Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE. 2018;13:e0206239.
Wang TT, Lee CY, Lai LC, Tsai MH, Lu TP, Chuang EY. anamiR: integrated analysis of MicroRNA and gene expression profiling. BMC Bioinforma. 2019;20:239.
Stelekati E, Cai Z, Manne S, Chen Z, Beltra JC, Buchness LA, et al. MicroRNA-29a attenuates CD8 T cell exhaustion and induces memory-like CD8 T cells during chronic infection. Proc Natl Acad Sci USA. 2022;119:e2106083119.
Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–44.
Juedes AE, Rodrigo E, Togher L, Glimcher LH, von Herrath MG. T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J Exp Med. 2004;199:1153–62.
Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–3.
Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci. 2003;100:15818–23.
Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324.
van Wijnen AJ, Bagheri L, Badreldin AA, Larson AN, Dudakovic A, Thaler R, et al. Biological functions of chromobox (CBX) proteins in stem cell self-renewal, lineage-commitment, cancer and development. Bone. 2021;143:115659.
Pagan JK, Arnold J, Hanchard KJ, Kumar R, Bruno T, Jones MJK, et al. A novel corepressor, BCoR-L1, represses transcription through an interaction with CtBP. J Biol Chem. 2007;282:15248–57.
You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–8.
Cui H, Wang L, Gong P, Zhao C, Zhang S, Zhang K, et al. Deregulation between miR-29b/c and DNMT3A Is Associated with Epigenetic Silencing of the CDH1 Gene, Affecting Cell Migration and Invasion in Gastric Cancer. PLoS One. 2015;10:e0123926.
Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA. 2007;104:15805–10.
Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA. miR-29a and miR-29b Contribute to Pancreatic β-Cell-Specific Silencing of Monocarboxylate Transporter 1 (Mct1)▿. Mol Cell Biol. 2011;31:3182–94.
Choi H, Song H, Jung YW. The Roles of CCR7 for the Homing of Memory CD8+ T Cells into Their Survival Niches. Immune Netw. 2020;20:e20.
Hwang SS, Lim J, Yu Z, Kong P, Sefik E, Xu H, et al. mRNA destabilization by BTG1 and BTG2 maintains T cell quiescence. Science. 2020;367:1255–60.
Priatel JJ, Chen X, Huang YH, Chow MT, Zenewicz LA, Coughlin JJ, et al. RasGRP1 regulates antigen-induced developmental programming by naive CD8 T cells. J Immunol Balt Md 1950. 2010;184:666–76.
Letafati A, Najafi S, Mottahedi M, Karimzadeh M, Shahini A, Garousi S, et al. MicroRNA let-7 and viral infections: focus on mechanisms of action. Cell Mol Biol Lett. 2022;27:14.
Su JL, Chen PS, Johansson G, Kuo ML. Function and regulation of let-7 family microRNAs. MicroRNA Shariqah U Arab Emir. 2012;1:34–9.
Guo Y, Alexander K, Clark AG, Grimson A, Yu H. Integrated network analysis reveals distinct regulatory roles of transcription factors and microRNAs. RNA. 2016;22:1663–72.
Macchi C, Moregola A, Greco MF, Svecla M, Bonacina F, Dhup S, et al. Monocarboxylate transporter 1 deficiency impacts CD8+ T lymphocytes proliferation and recruitment to adipose tissue during obesity. iScience. 2022;25:104435.
Bossel Ben-Moshe N, Avraham R, Kedmi M, Zeisel A, Yitzhaky A, Yarden Y, et al. Context-specific microRNA analysis: identification of functional microRNAs and their mRNA targets. Nucleic Acids Res. 2012;40:10614–27.
Cantini L, Isella C, Petti C, Picco G, Chiola S, Ficarra E, et al. MicroRNA-mRNA interactions underlying colorectal cancer molecular subtypes. Nat Commun. 2015;6:8878.
van Iterson M, Bervoets S, de Meijer EJ, Buermans HP, ’t Hoen PAC, Menezes RX, et al. Integrated analysis of microRNA and mRNA expression: adding biological significance to microRNA target predictions. Nucleic Acids Res. 2013;41:e146.
Nam S, Li M, Choi K, Balch C, Kim S, Nephew KP. MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 2009;37:W356–362.
Ning B, Spira T, Beane JE, Lenburg ME. Differential regulation analysis quantifies mirna regulatory roles and context-specific targets [Internet]. bioRxiv. 2022. cited 2024 Oct 9]. p. 2022.07.24.501303. Available from: https://www.biorxiv.org/content/10.1101/2022.07.24.501303v1.
Wells AC, Hioki KA, Angelou CC, Lynch AC, Liang X, Ryan DJ, et al. Let-7 enhances murine anti-tumor CD8 T cell responses by promoting memory and antagonizing terminal differentiation. Nat Commun. 2023;14:5585.
Zhang Z, Cao Y, Zhai Y, Ma X, An X, Zhang S, et al. MicroRNA-29b regulates DNA methylation by targeting Dnmt3a/3b and Tet1/2/3 in porcine early embryo development. Dev Growth Differ. 2018;60:197–204.
Swahari V, Nakamura A, Hollville E, Stroud H, Simon JM, Ptacek TS, et al. MicroRNA-29 is an essential regulator of brain maturation through regulation of CH methylation. Cell Rep. 2021;35:108946.
Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CEA, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–8.
Amodio N, Stamato MA, Gullà AM, Morelli E, Romeo E, Raimondi L, et al. Therapeutic targeting of miR-29b/HDAC4 epigenetic loop in multiple myeloma. Mol Cancer Ther. 2016;15:1364–75.
Mullany LE, Herrick JS, Wolff RK, Stevens JR, Samowitz W, Slattery ML. MicroRNA-transcription factor interactions and their combined effect on target gene expression in colon cancer cases. Genes Chromosomes Cancer. 2018;57:192–202.
Qin G, Mallik S, Mitra R, Li A, Jia P, Eischen CM, et al. MicroRNA and transcription factor co-regulatory networks and subtype classification of seminoma and non-seminoma in testicular germ cell tumors. Sci Rep. 2020;10:852.
Lin Y, Sibanda VL, Zhang HM, Hu H, Liu H, Guo AY. MiRNA and TF co-regulatory network analysis for the pathology and recurrence of myocardial infarction. Sci Rep. 2015;5:9653.
Zhang Z, Zou Z, Zhang H, Zhang DM. Regulatory network analysis based on integrated miRNA-TF reveals key genes in heart failure. Sci Rep. 2024;14:13896.
Zhang XM, Guo L, Chi MH, Sun HM, Chen XW. Identification of active miRNA and transcription factor regulatory pathways in human obesity-related inflammation. BMC Bioinforma. 2015;16:76.
Acknowledgements
We thank the Cornell Biotechnology Resource Center (BRC) Genomics and Transcriptional Regulation and Gene Expression (RRID: SCR_021727; SCR_022532) facilities for support.
Author information
Authors and Affiliations
Contributions
Conceptualization HZ and AG; Experiments DGS, NLS, CD, KYM, CT, and EMW; Data analysis HZ; Formal analysis and visualization HZ; Supervision AG and BDR; Writing HZ and AG.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All methods were performed in accordance with the relevant guidelines and regulations. Research was performed under MUA Number: IBC16034-02 from the Cornell Institutional Biosafety Committee (to AG). All protocols regarding animal use were reviewed and approved by the Institutional Animal Care and Use Committee at Cornell University (IACUC Number: 2011-0090 to BDR).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, H., Ganapathi Sankaran, D., Smith, N.L. et al. An efficient framework to decipher microRNA regulatory programs applied to T cells. Genes Immun (2025). https://doi.org/10.1038/s41435-025-00351-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41435-025-00351-5
