
Abstract
Src Homology 3 Domain-containing Adaptor Protein 3 (SASH3) deficiency is an X-linked immune disorder. Here we identified a male case with a pathogenic SASH3 variant (c.1039C>T [p.Arg347Cys]) who presented with osteogenesis imperfecta, intellectual disability and recurrent infections. While immunological features in this case were characterized, further studies are needed to determine the association between the SASH3 variant and the skeletal or neurological manifestations.
Similar content being viewed by others

Src Homology 3 Domain-containing Adaptor Protein 3 (SASH3), also called SH3-domain-containing lymphocyte protein (SLY), is involved mainly in immune cell signaling and T cell activation. It functions as an adaptor protein that helps to regulate intracellular signaling pathways in the lymphocytes1. Delmonte et al.2 first reported that pathogenic variants of SASH3 are associated with X-linked immune dysregulation disorders, and the disease concept is acquired. The SASH3 gene was identified as the causative gene of immune dysregulation disorders in a study involving 1505 individuals from 1000 families with suspected or known inborn errors of immunity3. Here, we encountered a case of short stature due to osteogenesis imperfecta and intellectual disability. A pathogenic variant of SASH3 was identified in the Initiative on Rare and Undiagnosed Diseases (IRUD)4, led by the Japan Agency for Medical Research and Development. Thus, we report the case of a male patient with SASH3 deficiency and discuss the clinical manifestations.
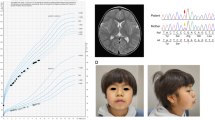
The patient was born via cesarean section at 39 weeks and 1 day of gestation. At birth, the patient’s height, weight and Apgar score were 46.0 cm (–1.5 standard deviation (s.d.)), 2556 g (–1.6 s.d.) and 9/10, respectively. The patient had no family history of hereditary disorders. The patient presented with a cleft lip and palate immediately after birth and hypertrophic pyloric stenosis. At the age of 2 years, the patient presented with a soft mass in the thenar region of the right thumb. Furthermore, the patient exhibited long bone dysplasia, reduced bone density, short stature, right renal agenesis and definite speech delay from early childhood (Fig. 1A–D). The patient’s developmental quotients (DQs) on the Kyoto Scale of Psychological Development at age 2 years and 8 months was 63 (total DQ) (postural-motor DQ, 54; cognitive-adaptive DQ, 70; and language-social DQ, 42). Between the ages of 5 and 7 years, the patient was hospitalized five times for infections such as bacterial pneumonia, bacterial cellulitis and rotavirus gastroenteritis. In each instance of hospitalization, the patient was admitted within 1 week and recovered with intravenous antibiotic therapy. Notably, despite the bacterial infections, the white blood cell counts at the time of admission remained low, ranging from 1300 to 5100/μl.

A Computed tomography at 1 year and 5 months of age. Bilateral hypoplasia of the diaphyseal ends of the femurs was observed. B X-ray of the hands at 2 years and 5 months of age. Underdevelopment of the finger bones in both hands was noted. C X-ray of the legs at 10 years of age. Shortening of the long bones and enlargement of the diaphyseal ends of the long bones are observed. D Growth curve. Marked growth retardation was observed in this patient. E Sanger sequencing. The patient has a hemizygous missense variant (c.1039C>T [p.Arg347Cys]) in the SASH3 gene. This pathogenic variant was inherited from the mother, who carries the same variant in the heterozygous state.
At 14 years of age, the patient was diagnosed with intellectual disability (total intelligence quotient 67 on the Wechsler Intelligence Scale for Children III test), long bone dysplasia with impaired endochondral ossification, persistent short stature and hearing loss. Although the range of motion in the hip, knee and ankle joints is currently limited, the patient is able to walk independently.
The patient was registered with IRUD3, and whole-exome sequencing identified a hemizygous missense variant (c.1039C>T [p.Arg347Cys]) in SASH3 (MIM *300441) (Fig. 1E and Supplementary Data 1). Since then, we investigated the patient’s immune function at 16 years of age (Fig. 2 and Supplementary Data 2). Laboratory tests revealed mildly decreased white blood cell counts. Immunoglobulin levels revealed IgG and IgA within the age-matched normal range, whereas IgM levels were decreased. Lymphocyte subset analysis showed a notable T cell reduction: CD3+ T cells accounted for 46.2%, CD4+ T cells for 15.2% and CD8+ T cells for 23.2%, indicating a substantial decrease in CD3+ and CD4+ T cells5, consistent with previous reports2,6,7 (Fig. 2). By contrast, CD19+ B cells comprised 24.2% and CD56+ natural killer (NK) cells 25.6%, with no decrease observed, differing from previously reported cases (Fig. 2). However, further analysis of B cells demonstrated a considerable reduction in IgM memory B cells (CD19+CD27+IgD+) at 1.6% and switched memory B cells (CD19+CD27+IgD−) at 2.4% (Fig. 2). Although the total B cell count and IgG levels did not decrease, these results indicated a clear impairment in B cell differentiation.

A Representative dot plots of T cell subpopulations, showing CD3, CD4 and CD8 expression profiles in peripheral blood lymphocytes B Representative dot plots of B and NK cell subpopulations, displaying CD3, CD19 and CD56 expression in peripheral blood lymphocytes. C Four-color flow cytometry analysis using IgD, CD27, CD45 and CD19 to evaluate B cell maturation and memory status. P1 and P2 denote the levels of all peripheral blood lymphocytes.
Thus, the patient was diagnosed with combined immunodeficiency owing to functional abnormalities in T and B cells. The immune function in this patient with SASH3 deficiency was generally consistent with the clinical features of SASH3 deficiency reported in previous studies2,6,7 (Supplementary Data 3). SASH3 deficiency is typically characterized by three main findings: cytopenia affecting one or more blood cell lineages (including white blood cells such as neutrophils and lymphocytes, red blood cells or platelets), hypogammaglobulinemia and reduced numbers of lymphocyte subsets (T cells, B cells or NK cells) (Supplementary Data 3).
This patient exhibited cytopenia and T cell reduction, consistent with prior findings. However, B cell counts remained within normal range. Although mild leukopenia was present, hypogammaglobulinemia was not observed, making immunodeficiency less clinically apparent. Moreover, this case had no history of recurrent or severe infections, including opportunistic infections, and the clinical course did not suggest overt immunodeficiency.
This case was conclusively diagnosed as SASH3 deficiency via IRUD; however, the disease was not suspected before undergoing IRUD. The patient’s primary clinical manifestations were osteogenesis imperfecta and intellectual disability.
According to Delmonte et al.2, patients with SASH3 deficiency demonstrate CD4+ T cell lymphopenia, reduced T cell proliferation, impaired cell cycle progression and elevated T cell apoptosis in response to mitogens. Moreover, five previously reported cases of SASH3 deficiency recapitulate several of the immunological defects described in two different Sly1-deficient mouse models (Sly1Δ/Δ and Sly1−/−)8,9 (Supplementary Data 3). Specifically, (1) the absence of the SLY1 (SASH3) protein influences CD4+ T cell development, T cell proliferation and cytokine production. (2) SLY1 functions as an anti-apoptotic factor necessary for thymocyte development. (3) Antibody responses to T-dependent and T-independent antigens are impaired. Consequently, B cell differentiation and maturation are impaired.
So far, only four types of pathogenic variant (three nonsense variants: c.505C>T [p.Gln169*], c.733C>T [p.Arg245*] and c.862C>T [p.Arg288*], and one missense variant: c.1039C>T [p.Arg347Cys]) have been reported globally in patients with SASH3 deficiency (Supplementary Data 3). None of these variants is registered in the gnomAD10 or the Tohoku Medical Megabank11 genome databases. In silico analyses—including CADD (score: 25.0; deleterious), SIFT (deleterious), PolyPhen-212 (probably damaging) and MutationTaster (deleterious)—consistently suggest that the c.1039C>T [p.Arg347Cys] variant is pathogenic. According to the guidelines of American College of Medical Genetics and Genomics and Association for Molecular Pathology, this variant is classified as likely pathogenic based on the criteria PS1, PM2 and PP313.
Furthermore, Delmonte et al.2 investigated sequence motifs surrounding Arg347, a highly conserved residue from Xenopus to humans, and identified a putative protein-kinase-A-binding motif using a three-dimensional model of SASH3. The wild-type SASH3 protein has been shown to be phosphorylated at Ser349, supporting the functional significance of this motif. Therefore, the p.Arg347Cys amino acid substitution may impair SASH3 function by disrupting the Ser349 phosphorylation site, a key regulatory element.
Typically, the disease manifests in childhood2,6; however, diagnosis is often delayed. SASH3 deficiency may represent a mild or moderate form of this condition, although SASH3 is the causative gene of combined immune deficiency. In addition, the unique clinical manifestations noted in this patient, metaphyseal dysplasia and intellectual impairment, have not been reported in all individuals with SASH3 deficiency. Whole-exome sequencing conducted using IRUD did not detect pathogenic variants in genes linked to osteogenesis imperfecta or intellectual disabilities. The genetic basis for these manifestations remains elusive.
In conclusion, we presented a case of SASH3 deficiency characterized by combined immune deficiency, metaphyseal dysplasia and intellectual impairment. Despite recurrent viral and bacterial infections during early life, the patient’s immune deficiency was evident from childhood but was not life-threatening.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3539.
Change history
26 September 2025
References 5 and 13 style have been updated.
References
Beer, S., Simins, A. B., Schuster, A. & Holzmann, B. Molecular cloning and characterization of a novel SH3 protein (SLY) preferentially expressed in lymphoid cells. Biochim. Biophys. Acta https://doi.org/10.1016/S0167-4781(01)00242-1 (2001).
Delmonte, O. M., Bergerson, J., Kawai, T., Kuehn, H. S., McDermott, D. H., Cortese, I. et al. SASH3 variants cause a novel form of X-linked combined immunodeficiency with immune dysregulation. Blood https://doi.org/10.1182/blood.2020008629 (2021).
Similuk, M. N., Yan, J., Ghosh, R., Oler, A. J., Franco, L. M., Setzer, M. R. et al. Clinical exome sequencing of 1000 families with complex immune phenotypes: Toward comprehensive genomic evaluations. J. Allergy Clin. Immunol. https://doi.org/10.1016/j.jaci.2022.06.009 (2022).
Takahashi, Y. & Mizusawa, H. Initiative on rare and undiagnosed disease in Japan. JMA J. https://doi.org/10.31662/jmaj.2021-0003 (2021).
Ding, Y., Zhou, L., Xia, Y., Wang, W., Wang, Y., Li, L. et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China. J. Allergy Clin. Immunol. https://doi.org/10.1016/j.jaci.2018.04.022 (2018).
Labrador-Horrillo, M., Franco-Jarava, C., Garcia-Prat, M., Parra-Martínez, A., Antolín, M., Salgado-Perandrés, S. et al. Case report: X-Linked SASH3 deficiency presenting as a common variable immunodeficiency. Front. Immunol. https://doi.org/10.3389/fimmu.2022.881206 (2022).
Novak, W., Berner, J., Svaton, M., Jimenez-Heredia, R., Segarra-Roca, A., Frohne, A. et al. Evans syndrome caused by a deleterious mutation affecting the adaptor protein SASH3. Br. J. Haematol. https://doi.org/10.1111/bjh.19061 (2023).
Beer, S., Scheikl, T., Reis, B., Hüser, N., Pfeffer, K., Holzmann et al. Impaired immune responses and prolonged allograft survival in Sly1 mutant mice. Mol. Cell Biol. https://doi.org/10.1128/MCB.25.21.9646-9660.2005 (2005).
Reis, B., Pfeffer, K. & Beer-Hammer, S. The orphan adapter protein SLY1 as a novel anti-apoptotic protein required for thymocyte development. BMC Immunol. https://doi.org/10.1186/1471-2172-10-38 (2009).
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q. et al. Genome Aggregation Database Consortium. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature https://doi.org/10.1038/s41586-020-2308-7 (2020).
Tadaka, S., Kawashima, J., Hishinuma, E., Saito, S., Okamura, Y., Otsuki, A. et al. jMorp: Japanese multi-omics reference panel update report 2023. Nucleic Acids Res. https://doi.org/10.1093/nar/gkad978 (2024).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging messense mutations. Nat. Methods https://doi.org/10.1038/nmeth0410-248 (2010).
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. https://doi.org/10.1038/gim.2015.30 (2015).
Acknowledgements
We thank all the staff members in the Department of Pediatrics at Kumamoto University Hospital for their help with clinical practice. We are also grateful to S. Shigeta for her help with the IRUD.
Funding
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants-in-Aid for scientific research under the grant numbers JP23K07316 (J.K.), JP23K27568 (T. Mizuguchi) and JP24K02230 (N.M.), the Japan Agency for Medical Research and Development (AMED) under the grant numbers JP24ek0109674, JP24ek0109760, JP24ek0109617, JP24ek0109648 and JP24ek0109677 (N.M.) and the Takeda Science Foundation (T. Mizuguchi and N.M.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
The Ethics Committee of the Faculty of Life Sciences at Kumamoto University approved this study (no. 1574 (Genome No. 382)). Written informed consent was obtained from the patient’s legal guardian for the publication of their medical case details and any accompanying images.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kido, J., Mizukami, T., Misumi, Y. et al. Osteogenesis imperfecta, intellectual disability and recurrent infections in a male with a pathogenic SASH3 variant. Hum Genome Var 12, 19 (2025). https://doi.org/10.1038/s41439-025-00323-1
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41439-025-00323-1
