
Abstract
Changes in epigenetic states can allow individuals to cope with environmental changes. If such changes are heritable, this may lead to epigenetic adaptation. Thus, it is likely that in sessile organisms such as plants, part of the spatial epigenetic variation found across individuals will reflect the environmental heterogeneity within populations. The departure of the spatial epigenetic structure from the baseline genetic variation can help in understanding the value of epigenetic regulation in species with different breadth of optimal environmental requirements. Here, we hypothesise that in plants with narrow environmental requirements, epigenetic variability should be less structured in space given the lower variability in suitable environmental conditions. We performed a multispecies study that considered seven pairs of congeneric plant species, each encompassing a narrow endemic with habitat specialisation and a widespread species. In three populations per species we used AFLP and methylation-sensitive AFLP markers to characterise the spatial genetic and epigenetic structures. Narrow endemics showed a significantly lower epigenetic than genetic differentiation between populations. Within populations, epigenetic variation was less spatially structured than genetic variation, mainly in narrow endemics. In these species, structural equation models revealed that such pattern was associated to a lack of correlation between epigenetic and genetic information. Altogether, these results show a greater decoupling of the spatial epigenetic variation from the baseline spatial genetic pattern in endemic species. These findings highlight the value of studying genetic and epigenetic spatial variation to better understand habitat specialisation in plants.
Similar content being viewed by others
Introduction
In sessile organisms such as flowering plants, the distribution of genetic information in space—known as spatial genetic structure (SGS)–, reflects predominant dispersal of pollen and seeds over short distances. Usually, these processes result in a SGS characterised by an exponential decay of genetic relatedness with distance (Wright 1943; Epperson 2005; Rousset 2000). Characterising the SGS both among and within populations has been important in evolutionary ecology studies because of its retrospective ability to infer historical eco-evolutionary processes. The variation of genetic relatedness with spatial distance can reflect differences in gene flow through seed dispersal (Hardy et al. 2006; Hamrick and Trapnell 2011) or mating system (Vekemans and Hardy 2004) or can result from associations between genetic variability and environmental factors (e.g., Valverde et al. 2016; Osuna-Mascaró et al. 2023; Fig. 1a). The SGS of plant populations therefore represents the spatial template of genetic variation that defines their evolutionary potential. In addition, recent studies support that plants can react to external environmental factors by adjusting their phenotype through epigenetic marks without showing changes in their DNA sequence (e.g., Lira-Medeiros et al. 2010; Herrera and Bazaga 2011, 2016; Zhang et al. 2012; Wu et al. 2013; Nicotra et al. 2015; Kim et al. 2016; Wilschut et al. 2016; Lehmair et al. 2022). Epigenetic changes are therefore a potential source of phenotypic variation upon which selection can act (Herrera et al. 2016) and might play an important role in the fate of populations, especially in harsh environments (Richards 2006; Jablonka and Raz 2009; Balao et al. 2018; Medrano et al. 2020). The complementary analysis of the SGS and the spatial epigenetic structure (SEGS) can provide essential information about the processes that shape natural epigenetic variation in nature (Herrera et al. 2017). However, studies comparing SGS and SEGS are still relatively rare (see e.g., Lele et al. 2018; Guan et al. 2020; Shen et al. 2021).

a Relationships defining SGS and SEGS. SGS results from reproductive and dispersive plant characteristics and from potential genetic-environment associations (i). SEGS is derived from the previous baseline SGS and can be modified depending on the dependence of epigenetic marks on genetic information and their transgenerational inheritance (ii) and on whether these respond to spatial variation in the environment (iii). b Hypothetical case of how a spatial environmental gradient defined by some relevant environmental feature (solid black line) can determine the spatial distribution of individuals (symbols along the X-axis) of two species with contrasting niche breadths (blue circles, broad niche breadth; red triangles, narrow niche breadth). Note that the coloured area represents the narrow niche breadth. c Expected SEGS when epigenetic variability is generally robust to variation in the environment. d Expected SEGS when epigenetic variability responds to the environment. When such response exists, SEGS might be differently modified depending on the environmental heterogeneity and the species niche breadth.
Methylation of DNA cytosine residues is one of the most studied epigenetic mechanisms in plants (Feng et al. 2010). Methylation marks can be reset between generations, but they can also be inherited (Richards 2006; Turner 2009; Verhoeven et al. 2010; Johannes and Schmitz 2019, Yao et al. 2023). Heritable epigenetic information is dispersed by the same means as genetic information, mainly through seeds and pollen, and thus the spatial distribution of both could be affected by migration and drift in a similar fashion (Slatkin 1993; Herrera et al. 2016). However, in addition to genetically-determined and spontaneous methylation variants, epimutations can be directly induced by environmental changes, adjusting the phenotype of a plant to such changes (Gao et al. 2010; Lira-Medeiros et al. 2010; Verhoeven et al. 2010, 2016; Noshay and Springer 2021; Van Antro et al. 2023). Therefore, the similarity of the environmental conditions of an individual to that of its progenitors might modulate the similarity in such epigenetic marks: under similar environmental conditions, the environmentally induced methylation variants would be maintained, whereas under contrasting environmental conditions these might vary (Ibañez et al. 2021). This correlation between environmental similarity and epigenetic similarity have been observed in experimental populations (e.g., Verhoeven et al. 2010; Rendina González et al. 2018; Xue et al. 2022) but also between natural populations experiencing similar habitat conditions (Paun et al. 2010; Schulz et al. 2014; Lehmair et al. 2022), indicating that the SEGS should be the result of a mixture of neutral and non neutral processes (Fig. 1a).
The simultaneous study of the genetic and epigenetic spatial structures of a group of sampled individuals, as proposed by Herrera et al. (2016), permits exploring the epigenetic deviation from a spatial genetic baseline null model. This approximation envisions two main scenarios. In the equilibrium between gene flow and epigenetic resetting, SGS and SEGS might show similar patterns if dispersal is the only process operating or if epigenetic reset between generations is minimal (Fig. 1c). This scenario should imply a high genetic-epigenetic correlation between individuals. If, on the contrary, environmentally driven epigenetic readjustment occurs, the SEGS will deviate from the SGS implying a decoupling of the genetic-epigenetic correlation (Fig. 1d). As environmental similarity usually diminishes with distance (Fig. 1b; Legendre 1993), environmentally induced epigenetic variability should result in a steeper decline of epigenetic similarity with distance (stronger SEGS than the baseline SGS). However, here we propose that the strength of SEGS might also depend on the range of suitable environmental requirements of each species (niche breadth), and on the spatial distribution of suitable environmental conditions (i.e., environmental heterogeneity; Fig. 1b). For instance, for a plant species with narrow environmental requirements, the higher homogeneity between suitable growing sites could result in higher epigenetic similarity between plants. This, in turn, could lead to a more gradual decline in epigenetic similarity with distance that could eventually result in the opposite pattern: a weaker SEGS than the baseline SGS (Fig. 1d).
Exploring the spatial-genetic-epigenetic relationship may especially provide insights in the ecology and evolution of species with narrow environmental requirements (narrow endemics sensu Rabinowitz 1981). Their very specific environmental requirements typically limit their distribution to small and isolated populations (reviewed in Slatyer et al. 2013). Theoretically, these circumstances would lead to a genetic impoverishment of their populations through drift and inbreeding (Barrett and Kohn 1991; Ellstrand and Elam 1993; Gitzendanner and Soltis 2000). Interestingly, several studies indicate that narrow endemics from the Mediterranean depart from this prediction, showing unexpectedly high population genetic (Fernández-Mazuecos et al. 2014; Jiménez-Mejías et al. 2015; Forrest et al. 2017; Medrano et al. 2020) and epigenetic diversities (e.g., Herrera and Bazaga 2010; Medrano et al. 2014, 2020). In addition, Mediterranean narrow endemics tend to show a positive correlation between genetic and epigenetic diversity, but populations with low genetic diversity still have a relatively high epigenetic diversity (Medrano et al. 2020). These findings indicate that epigenetics might act as a compensatory mechanism to create phenotypic variability. But they also suggest that the spatial correlation between genetic and epigenetic information across individuals might not behave similarly in narrow and widespread species, which could result in contrasting patterns of SGS and SEGS.
Based on the shared characteristics among narrow endemics (usually small, isolated populations with a narrow ecological niche) we can expect differences with widespread congeners in the SGS and SEGS. (1) Higher SGS. In smaller populations, an increased mating between nearby relatives is predicted, which can be further enhanced by an aggregated distribution of individuals (Doligez et al. 1998; Lara-Romero et al. 2016). This might lead to a generalised higher SGS as previously observed in peripheral, smaller populations (e.g., Gapare and Aitken 2005; Eckstein et al. 2006; De-Lucas et al. 2009; Pandey and Rajora 2012). (2) Weaker SEGS with respect to the baseline SGS. Narrow endemics inhabit highly specific habitats such as rocky outcrops that are often spatially dispersed at scales that can vary from cm to km (Lavergne et al. 2004; Thompson et al. 2005) and that condition the distribution of individuals and populations to such microenvironments (e.g., Luzuriaga et al. 2015; Schouten and Houseman 2019; Pescador et al. 2020). If part of the epigenetic variation is environmentally responsive, the higher environmental homogeneity among sites where these species grow might reflect a lower epigenetic variance in space. This should consequently result in a decoupling of the epigenetic variation from the genetic variation. In contrast, the broader niche breadth of widespread species allows for the colonisation of different and more heterogeneous habitats (Slatyer et al. 2013) which might reflect stronger SEGS, i.e., higher epigenetic variance in space.
Here, we test whether in species with greater habitat specificity SEGS is weaker than the baseline SGS. We explore this hypothesis in the seven pairs of congeneric plant species previously studied in Medrano et al. (2020) that consists of a narrow endemic and a widespread species that inhabit the Sierra de Cazorla mountain range (SE Spain). First, we compare the genetic and epigenetic variation across populations. Then, at the population level, we use the framework of Herrera et al. (2016) and the model presented in Fig. 1a to test if SEGS differs from the SGS more in narrow endemics. An understanding of the sign and intensity of the deviation of SEGS from the SGS baseline in these two groups of species will provide a better understanding of the role of epigenetics in the adaptation to harsh environments.
Materials and methods
Study species and field sampling
This study was carried out in the Sierras de Cazorla, Segura y Las Villas Natural Park (37° 32’ 00”–38° 41’ 30” N; 2° 26’ 30”–3° 07’ 00” W; 600–2107 m a.s.l; SE Spain). We chose seven pairs of congeneric plant species from seven different families. Each congeneric pair consisted of a narrow endemic from the Baetic Range (R, hereafter) and a widely distributed species (Widespread; W, hereafter), all inhabiting this mountain range. These species correspond to those studied in Medrano et al. (2020): Fabaceae, Anthyllis ramburii (R) and A. vulneraria (W); Ranunculaceae, Aquilegia pyrenaica subsp. cazorlensis (R) and A. vulgaris (W); Convolvulaceae, Convolvulus boissieri (R) and C. arvensis (W); Geraniaceae, Erodium cazorlanum (R) and E. cicutarium (W); Thymelaeaceae, Daphne oleoides (R) and D. laureola (W); Lamiaceae, Teucrium rotundifolium (R) and T. similatum (W); Violaceae, Viola cazorlensis (R) and V. odorata (W). All species are characterised by dispersing their seeds mainly at short distances and for being pollinated by insects. The classification as narrow endemic or widespread species follows criteria of geographic distribution range, population density and size, and niche breadth. Narrow endemics are those species with a restricted geographic distribution, sparse and small populations, and associated to specific stressing microenvironments. Widespread species are those with a broad geographic range and with large populations associated to a wider niche breadth. We relied on Blanca et al. (2009) and our own familiarity with population abundance within the study area (see Table S1 for details).
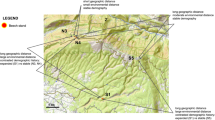
For each species we chose three populations based on their accessibility (Fig. 2; see Medrano et al. 2020 for exact locations). From April to June we sampled 23 to 40 widely spaced flowering individuals per population. Sampling in the larger populations was carried out in a representative area to minimise variability between sampling areas (mean of maximum distance between individuals from a population = 181.98 ± 195.49 metres). The chosen plants were spatially located using a GPS device (Garmin GPSMAP 64s). Spatial data for three of the populations studied that were located in canyons or under steep cliffs (two of Aquilegia cazorlensis (R) and one of Anthyllis ramburii (R)) could not be obtained, so these populations were discarded from the fine-scale spatial analyses. From each plant we collected 5–6 new fully expanded leaves with no sign of senescence or disease that were immediately dried in silica gel for further analyses.

For each of the studied genus the approximate location of the sampled populations of the narrow endemic and widespread congener are shown with purple squares and green circles respectively. The delimited area depicts the limits of the natural park.
Laboratory procedures
We used amplified fragment length polymorphism (AFLP; Vos et al. 1995) and the related methylation-sensitive amplified polymorphism technique (MSAP; Reyna-López et al. 1997; Fulneček and Kovařík 2014) to obtain the genetic and epigenetic fingerprint of each of the 1088 sampled individuals. The laboratory procedures and primer combinations are detailed in Medrano et al. (2020) and in Table S2. AFLP and MSAP band scoring resulted in two binary matrices, one depicting the presence or absence of each AFLP marker and another depicting the presence or absence of each hemi- or fully methylated epiloci (Schulz et al. 2014). We assessed the repeatability of banding patterns for each species by repeating the entire AFLP/MSAP protocol in a number of randomly selected samples (8.6–17.6% of samples per species for AFLP; 13.3–29.3% of samples for MSAP; see Medrano et al. 2020 for further details). We also discarded non-informative monomorphic loci that showed less than 5% of variability across samples. This yielded a variable number of polymorphic loci that varied from 86 (Daphne oleoides) to 418 (Teucrium similatum) for AFLP markers, and from 95 (Daphne oleoides) to 213 (Teucrium similatum) for MSAP markers (see Table S3 and Medrano et al. 2020 for further information).
Although ALFP and MSAP markers are dominant, and—unlike co-dominant markers—do not permit distinguishing between homozygous and heterozygous states (Paun and Schönswetter 2012), these techniques have important advantages when working with several species and large sample sizes. First, they allow reliable detection of genome-wide variants of genetic and epigenetic signatures in non-model species lacking genomic resources (Schrey et al. 2013; Medrano et al. 2014, 2020; Herrera et al. 2016; Wilschut et al. 2016; Thiebaut et al. 2019). And second, both techniques, AFLP and MSAP, detect anonymous markers using a similar procedure, making them mutually comparable.
Genetic and epigenetic population structure
To assess the genetic and epigenetic structure at a regional scale, we first estimated the genetic inbreeding coefficient of each population using the Markov Chain Monte Carlo procedure implemented in the I4A software (Chybicki et al. 2011). Posterior distributions were obtained using 50,000 sampling steps and 10,000 burning steps. This Bayesian approach provides robust estimates of population inbreeding using dominant markers (Chybicki et al. 2011; Stone et al. 2019; García-Castaño et al. 2021). Next, for each species we calculated the genetic and epigenetic divergence between populations using Wright’s unbiased fixation index FST (Wright 1951) and considering the average of the previously calculated population inbreeding coefficients. These calculations were performed with the Bayesian method implemented in the AFLP-SURV software (Vekemans et al. 2002) using a non-uniform prior distribution. Estimated FST values were compared to a null distribution of values obtained after 1000 permutations of individuals across populations that depicted a complete absence of structure among populations. An additional hierarchical analysis of molecular variance (AMOVA) was performed to partition the genetic and epigenetic variance between and within populations in each plant species using the R package ade4 (Dray and Dufour 2007).
For each group of species (narrow endemics and widespread species), we compared the genetic and epigenetic FST values using a non-parametric Wilcoxon paired test. To be confident of our results these analyses were repeated for FST values that were calculated considering inbreeding estimates of 0 (panmixia) and 1 (autogamy) that encompass the maximum range of variation.
Fine-scale spatial genetic and epigenetic structure
To study the spatial genetic and epigenetic structure within each of the sampled populations we calculated the genetic relatedness and epigenetic similarity between each pair of individuals using the kinship coefficient proposed by Hardy (2003) for dominant markers. The calculation of this coefficient included the required estimates of inbreeding coefficient for each population. This kinship coefficient expresses the degree of similarity between individuals relative to the average similarity found in the population. Thus, positive and negative values depict respectively higher and lower relatedness than random individuals from the population. We visually explored the fine-scale genetic and epigenetic spatial structures (SGS and SEGS) by calculating the average kinship coefficients (FN) for six distance classes that showed even number of pairwise distances and that avoided spatial gaps. The significance of each FN with respect to the expected in the absence of structure was tested using 1000 permutation and two-sided tests. These calculations were performed using the SPAGeDi software (Hardy and Vekemans 2002).
We compared SGS and SEGS in populations of narrow endemics and widespread species independently. For each population we calculated the slope of the linear relationship between the logarithm of the spatial separation between each pair of individuals and their (epi)genetic kinship coefficients. Next, we applied mixed-effects models using the lme4 package in R (Bates et al. 2015) to explore whether there was any generalised trend in the relationship between SGS and SEGS. The slopes of the log10 (distance)-kinship relationship were used as dependent variable and modelled as a function of the type of marker (AFLP or MSAP). Population was included as a random factor to account for the paired nature of data. To control for potential effects of the spatial distribution of sampled individuals (Doligez et al. 1998), we included the coefficient of variation and the maximum value of the pairwise spatial distances of each population as covariates (Nagamitsu et al. 2019) (R syntax: lmer(log.slope ~ distance.cv + scale(distance.max) + marker.type + (1|population), data)).
Finally, we examined the extent to which the deviation of SEGS from SGS differed between populations of endemic and widespread species. The differences in slope between SEGS and SGS were used as response variable in a mixed-effects model. Plant distribution (narrow endemic versus widespread) was used as predictory variable and the coefficient of variation and the maximum value of the pairwise spatial distances as covariates. Genus was included as a random intercept effect to account for variation among pairs of congeneric species (R syntax: lmer(difference.in.log.slope ~ distance.cv + scale(distance.max) + distribution.type + (1|genus), data)).
Spatial-genetic-epigenetic relationships
We explored the differences between narrow endemics and widespread congeners in the relationship between spatial distance, genetic relatedness and epigenetic similarity. We first defined a causal model that relates the previous three variables and that includes the following direct causal effects (Fig. 1a): (i) the relationship between the log10 (distance) and genetic relatedness, which typically denotes gene dispersal (Wright 1943); (ii) the relationship between genetic relatedness and epigenetic similarity, which reflects the dependence epigenetic marks on genetic identity (Noshay and Springer 2021); and (iii) the relationship between the log10 (distance) and epigenetic similarity, which might describe the putative dependence of environmentally induced epigenetic marks with space (Herrera et al. 2016). This model allowed us to calculate the indirect relationship between spatial distance and epigenetic similarity through genetic relatedness (i + ii) which might reflect the epigenetic reset across space. We fitted this model to each of the study populations separately using structural equation models using the piecewiseSEM R package (Lefcheck 2016). This approximation allowed us to calculate the previously detailed direct and indirect relationships as standardised estimates and their respective confidence intervals and significance after 1000 bootstrap steps using the semEff R package (Murphy 2022). As a result, for each relationship we obtained a set of estimates, each from one of the studied populations.
We tested the differences between endemics and widespread congeners in the previous direct and indirect relationships by means of mixed-effects linear models. Genus was included as random intercept effect in both models. To control for the spatial distribution of plants, all models included the coefficient of variation and maximum distances as covariates (R syntax: lmer(effect ~ distance.cv + scale(distance.max) + distribution.type + (1|genus), data)). Models yielding singular fits due to low variance in the random effects were modelled using simple linear models (R syntax: lm(effect ~ distance.cv + scale(distance.max) + distribution.type, data)).
Results
Genetic and epigenetic population structure
Species-averaged inbreeding estimates ranged between 0.04 (Viola cazorlensis (R)) and 0.96 (both species of Teucrium; Table S3). Most species showed small although significant FST values for genetic and epigenetic markers (FST ≤ 0.27 and FST < 0.37 for genetic and epigenetic, respectively; Table S4). These results are in agreement with those from the AMOVA analyses, that showed significant population differentiation although most of the genetic and epigenetic variance occurred within population (range of total variance occurring within populations = 62.5–99.1% for AFLP and 51.1–94.3% for MSAP; Table S5). In most species, the variation attributable to differences between populations was greater for genetic than for epigenetic markers (Fig. 3). However, only in narrow endemics genetic FST was significantly higher than epigenetic FST (V = 28, p = 0.016 in endemics; V = 21, p = 0.297 in widespread species). These comparisons were consistent when considering inbreeding coefficients of 0 (V = 28, p = 0.016 in endemics; V = 20, p = 0.375 in widespread) or 1 (V = 27, p = 0.031 in endemics; V = 21, p = 0.297 in widespread), thus justifying the performance of our inbreeding estimates in these analyses.

For each study species the genetic (AFLP) divergence between populations is plotted against the methylation-sensitive epigenetic (MSAP) divergence, both calculated as the Wright fixation index (FST). Error bars span FST values calculated using inbreeding coefficients of 0 and 1, while points represent values of FST calculated using the inbreeding estimates after the method proposed by Chybicki et al. (2011). The dashed line is used to contrast cases where the differentiation is relatively greater for genetic markers (lower area) or for epigenetic markers (upper area). Results from Wilcoxon paired tests for the differences between genetic and epigenetic FST values are shown in each panel.
Fine-scale genetic and epigenetic spatial structure
Populations showed a varied spatial distribution of the sampled plants (see examples in Fig. 4) that resulted in a high heterogeneity in the distribution of distances between plants (CV range = 0.48–1.17; see Figs. 4 and S1 and Table S3), with 8 and 5 populations of endemic and widespread species respectively showing a multimodal distribution of spatial distances (Hartigans’ multimodality test p < 0.05). A total of 24 populations displayed a significant decrease of genetic relatedness with log10 (distance) (b = −0.11–−0.01, p ≤ 0.047), while for epigenetic markers this relationship was significant and negative in 14 populations (b = −0.05–−0.01, p ≤ 0.034; Fig. S1; Table S6).

These populations have been arbitrarily chosen to exemplify the variability found across populations. Left panels show the spatial distribution of sampled plants. Middle panels depict the distribution of spatial distances between plants and show information about its variability (CV) and multimodality (Hartigan’s D and associated p value). Right panels show the slopes of the relationship between the logarithm of the spatial distance and the genetic relatedness (SGS, grey lines) and between the logarithm of the spatial distance and the epigenetic similarity (SEGS, red lines). Dashed lines depict non-significant relationships. Figure S1 provides this information for all the sampled populations.
Narrow endemics showed overall significant differences between SEGS and SGS (t = 2.16, p = 0.039; Table S7; Fig. 5). Specifically, a weaker SEGS pattern than the baseline SGS, i.e., the across populations average slope of the log10 (spatial distance)-epigenetic similarity was more gradual (marginal mean = −0.009 ± 0.002) than the slope of the log10 (spatial distance)-genetic relatedness (marginal mean = 0.016 ± 0.002). This comparison was not significant in widespread species (t = 1.68, p = 0.094; Fig. 5). Yet, average SEGS was weaker (marginal mean = −0.019 ± 0.005) than SGS (marginal mean = −0.030 ± 0.005). Finally, the deviation of the SEGS from the baseline SGS did not differed significantly between narrow endemics and widespread species (t = 1.58, p = 0.113; Table S8).

The panels summarise the slopes of the relationship between the logarithm of the spatial distance and the genetic relatedness (SGS) and between the logarithm of the spatial distance and the epigenetic similarity (SEGS). Points within the grey areas denote populations where epigenetic similarity decreases with distance more gradually than genetic relatedness (SEGS < SGS) (and the opposite for white areas; SEGS > SGS). Error bars span ±1 SE after a jackknifing procedure over loci. Statistics at each panel refer to the term defining the type of marker in the mixed model and thus reflect overall differences between genetic and epigenetic slopes.
Spatial-genetic-epigenetic relationships
Structural equation models showed a positive and significant relationship between genetic relatedness and epigenetic similarity in 64% of the populations, most of them from widespread species (7 in endemics, 18 in widespread). It also revealed that spatial distance and epigenetic kinship were significantly correlated only in nine out of the 39 studied populations (Table S9), although this relationship was negative in most cases. In contrast, the indirect relationship between spatial distance and epigenetic kinship—mediated through genetic kinship–, was negative and significant in 41% of populations, most of them from widespread species (4 in endemics, 12 in widespread).
Models showed that the relationship between genetic relatedness and epigenetic similarity and the indirect relationship between spatial distance and epigenetic similarity significantly differed between narrow endemics and widespread species (t = 5.74, p < 0.001 and t = −3.48, p = 0.001, respectively; Table S10; see Fig. 6). Estimated marginal means indicated a four-fold higher effect of genetic relatedness on epigenetic similarity in widespread species that in their endemic congeners (0.094 ± 0.037 in endemics; 0.347 ± 0.034 in widespread, Fig. 6). The indirect relationship between spatial distance and epigenetic similarity was 18 times stronger in widespread species (−0.004 ± 0.018 in endemics; −0.070 ± 0.017 in widespread, Fig. 6). In contrasts, the direct relationship of spatial distance with both epigenetic similarity and genetic relatedness showed no differences between narrow and widespread (t = −1.69, p = 0.092 and t = 1.08, p = 0.289; Table S10; Fig. 6).

a Structural equation models. Arrows depict direct relationships. For each group of species, the mean value between populations (±standard error) of each direct relationship is shown. The size and colour of arrows relates to the magnitude and sign (blue, positive; red, negative) of such value. b Comparison between groups of species in each of the direct and indirect relationships. Each relationship is presented in a separate panel showing population estimates for both groups of species. Each genus is represented by a colour (see legend in Figs. 3 and 5). Marginal means of each group of species and their associated standard error are shown where significant differences between groups were found.
Discussion
This research illustrates the broad variety of spatial structuring of genetic and epigenetic variation in natural populations of Mediterranean montane areas. Using a multispecies comparative approach with a set of seven pairs of congeneric species differing in their range of geographic distribution and habitat specialisation, we tested the specific hypothesis that in species with greater habitat specificity, SEGS should be weaker than the SGS. Our results showed that epigenetic variability was less structured in space than genetic variability both between and within populations of narrow endemisms. Within populations of narrow endemics, such differences reflect the lack of correlation between epigenetic and genetic variation. In contrast, in widespread species, we found a higher variability across species in the SEGS and a stronger and significant genetic-epigenetic correlation.
Genetic and epigenetic population structure
Our sampling scheme showed low genetic differentiation between populations in the studied species, with most of the genetic variance found within populations. Wright fixation index ranged between 0.02 and 0.24 in narrow endemics and between 0.01 and 0.27 in widespread species. These values lay within the lower part of the range reported for 86 insect-pollinated temperate plant species (average = 0.21; 95% CI = 0.01–0.51; Gamba and Muchhala 2020 and references therein). Caution should be taken when interpreting these parameters because an analysis of three populations per species might not be representative of population differentiation in the full geographic distribution range (e.g., Castilla et al. 2012 for Daphne laureola; Daco et al. 2022 for A. vulneraria; but see Herrera and Bazaga 2008 for Viola cazorlensis). However, the FST values obtained through the multispecies framework and population level approach can provide representative data for the particular geographic area in which the sampling was conducted. Specifically, it suggests that in our study area, the magnitude of population differentiation of endemic and widespread species could be similar, although future studies with more species and populations will be required to support these results.
Epigenetic differentiation was also low (FST range = 0.012–0.159), except for A. vulgaris with Fst = 0.367. But more importantly, it was lower than genetic differentiation in most of the species studied. Especially in narrow endemics, epigenetic differentiation was significantly lower than genetic differentiation, while widely distributed species did not show a significant trend across species or populations. The few studies on epigenetic differentiation of populations point to a lack of common pattern in the comparison between epigenetic and genetic differentiation (e.g., Wu et al. 2013; Herrera et al. 2017versus Avramidou et al. 2015; Lele et al. 2018). However, in most studies where there is higher epigenetic than genetic differentiation among populations, the former is associated with environmental factors. Among these studies, it is worth highlighting those that show an association with population soil variables such as moisture, pH or chemical composition (Lira-Medeiros et al. 2010; Shen et al. 2021; Lehmair et al. 2022), which together with experimental studies controlling for soil variables (reviewed in Alonso et al. 2019), add to the evidence of the ubiquitous effect of soil characteristics and habitat features on epigenetic variation (see also Paun et al. 2010, Schulz et al. 2014). In our study, endemic species are characterised by having a narrow niche breadth, with suitable habitat sparsely distributed in space, resulting in a patchy distribution. The similarity in the microenvironmental conditions of individuals of each of these species would lead to a convergence of methylation patterns. This would explain the lower epigenetic differentiation between endemic populations, especially in those species with higher genetic differentiation among populations (Fst > 0.1) where the evidence of lower gene flow contrasts with higher similarities in epigenetic signatures. This decoupling is not so evident in widely distributed species among which there are also cases with opposite patterns (e.g., Aquilegia vulgaris) that might reflect a broader environmental niche. This heterogeneity demonstrates that studies that consider more populations are needed to investigate the spatial (epi)genetic patterns of specific species.
Fine-scale genetic structure within populations
Within most of the populations studied, the genetic variation was spatially structured, showing a significant decrease with spatial distance. This fine-scale SGS is expected for sessile organisms with short-distance dispersal mechanisms (Vekemans and Hardy 2004) such as those shared among the studied species. Contrary to our expectations, narrow endemics and widespread species did not show global differences in the fine-scale SGS. This finding is somehow surprising given that narrow endemics are usually comprised of smaller and isolated populations. Under such circumstances, with spatially restricted pollen flow and seed dispersal, kinship between close individuals is expected to increase and, consequently, the strength of fine-scale SGS (Vekemans and Hardy 2004; Gapare and Aitken 2005; Volis et al. 2016). A plausible explanation of the lack of differentiation in SGS could be the fact that some of the sampled widespread populations were small (i.e., Viola odorata). Another explanation could lie in the different pollinators visiting the species studied. For instance, the endemic Viola cazorlensis is mainly pollinated by the hawkmoth Macroglossum stellatarum which is able to fly long distances and thus create a flow of pollen between individuals located far apart within the same population. Reproductive patterns are key in generating genetic spatial variation (Ennos 2001; Vekemans and Hardy 2004; Gamba and Muchhala 2022). In the case of our Mediterranean endemic species, it is likely that the increase in foraging types that comes with increased pollinator diversities promotes greater pollen movement, which in the long run would relax the SGS. Our ongoing work is testing this hypothesis.
It is noteworthy to highlight that the number of loci can affect the accuracy of SGS detection (Cavers et al. 2005; Jump and Penuelas 2007). The phylogenetically diverse set of species considered here resulted in contrasting number of loci between species (from 86 in Daphne oleoides to 418 in Teucrium similatum). However, in our data there is a lack of correlation between the number of loci and the SGS slope per population (r = 0.16, p = 0.328) and between the number of loci and the range of SGS slope values per species (r = −0.14, p = 0.622). In addition, after subsampling 100 times each data set to the minimum number of loci present across species (see example in Fig. S2) we obtained virtually the same average log10 (distance)-genetic kinship slopes (Table S11) indicating that the patterns in our data are robust to variation in the number of markers. Nonetheless, caution must be taken when interpreting the results obtained for populations with the lowest number of loci (i.e., those of Daphne oleoides).
Patterns of spatial-genetic-epigenetic relationships
Epigenetic similarity within populations decreased with distance more gradually than genetic relatedness in most cases (i.e., steeper slope for genetic markers), indicating that fine-scale SEGS was in general weaker than the baseline spatial genetic variation. However, in widespread species there was a higher heterogeneity in the deviation of the SEGS from the baseline SGS (standard deviation = 0.03 in widespread species versus 0.01 in narrow endemics) which explains the lack of differences between narrow endemics and widespread species in the overall SEGS-SGS deviation. Several examples illustrate this higher heterogeneity in widespread species. For instance, opposite to the general trend, in all sampled populations of Viola odorata epigenetic variation was more spatially structured (stronger SEGS). In Anthyllis vulneraria and Daphne laureola, there was a lack of consistency in the spatial epigenetic patterns across populations. Such inconsistency can be attributed to multiple factors, ranging from environmental heterogeneity to different epigenetic responses, either independent of the environment, highly dependent on genetic variation or highly dependent on the environment, which emphasises the need of studying each species in detail.
In contrast, populations from narrow endemics showed a higher consistency across populations, with most populations showing a weaker SEGS than the baseline SGS. According to the original framework of Herrera et al. (2016), this could result from a prevalent non-environmentally responsive epigenetic reset. This reasoning is based on the fact that environmental variables usually exhibit a strong spatial autocorrelation (i.e., a decrease in environmental similarity with distance; Fig. 1b; Legendre 1993). Under such an assumption the framework predicts that while a stronger SEGS could result from a correlation with a spatially structured environmental variable, a weaker spatial epigenetic spatial structure -which is our case-would be the consequence of an epigenetic readjustment independent of the environment. However, we believe that this argument does not fit our particular case and that, similar to what we have previously discussed about the patterns found between populations, within-population epigenetic variation in endemic species could be explained by their habitat specificity and the spatial heterogeneity of the microenvironmental growing conditions, as we expose below (see Fig. 1).
In the studied populations of endemic species we found a low or even a lack of dependence of epigenetic variation on genetic variation, which contrasts with the highly significant genetic-epigenetic correlation found in widespread species. Although studies in Arabidopsis thaliana demonstrate an interdependence between genetic and epigenetic signatures (Zilberman et al. 2007; Dubin et al. 2015; Kawakatsu et al. 2016), other studies demonstrate a lack of such correlation in natural populations of non-model species (e.g., Herrera and Bazaga 2010; Richards et al. 2012; Schulz et al. 2014; Foust et al. 2016). Such contrasting findings might be due to higher rates of epigenetic spontaneous mutations in some species in comparison to that of A. thaliana. However, evidence of the existence of epimutations associated with the environment (e.g., Lira-Medeiros et al. 2010; Herrera and Bazaga 2011, 2016; Nicotra et al. 2015; Gáspár et al. 2018) suggest that environmental stochasticity can be a major determinant of epigenetic variation. Our finding of differences in genetic-epigenetic correlations associated with niche breadth (narrow endemics vs widespread species) suggests a decoupling of epigenetic variation from genetic variation in narrow endemics. The cause of this decoupling requires further work.
On top of this, structural equation models showed differences in fine-scale epigenetic spatial structure between endemics and widespread congeners. But more importantly, it demonstrated significant differences between endemic and widespread populations in the indirect effect of spatial distance on epigenetic similarity when mediated by the genetic relatedness of individuals. Specifically, the results indicate that in endemics the weak epigenetic spatial structure within populations is mainly due to the aforementioned lack of relationship between epigenetic and genetic variation at this scale. This finding contrasts with that in widespread species, where the greater epigenetic spatial structure is mediated by the close dependence of epigenetic variation on genetic relatedness. As a result, both groups of plants differed in the fine-scale SEGS deviation from the baseline SGS.
This further supports our rationale that the environmental requirements defining each group of studied species shape the epigenetic response in space. On the one hand, the endemic plants studied inhabit highly specific microsites, mainly dolomitic and rocky outcrops surrounded by a matrix of less favourable environmental conditions (e.g., soil conditions or increased interspecific competition), which is reflected in the aggregate spatial distribution of individuals. On the other hand, widely distributed species can inhabit a broader range of microsites, occupying a large fraction of space despite environmental heterogeneity. As a consequence of the higher specificity in endemic species, the environmental similarity between any pair of plants is expected to be higher than in widely distributed species. Assuming that part of epigenetic variation can respond to the environment, epigenetic changes in endemics should be lower, resulting in higher epigenetic similarity between individual plants. This would then explain the observed decoupling between spatial epigenetic and genetic structures.
To test this rationale, future work should characterise the microenvironmental conditions of individual plants and incorporate this as a direct effect on epigenetic similarity in the structural equation model (e.g., Herrera and Bazaga 2011; Lele et al. 2018). This piece of information is especially relevant given the effect that environmental dissimilarity can have on the spatial distribution of epigenetic variants (Herrera et al. 2017). In any case, our findings are in line with previous findings in which a decoupling of the genetic and epigenetic variation are accompanied by a correlation of epigenetic variation with environmental conditions (Lehmair et al. 2022) and highlight the importance of such correlation in determining the spatial pattern of genetic and epigenetic variation.
Data availability
All data are available in Figshare. https://doi.org/10.6084/m9.figshare.24898461.
References
Alonso C, Ramos-Cruz D, Becker C (2019) The role of plant epigenetics in biotic interactions. N Phytol 221(2):731–737
Avramidou EV, Ganopoulos IV, Doulis AG, Tsaftaris AS, Aravanopoulos FA (2015) Beyond population genetics: natural epigenetic variation in wild cherry (Prunus avium). Tree Genet Genomes 11(5):95
Balao F, Paun O, Alonso C (2018) Uncovering the contribution of epigenetics to plant phenotypic variation in Mediterranean ecosystems. Plant Biol 20:38–49
Barrett SCH, Kohn JR (1991) Genetic and evolutionary consequences of small population size. In: Falk DA, Holsinger KE (eds) Genetics and conservation of rare plants. Oxford University Press, Oxford, pp 3–30
Bates D, Maechler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67(1):1–48
Blanca G, Cabezudo B, Cueto M, Fernández López C, Morales Torres C (2009) Flora Vascular de Andalucía Oriental, 4 vols. Consejería de Medio Ambiente, Junta de Andalucía, Sevilla
Castilla AR, Alonso C, Herrera CM (2012) Genetic structure of the shrub Daphne laureola across the Baetic Ranges, a Mediterranean glacial refugium and biodiversity hotspot. Plant Biol 14:515–524
Cavers S, Degen B, Caron H, Lemes MR, Margis R, Salgueiro F et al. (2005) Optimal sampling strategy for estimation of spatial genetic structure in tree populations. Heredity 95:281–289
Chybicki IJ, Oleksa A, Burczyk J (2011) Increased inbreeding and strong kinship structure in Taxus baccata estimated from both AFLP and SSR data. Heredity 107:589–600
Daco L, Matthies D, Hermant S, Colling G (2022) Genetic diversity and differentiation of populations of Anthyllis vulneraria along elevational and latitudinal gradients. Eco Evol 12:e9167
De-Lucas AI, González-Martínez SC, Vendramin GG, Hidalgo E, Heuertz M (2009) Spatial genetic structure in continuous and fragmented populations of Pinus pinaster Aiton. Mol Ecol 18(22):4564–4576
Doligez A, Baril C, Joly HI (1998) Fine‐scale spatial genetic structure with nonuniform distribution of individuals. Genetics 148:905–919
Dray S, Dufour A (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Soft 22:1–20
Dubin MJ, Zhang P, Meng D, Remigereau M-S, Osborne EJ, Casale FP et al. (2015) DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. ELife 4:e05255
Eckstein RL, O’Neill RA, Danihelka J, Otte A, Köhler W (2006) Genetic structure among and within peripheral and central populations of three endangered floodplain violets. Mol Ecol 15:2367–2379
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Ennos R (2001) Inferences about spatial processes in plant populations from the analysis of molecular markers. In: Silvertown J, Antonovics J (eds) Integrating ecology and evolution in a spatial context. Blackwell Science, London, pp 45–72
Epperson BK (2005) Estimating dispersal from short distance spatial autocorrelation. Heredity 95:7–15
Feng S, Jacobsen SE, Reik W (2010) Epigenetic reprogramming in plant and animal development. Science 330:622–627
Fernández-Mazuecos M, Jiménez-Mejías P, Rotllan-Puig X, Vargas P (2014) Narrow endemics to Mediterranean islands: moderate genetic diversity but narrow climatic niche of the ancient, critically endangered Naufraga (Apiaceae). Perspect Plant Ecol Evol Syst 16(4):190–202
Forrest A, Escudero M, Heuertz M, Wilson Y, Cano E, Vargas P (2017) Testing the hypothesis of low genetic diversity and population structure in narrow endemic species: the endangered Antirrhinum charidemi (Plantaginaceae). Bot J Linn Soc 183:260–270
Foust CM, Preite V, Schrey AW, Alvarez M, Robertson MH, Verhoeven KJF et al. (2016) Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Mol Ecol 25(8):1639–1652
Fulneček J, Kovařík A (2014) How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet 15:2–9
Gamba D, Muchhala N (2020) Global patterns of population genetic differentiation in seed plants. Mol Ecol 29:3413–3428
Gamba D, Muchhala N (2022) Pollinator type strongly impacts gene flow within and among plant populations for six Neotropical species. Ecology 104(1):e3845
Gao L, Geng Y, Li B, Chen J, Yang J (2010) Genome-wide DNA methylation alterations of Alternanthera philoxeroides in natural and manipulated habitats: Implications for epigenetic regulation of rapid responses to environmental fluctuation and phenotypic variation. Plant Cell Environ 33:1820–1827
Gapare WJ, Aitken SN (2005) Strong spatial genetic structure in peripheral but not core populations of Sitka spruce [Picea sitchensis (Bong.) Carr.]. Mol Ecol 14:2659–2667
García-Castaño JL, Balao F, Lorenzo MT, Vela E, Hadjadj-Aoul S, Mifsud S et al. (2021) A complex genetic structure of Tetraclinis articulata (Cupressaceae) in the western Mediterranean. Bot J Linn Soc 197(3):420–438
Gáspár B, Bossdorf O, Durka W (2018) Structure, stability and ecological significance of natural epigenetic variation: a large-scale survey in Plantago lanceolata. N. Phytol 221:1585–1596
Gitzendanner MA, Soltis PS (2000) Patterns of genetic variation in rare and widespread plant congeners. Am J Bot 87(6):783–792
Guan Y, Qu P, Lu S, Crabbe M, Zhang T, Geng Y (2020) Spatial genetic and epigenetic structure of Thlaspi arvense (field pennycress) in China. Genes Genet Syst 95(5):225–234
Hamrick JL, Trapnell DW (2011) Using population genetic analyses to understand seed dispersal patterns. Acta Oecol 37:641–649
Hardy OJ (2003) Estimation of pairwise relatedness between individuals and characterisation of isolation by distance processes using dominant genetic markers. Mol Ecol 12:1577–1588
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hardy OJ, Maggia L, Bandou E, Breyne P, Caron H, Chevallier MH et al. (2006) Fine-scale genetic structure and gene dispersal inferences in 10 Neotropical tree species. Mol Ecol 15:559–571
Herrera CM, Bazaga P (2008) Population-genomic approach reveals adaptive floral divergence in discrete populations of a hawk moth-pollinated violet. Mol Ecol 17(24):5378–5390
Herrera CM, Bazaga P (2010) Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. N. Phytol 187(3):867–876
Herrera CM, Bazaga P (2011) Untangling individual variation in natural populations: ecological, genetic and epigenetic correlates of long-term inequality in herbivory. Mol Ecol 20:1675–1688
Herrera CM, Bazaga P (2016) Genetic and epigenetic divergence between disturbed and undisturbed subpopulations of a Mediterranean shrub: a 20-year field experiment. Ecol Evol 6(11):3832–3847
Herrera CM, Medrano M, Bazaga P (2016) Comparative spatial genetics and epigenetics of plant populations: Heuristic value and a proof of concept. Mol Ecol 25:1653–1664
Herrera CM, Medrano M, Bazaga P (2017) Comparative epigenetic and genetic spatial structure of the perennial herb Helleborus foetidus: Isolation by environment, isolation by distance, and functional trait divergence. Am J Bot 104(8):1195–1204
Ibañez VN, Masuelli RW, Marfil CF (2021) Environmentally induced phenotypic plasticity and DNA methylation changes in a wild potato growing in two contrasting Andean experimental gardens. Heredity 126:50–62
Jablonka E, Raz G (2009) Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol 84(2):131–177
Jiménez‐Mejías P, Fernández‐Mazuecos M, Amat ME, Vargas P (2015) Narrow endemics in European mountains: high genetic diversity within the monospecific genus Pseudomisopates (Plantaginaceae) despite isolation since the late Pleistocene. J Biogeogr 42(8):1455–1468
Johannes F, Schmitz RJ (2019) Spontaneous epimutations in plants. N. Phytol 221:1253–1259
Jump AS, Penuelas J (2007) Extensive spatial genetic structure revealed by AFLP but not SSR molecular markers in the wind‐pollinated tree, Fagus sylvatica. Mol Ecol 16(5):925–936
Kawakatsu T, Huang S-SC, Jupe F, Sasaki E, Schmitz RJ, Urich MA et al. (2016) Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 166:492–505
Kim NS, Im MJ, Nkongolo K (2016) Determination of DNA methylation associated with Acer rubrum (red maple) adaptation to metals: Analysis of global DNA modifications and methylation‐sensitive amplified polymorphism. Ecol Evol 6(16):5749–5760
Lara-Romero, García-Fernandez C, Robledo-Arnuncio A, Roumet JJ, Morente-Lopez M, Lopez-Gil A et al. (2016) Individual spatial aggregation correlates with between-population variation in fine-scale genetic structure of Silene ciliata (Caryophyllaceae). Heredity 116:417–423
Lavergne S, Thompson JD, Garnier E, Debussche M (2004) The biology and ecology of narrow endemic and widespread plants: a comparative study of trait variation in 20 congeneric pairs. Oikos 107(3):505–518
Lefcheck JS (2016) piecewiseSEM: Piecewise structural equation modeling in R for ecology, evolution, and systematics. Methods Ecol Evol 7(5):573–579
Legendre P (1993) Spatial autocorrelation: trouble or new paradigm? Ecology 74:1659–1673
Lehmair TA, Poschlod P, Reisch C (2022) The impact of environment on genetic and epigenetic variation in Trifolium pratensepopulations from two contrasting semi-naturalgrasslands. R Soc Open Sci 9:211406
Lele L, Ning D, Cuiping P, Xiao G, Weihua G (2018) Genetic and epigenetic variations associated with adaptation to heterogeneous habitat conditions in a deciduous shrub. Ecol Evol 8(5):2594–2606
Lira-Medeiros CF, Parisod C, Fernandes RA, Mata CS, Cardoso MA, Ferreira PCG (2010) Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 5:1–8
Luzuriaga AL, González JM, Escudero A (2015) Annual plant community assembly in edaphically heterogeneous environments. J Veg Sci 26(5):866–875
Medrano M, Herrera CM, Bazaga P (2014) Epigenetic variation predicts regional and local intraspecific functional diversity in a perennial herb. Mol Ecol 23:4926–4938
Medrano M, Alonso C, Bazaga P, López E, Herrera CM (2020) Comparative genetic and epigenetic diversity in pairs of sympatric, closely related plants with contrasting distribution ranges in south-eastern iberian mountains. AoB Plants 12(3):plaa013
Murphy MV (2022) semEff: automatic calculation of effects for Piecewise Structural Equation Models. R package version 0.6.1. https://CRAN.R-project.org/package=semEff
Nagamitsu T, Shuri K, Kikuchi S, Koike S, Naoe S, Masaki T (2019) Multiscale spatial genetic structure within and between populations of wild cherry trees in nuclear genotypes and chloroplast haplotypes. Ecol Evol 9(19):11266–11276
Nicotra AB, Segal DL, Hoyle GL, Schrey AW, Verhoeven KJF, Richards CL (2015) Adaptive plasticity and epigenetic variation in response to warming in an Alpine plant. Ecol Evol 5:634–647
Noshay JM, Springer NM (2021) Stories that can’t be told by SNPs; DNA methylation variation in plant populations. Curr Opin Plant Biol 61:101989
Osuna‐Mascaró C, Agneray AC, Galland LM, Leger EA, Parchman TL (2023) Fine‐scale spatial genetic structure in a locally abundant native bunchgrass (Achnatherum thurberianum) including distinct lineages revealed within seed transfer zones. Evol Appl 16(5):979–996
Pandey M, Rajora OP (2012) Higher fine-scale genetic structure in peripheral than in core populations of a long-lived and mixed-mating conifer - eastern white cedar (Thuja occidentalis L.). BMC Evol Biol 12:1–14
Paun O, Bateman RM, Fay MF, Hedren M, Civeyrel L, Chase MW (2010) Stable epigenetic effects impact adaptation in allopolyploid orchids (Dactylorhiza: Orchidaceae). Mol Biol Evol 27(11):2465–2473
Paun O, Schönswetter P (2012) Amplified fragment length polymorphism: an invaluable fingerprinting technique for genomic, transcriptomic, and epigenetic studies. Methods Mol Biol 862:75–87
Pescador DS, Cruz M, Chacón‐Labella J, Pavón‐García J, Escudero A (2020) Tales from the underground: soil heterogeneity and not only above‐ground plant interactions explain fine‐scale species patterns in a Mediterranean dwarf‐shrubland. J Veg Sci 31(3):497–508
Rabinowitz D (1981) Seven forms of rarity. In: Synge H (ed) Biological aspects of rare plant conservation. John Wiley and Sons, New York, pp 205–217
Rendina González AP, Preite V, Verhoeven KJ, Latzel V (2018) Transgenerational effects and epigenetic memory in the clonal plant Trifolium repens. Front Plant Sci 9:1677
Reyna-López GE, Simpson J, Ruiz-Herrera J (1997) Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 253(6):703–710
Richards EJ (2006) Inherited epigenetic variation –revisiting soft inheritance. Nat Rev Genet 7:395–401
Richards CL, Schrey AW, Pigliucci M (2012) Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol Lett 15(9):1016–1025
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62
Schouten OS, Houseman GR (2019) Effect of soil heterogeneity and endogenous processes on plant spatial structure. Ecology 100:e02837
Schrey AW, Alvarez M, Foust CM, Kilvitis HJ, Lee JD, Liebl AL et al. (2013) Ecological epigenetics: beyond MS-AFLP. Integr Comp Biol 53:340–350
Schulz B, Eckstein RL, Durka W (2014) Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol Ecol 23:3523–3537
Shen J, Wang Z, Su Y, Wang T (2021) Associations between population epigenetic differentiation and environmental factors in the exotic weed mile-a-minute (Mikania micrantha). Weed Sci 69(3):307–332
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279
Slatyer RA, Hirst M, Sexton JP (2013) Niche breadth predicts geographical range size: a general ecological pattern. Ecol Lett 16:1104–1114
Stone BW, Ward A, Farenwald M, Lutz AW, Wolfe AD (2019) Genetic diversity and population structure in Cary’s Beardtongue Penstemon caryi (Plantaginaceae), a rare plant endemic to the eastern Rocky Mountains of Wyoming and Montana. Conserv Genet 20:1149–1161
Thiebaut F, Hemerly AS, Ferreira PCG (2019) A role for epigenetic regulation in the adaptation and stress responses of non-model plants. Front Plant Sci 10:246
Thompson JD, Lavergne S, Affre L, Gaudeul M, Debussche M (2005) Ecological differentiation of Mediterranean endemic plants. Taxon 54(4):967–976
Turner BM (2009) Epigenetic responses to environmental change and their evolutionary implications. Philos Trans R Soc Lond B Biol Sci 364:3403–3418
Valverde J, Gómez JM, García C, Sharbel TF, Jiménez MN, Perfectti F (2016) Inter-annual maintenance of the fine-scale genetic structure in a biennial plant. Sci Rep. 24(6):37712
Van Antro M, Prelovsek S, Ivanovic S, Gawehns F, Wagemaker NC, Mysara M et al. (2023) DNA methylation in clonal duckweed (Lemna minor L.) lineages reflects current and historical environmental exposures. Mol Ecol 32(2):428–443
Vekemans X, Beauwens T, Lemaire M, Roldan-Ruiz I (2002) Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol Ecol 11:139–151
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935
Verhoeven KJF, Jansen JJ, van Dijk PJ, Biere A (2010) Stress-induced DNA methylation changes and their heritability in asexual dandelions. N. Phytol 185:1108–1118
Verhoeven KJF, Von Holdt BM, Sork VL (2016) Epigenetics in ecology and evolution: what we know and what we need to know. Mol Ecol 25:1631–1638
Volis S, Ormanbekova D, Shulgina I (2016) Fine-scale spatial genetic structure in predominantly selfing plants with limited seed dispersal: a rule or exception? Plant Divers 38(2):59–64
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M et al. (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21):4407–4414
Wilschut RA, Oplaat C, Snoek LB, Kirschner J, Verhoeven KJF (2016) Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Mol Ecol 25(8):1759–1768
Wright S (1943) Isolation by distance. Genetics 28(2):114–138
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Wu WQ, Yi MR, Wang XF, Ma L, Jiang L, Li XW et al. (2013) Genetic and epigenetic differentiation between natural Betula ermanii (Betulaceae) populations inhabiting contrasting habitats. Tree Genet Genomes 9:1321–1328
Xue W, Huang L, Yu FH, Bezemer TM (2022) Light condition experienced by parent plants influences the response of offspring to light via both parental effects and soil legacy effects. Funct Ecol 36(10):2434–2444
Yao N, Zhang Z, Yu L, Hazarika R, Yu C, Jang H et al. (2023) An evolutionary epigenetic clock in plants. Science 381(6665):1440–1445
Zhang Y-Y, Fischer M, Colot V, Bossdorf O (2012) Epigenetic variation creates potential for evolution of plant phenotypic plasticity. N. Phytol 197(1):314–322
Zilberman D, Gehring M, Tran R, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39:61–69
Acknowledgements
We are very grateful to Pilar Bazaga and Esmeralda López-Perea for laboratory assistance. Noelia Zara for her help in sampling Erodium individuals. We thank the Consejería de Medio Ambiente, Junta de Andalucía, for authorising this research. This work was supported by the Consejería de Innovación Ciencia y Empresa, Junta de Andalucía [P18-FR-4413] and the Ministerio de Ciencia e Innovación, Spanish Government [PID2019-104365GB-I00/AEI/10.13039/501100011033 and SUMHAL-LIFEWATCH-2019-09-CSIC-4, through European Regional Development Funds POPE 2014-2020].
Author information
Authors and Affiliations
Contributions
CA, CMH and MM designed the methodology and collected the data; CA, CMH, MM and JV elaborated the hypotheses; JV analysed the data and led the writing of the manuscript. All authors contributed to the revision of the manuscript and gave their final approval for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Marc Stift.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Valverde, J., Medrano, M., Herrera, C.M. et al. Comparative epigenetic and genetic spatial structure in Mediterranean mountain plants: a multispecies study. Heredity 132, 106–116 (2024). https://doi.org/10.1038/s41437-024-00668-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41437-024-00668-3
