
Abstract
The powerful insecticidal and multi-drug-resistance-reversing activities displayed by the stemofoline group of alkaloids render them promising lead structures for further development as commercial agents in agriculture and medicine. However, concise, enantioselective total syntheses of stemofoline alkaloids remain a formidable challenge due to their structural complexity. We disclose herein the enantioselective total syntheses of four stemofoline alkaloids, including (+)-stemofoline, (+)-isostemofoline, (+)-stemoburkilline, and (+)-(11S,12R)-dihydrostemofoline, in just 19 steps. Our strategy relies on a biogenetic hypothesis, which postulates that stemoburkilline and dihydrostemofolines are biogenetic precursors of stemofoline and isostemofoline. Other highlights of our approach are the use of Horner–Wadsworth–Emmons reaction to connect the two segments of the molecule, an improved protocol allowing gram-scale access to the tetracyclic cage-type core, and a Cu-catalyzed direct and versatile nucleophilic alkylation reaction on an anti-Bredt iminium ion. The synthetic techniques that we developed could also be extended to the preparation of other Stemona alkaloids.
Similar content being viewed by others
Introduction
Nature continues to serve as an invaluable source for the development of pharmaceutical drugs1, agrochemical agents2, and biomedical probes3. In this context, efficient total synthesis of these bioactive natural products is of crucial importance4. With significant advances in synthetic methodologies and strategies, many artful instances of total synthesis have been documented in the last few decades4. However, most of these reported studies involved only one target molecule5. Considering the huge number of biologically interesting natural products, there is clearly unmet need in developing novel synthetic strategies that can branch out toward multiple target compounds. In recent years, multi-target-oriented synthetic approaches, such as unified strategy6,7,8,9, collective synthesis10,11, and diversity-oriented synthesis12, have emerged as a frontier. It should be noted that many of these methods are biomimetic or bioinspired in that they rely on known biogenetic routes or hypotheses. But for those natural products whose biogenetic pathways are unknown or have not been investigated in detail, it is necessary to develop biogenetic hypotheses13,14.
The Stemona alkaloids are a class of structurally diverse natural products that act as the principle active constituents in Stemona plants (Stemonaceae)15,16,17,18,19,20. Known as “Bai Bu” in traditional Chinese medicine, Stemona plants have been used in East Asia for thousands of years as insecticides and anti-cough agents15,16,19,20. To date, more than 215 Stemona alkaloids have been isolated15, classified by Pilli19,20 into eight structural groups (see Supplementary Fig. 1). The attractive bioactivities15,16 and unique polycyclic structures of these alkaloids have led to intense phytochemical, synthetic, and biomedical investigations, and many innovative total syntheses have been achieved16,18,19,20,21,22,23,24.
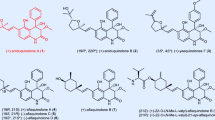
The stemofoline group of alkaloids (Fig. 1) are a highly versatile set of compounds that represent promising lead structures for agricultural and medicinal applications15,16. Stemofoline (1), the parent member of the stemofoline group, was isolated first from Stemona japonica by Irie and coworkers in 197025, and then from several other Stemona species. Its structure, including the absolute configuration, was determined by single-crystal X-ray crystallographic analysis25. To date, more than twenty members of the stemofoline group have been isolated and structurally determined, including isostemofoline (2)26, didehydrostemofoline (3)27,28, isodidehydrostemofoline (4)27, methoxystemofoline (5, suggested structure)29, methylstemofoline (6)30, stemoburkilline (7)31,32, and (11S,12R)-dihydrostemofoline (8)31 (Fig. 1). The high insecticidal activity33 of stemofoline (1) was first reported in 1978, according to which the compound acts as a potent agonist of the insect nicotinic acetylcholine receptors (nAChR) (EC50 = 1.7 nM) and is associated with acetylcholinesterase (AChE) inhibition15. Ye and coworkers have established that the cage-type moiety of stemofoline is pivotal to its insecticidal activity26. Notably, by using stemofoline (1) as a lead, synthetic cyanotropanes have been developed as a class of commercial insecticides2. Moreover, stemofoline (1) has been found to alleviate inflammation34 and reverse multidrug resistance of certain types of cancer15,35,36,37. Indeed, it has been shown that stemofoline (1) increases the sensitivity of patients toward anticancer drugs such as vinblastine, paclitaxel, and doxorubicin15,35. Once again, this desirable properties can be attributed to the core cage structure with nonpolar side chains35.

a Z-Series: (+)-stemofoline (1); (+)-didehydrostemofoline (3); (+)-methylstemofoline (6). b E-Series: (+)-isostemofoline (2); (+)-isodidehydrostemofoline (4); (+)-methoxystemofoline (5) (revised structure). c 11,12-Dihydro series: (+)-stemoburkilline (7); (+)-(11S, 12R)-dihydrostemofoline (8).
Much efforts have been devoted to the synthesis of stemofoline alkaloids. Starting from stemofoline38 and (11Z)-1′,2′-didehydrostemofoline39,40, several semisynthetic stemofoline alkaloids and analogues have been prepared and screened by Ye et al. and Pyne et al., respectively. Although tremendous efforts have been devoted to the total synthesis of these alkaloids41,42,43,44,45,46,47, successful examples remain rare, including racemic total syntheses of isostemofoline (2) by Kende48 and didehydrostemofoline (asparagamine A, 3) and isodidehydrostemofoline (4) by Overman49, and enantioselective formal total syntheses of didehydrostemofoline and isodidehydrostemofoline by Martin50. Notably, although stemofoline (1) displays remarkable bioactivities, and has proven to be a promising lead for the development of insecticide and anticancer agents, its total synthesis has not yet been achieved.
As a part of our efforts to achieve efficient total synthesis of structurally complex, enantiopure alkaloids51, we have been focused in the last decade on developing versatile methodologies for the direct transformation of amides52,53. We have recently reported a method for the construction of tropinone ring systems54 and applied it to the total synthesis of (+)-methoxystemofoline (5)55. We disclose herein biogenetic hypothesis-based syntheses of (+)-stemofoline (1), (+)-isostemofoline (2), (+)-stemoburkilline (7), (11S,12R)-dihydrostemofoline (8), and diastereomer 9.
Results
Biogenetic hypothesis
Structurally, stemofoline alkaloids consisted of a tetronate moiety connected through an ene diether (C11–C12) to a cage-like aza-pentacyclic ring system. Both the enantioselective construction of the cage-like aza-pentacyclic ring system56 and the stereoselective formation of the ene diether moiety are challenging48,49,55. Indeed, our previous total synthesis was lengthy and contained several steps of reaction with low chemo-, regio-, and/or diastereoselectivity55. In addition, the stemofoline alkaloids display great variation in the oxidation state and connectivity at C11, the stereochemistry of the ene diether connector, and substitution at the bridgehead carbon C3. Taking these factors into consideration, we wanted to develop a concise, versatile synthetic strategy. Although a bioinspired approach is highly desirable57, and indeed there are already several hypotheses on the origin of the central building blocks in Stemona alkaloids15,16,17,28, the basic biogenetic steps in the formation of Stemona alkaloids remain unknown15. In the search for a biogenetic relationship between the stemofoline group of alkaloids, we have noted that isostemofoline (2)26, stemoburkilline (7), and (11S,12R)-dihydrostemofoline (8)31,32 were congeners of stemofoline (1), and Pyne et al. also observed that (11S,12S)-dihydrostemofoline (9) (Fig. 2a) could be converted into a mixture of stemoburkilline (7) and (11S,12R)-dihydrostemofoline (8) upon treatment with DBU32. Moreover, Ye and coworkers26 have reported the co-existence of stemofoline (1) and isostemofoline (2), the latter as a minor congener, in S. japonica, whereas Jiwajinda et al. has detected equal amounts of 11Z-didehydrostemofoline (3) and 11E-didehydrostemofoline (4) in S. collinsae27. Combining these findings with ours, we hypothesized that the known compounds stemoburkilline (7) and (11S,12R)-dihydrostemofoline (8), and possibly the unknown compound (11S,12S)-dihydrostemofoline (9), are plausible biogenetic precursors of stemofoline (1) and isostemofoline (2) (Fig. 2a).

a Our biogenetic hypothesis. b Retrosynthetic analysis based on our biogenetic hypothesis.
Retrosynthetic analysis
Guided by our biogenetic hypothesis (Fig. 2a), we conducted a retrosynthetic analysis of stemofoline (1) as depicted in Fig. 2b. The (Z)-5-(dihydrofuran-2(3H)-ylidene)-4-methoxy-3-methylfuran-2(5H)-one [(Z)-ene diether moiety could be constructed through C–H oxygenation of stemoburkilline (7). The functionalized C11–C12 alkene moiety in 7 could be assembled through Horner–Wadsworth–Emmons-type reaction of diethyl (3-methoxy-4-methyl-5-oxo-2,5-dihydrofuran-2-yl)phosphonate (10) with the pentacyclic cage-type hemiacetal 11. Other key issues to be addressed were: (1) the O-debenzylation of 13 triggering double cyclization, (2) the direct, chemo-, regio-, and diastereoselective methylation at C10 to build a chiral canter, and (3) direct installation at C3 a butyl group or other alkyl groups needed for the synthesis of other stemofoline alkaloids and analogues.
Syntheses of stemoburkilline (7), its congeners 8, and 9
To implement our strategy, we first developed an improved five-step protocol to furnish the tropinone building block 16 on a multi-gram scale (Fig. 3). To this end, commercially available α-benzyloxy-γ-lactone (S)-18 was treated with O-silylated β-aminoethanol 19 in methanol at room temperature for 2–3 days, which afforded γ-hydroxyamide 20 in 90% yield. Swern oxidation of 20 generated a diastereomeric mixture of hemiaminals, which was then subjected without purification to NaH-mediated Horner–Wadsworth–Emmons reaction with dimethyl (2-oxopropyl)phosphonate (21) followed by cyclization to produce the desired cis-ketolactam 17 in 60% yield (over two steps), along with the trans-diastereomer in 21% yield. Conversion of 17 to bromo-tropinone building block 16 was achieved in 78% yield via a two-step keto-lactam cyclization-bromination cascade (TMSOTf, Et3N, CH2Cl2, 0 °C; Tf2O, DTBMP, ZnBr2, CH2Cl2, −78 °C to rt) that we previously developed54.

Reagents and conditions: a 19, MeOH, rt, 3 d, 90%; b (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C; c NaH, 21, THF, 0 °C to reflux, 60% (for 2 steps); d TMSOTf, Et3N, CH2Cl2, 0 °C, e Tf2O, DTBMP, ZnBr2, CH2Cl2, −78 °C to rt, 78% (for 2 steps from cis-17).
We next focused on the development of a direct and versatile method for the installation of an n-butyl group on the bridgehead carbon of 16. In our previous strategy55, three steps were required to introduce the 4-methoxybutyl group. A survey of literatures indicated that the bridgehead nitrogen in similar but simpler ring systems, such as 1-chloro-9-methyl-9-azabicyclo[3.3.l]nonane, enhances the rate of solvolysis dramatically58. However, attempted direct alkylation of a similar α-chloroamine with organolithium or Grignard reagent produced disappointing results59,60. Encouragingly, Kibayashi and coworkers have reported that bridged tricyclic N,O-acetals can be alkylated using Grignard reagents in the presence of Et2AlCl through SN1 reaction on bridgehead anti-Bredt iminium ion intermediates60,61. In light of these precedents, we opted to explore whether metal-catalyzed direct alkylation of 3-bromotropinone building block 16 with Grignard reagents was feasible. After extensive screening (cf. Table 1), we achieved gram-scale coupling of 16 with n-butyl magnesium bromide in THF to afford the desired butylated product 15 in 81% yield, with CuCl2 as a catalyst, TMEDA (N,N,N′,N′-tetramethylethylenediamine) as ligand and LiOMe as additive (Table 1, entry 12). According to the precedent set by Kibayashi, it is possible that the alkylation reaction involves an SN1 mechanism via the intermediacy of anti-Bredt bridgehead iminium ion A. Significantly, the method can be extended to other Grignard reagents, thereby allowing the introduction of other desired simple or functionalized alkyl groups at C3. In this manner, alkylated products 15a–15d (Fig. 4) could be prepared directly from 16 in good yields (75–85%).

Reagents and conditions: a CuCl2, TMEDA, LiOMe, RMgBr, THF, rt, 30 min.
To construct compound 14, which contains a tricyclic core with an ester-enone moiety (Fig. 5), tropinone 15 was successively treated with LDA and ethyl glyoxalate (22) to yield aldol adducts 23-1 and 23-2 in 71% yield with a diastereomeric ratio of 1.1:1, along with regioisomer 23a in 15% yield. The lack of diastereoselectivity was of no consequence as dehydration of 23-1 and 23-2 afforded the desired enone 24 as a single isomer in 72% yield. NOESY studies indicated that 24 adopted an E configuration (see Supplementary Fig. 3). From these experimental results, we deduced that the structures of two diastereomers 23-1 and 23-2 have the stereochemistries shown in Fig. 5.

Reagents and conditions: a LDA, ethyl glyoxalate (22), −78 °C, 1.5 h, 71% for 23 and 15% for 23a; (b) CDI, i-Pr2NEt, DMAP, CH2Cl2, 0 °C to rt, 4 h, 72%; c p-TsOH, acetone, 50 °C, 30 min, 96%; d CBr4, PPh3, CH2Cl2, 0 °C, 30 min, 95%; e EtONa, THF, 0 °C, 20 min, 95%; f MeLi, DMPU, Et2O, −40 °C, 20 min, 86%; g Pd/C, H2, EtOH, rt, 2 d, 87%; h p-TsOH, toluene, 85 °C, 2 h, 93% (83% for two steps from 13 without further purification).
Desilylation of 24 with p-TsOH, followed by bromination of the resultant alcohol 25, furnished tropinone halide 26 in an overall yield of 91.2% (for 2 steps; Fig. 5). Exposure of 26 to EtONa in THF resulted in the formation of tricyclic enone 14 in excellent yield (95%). Another key step for our strategy resided in the regio- and diastereoselective methylation at C10. To our delight, the intended conjugate addition was achieved by treating 14 with MeLi-DMPU in diethyl ether62, leading to 13 as a single regio- and diastereomer in 86% yield. Pd/C catalyzed O-debenzylation of 13 in ethanol was accompanied by tandem lactolization, which furnished the cage-like core structure 27 directly as a single diastereomer in 87% yield. Exposing 27 to p-TsOH in toluene produced the key pentacyclic lactone 12 in 93% yield. Importantly, neither the hydrogenation nor lactonization step required purification to yield 12 of sufficient purity for further reactions. The stereochemistry of 12 was determined by NOESY experiments (see Supplementary Fig. 6), which confirmed that the addition of MeLi to 14 was instrumental in creating the two vicinal chiral centers at C9 and C10.
After 12 was synthesized, it was reduced with DIBAL-H to afford hemiacetal 11 as an inseparable diastereomeric mixture in 78% yield (Fig. 6). Once again, the lack of diastereoselectivity did not matter because treatment of 11 with potassium t-butoxide and diethyl (3-methoxy-4-methyl-5-oxo-2,5-dihydrofuran-2-yl)phosphonate (10), prepared in one-pot from γ-bromotetronate63, resulted in the formation of stemoburkilline (7), (11S,12R)-dihydrostemofoline (8), and the diastereomeric (11S,12S)-dihydrostemofoline (9), in 27%, 24%, and 33% yield, respectively (combined yield: 84%). 1H and 13C nuclear magnetic resonance (NMR) and polarimetric data of 7–9 were largely consistent with previously reported values (see Supplementary Tables 5–10)31,32.

Reagents and conditions: a DIBAL-H, CH2Cl2, −78 °C, 30 min, 78%; b t-BuOK, 10, THF, 0 °C, 30 min, 84% (27% for 7; 24% for 8; 33% for 9); c LHDMS, THF, −78 °C, 1 h, 98%.
Given that the treatment of (11S,12S)-dihydrostemofoline (9) with DBU yielded a tautomeric mixture of 9, (11S,12R)-dihydrostemofoline (8), and stemoburkilline (7) in a ratio of 39:37:2432, we speculated that stemoburkilline (7) could be a kinetic tautomer and that, under kinetic conditions, it would be possible to selectively convert both 8 and 9 into 7. Indeed, treatment of a mixture of 8 and 9 with LHMDS resulted in the formation of stemoburkilline (7) in 98% yield (Fig. 6).
Total syntheses of stemofoline (1) and isostemofoline (2)
To complete the total synthesis of stemofoline, a one-step Wacker-type reaction of stemoburkilline (7) was explored. Unfortunately, none of the reaction conditions (Na2PdCl4, TBHP64,65; PdCl2, CuCl, O266,67; Pd(TFA)2, IMes68) (see Supplementary Table 1) that we tested yielded the desired final product stemofoline (1). In each instance, a complex mixture of by-products, decomposition of the starting material, or lack of any major reaction whatsoever, was observed.
Next, we envisioned a two-step dehydrogenation of 8 and 9 consisting of introduction of a leaving group at C12 followed elimination. However, oxygenation with IBX/DMSO or DBU/O2, halogenation with NIS/NBS, or methylsulfidation with MeSSMe, are all found to be disappointingly inadequate (see Supplementary Table 2). Attempted conversion of 8 and 9 to 1 (see Supplementary Table 3) through cyclization to form 29 followed by oxidation was also unsuccesful because treating a mixture of 8 and 9 with LHMDS and TMSCl resulted in no cyclization and exclusive formation of an O-TMS derivative 30 in 93% yield (Fig. 7a). We then turned our attention to the electrophilic cyclization of 7. However, 7 apparently showed no reaction with NIS and was thus mostly recovered (>95%), regardless of whether CH2Cl2, THF or MeCN was used as solvent, and whether the cyclization was performed at room temperature or under reflux. Running the reaction in the presence of a base (NEt3, DBU, NaOEt, or NaH) led to the formation of a mixture of 7–9 (see Supplementary Table 4). Interestingly, exposing 7 to I2/NaHCO3 in THF at room temperature produced the unexpected iodoetherification product 31 as a single diastereomer in 86% yield (Fig. 7b). A plausible mechanism for the diastereoselective formation of 31 is depicted in Fig. 7b69.

a Reaction of 8 and 9 with TMSCl. b Reaction of 7 with I2.
Finally, a dibromination tactic70 was explored (Fig. 8). To avoid possible interference of the hydroxyl group, 7–9 were converted to TMS-protected 30 in 95% yield. Compound 30 reacted with Br2 regioselectively at C11–C12 alkene to afford 32 as a diastereomeric mixture. Unexpectedly, subsequent treatment of this mixture with DBU in CH2Cl2 caused it to revert back to 30. Finally, we were pleased to find that treating 32 with TBAF in THF yielded stemofoline (1) directly in 50% yield, isostemofoline (2) in 10% yield, and stemoburkilline (7) in 12% yield. 1H and 13C NMR and polarimetric data of stemofoline (1) {[α]D25 +276 (c 1.0, MeOH); lit.25 [α]D25+273 (MeOH); lit.28 [α]D25 +270 (c 0.8, MeOH)}, and isostemofoline (2) {[α]D25 +102.1 (c 0.5, CHCl3)} were fully consistent with those reported in earlier studies (see Supplementary Tables 11–13), which confirmed not only the structures including relative and absolute stereochemistries of our synthetic stemofoline (1) and isostemofoline (2), but also those of stemoburkilline (7), (11S,12R)-dihydrostemofoline (8), and (11S,12S)-dihydrostemofoline (9).

Reagents and conditions: a LHMDS, TMSCl, THF, −78 °C, 1 h, 95%; b Br2, CH2Cl2, rt, 30 min; c TBAF, THF, 0 °C, 1 h, 50% for 1; 10% for 2; 12% for 7 (for 2 steps from 30).
Discussion
In summary, we have achieved the total synthesis of stemofoline (1) after it was first isolated and structurally characterized 50 years ago. Our method features a relatively concise (19 steps) route that leads to multiple biomedically relevant compounds in an enantioselective manner. Particularly, we were able to simultaneously obtain stemofoline (1) and isostemofoline (2), two other members belonging to the same group stemoburkilline (7), (11S,12R)-dihydrostemofoline (8), as well as the (11S,12S)-diastereomer 9 at step 16 of the total synthesis. We argue that our success could be, at least in part, contributed to, and confirm, our biogenetic hypothesis. Furthermore, the synthetic efficiency that we accomplished was also ensured by: (1) an improved three-step protocol that allowed the gram-scale synthesis of keto-lactam cis-17, (2) a Cu-catalyzed direct nucleophilic alkylation reaction of 3-bromotropinone 16, (3) a concise eight-step route for the efficient and selective construction of the pentacyclic core 12, (4) the successful use of Horner–Wadsworth–Emmons reaction to assemble the two segments of the target molecules, and (5) the convergent transformation of 7–9 via 30 to biomimetically forge stemofoline (1) and isostemofoline (2). The realization of this bioinspired approach allowed us to predict that (11S,12S)-dihydrostemofoline (9) might be a natural product yet to be discovered. The direct, flexible, and versatile introduction of different side chains at C3 of 16 to form 15a–15d opened an avenue for the synthesis of other stemofoline alkaloids such as methylstemofoline (6) and its analogues. Moreover, this strategy may also find application in the synthesis of other Stemona alkaloids.
Methods
General
All reactions were performed anhydrously under nitrogen atmosphere. All reagents were purchased from commercial suppliers without further purification. Solvent purification was conducted according to Purification of Laboratory Chemicals (Peerrin, D.D.; Armarego, W.L. and Perrins, D.R., Pergamon Press: Oxford, 1980). Yields were calculated based on the weights of chromatographically isolated products. Reactions were monitored by thin-layer chromatography (TLC) on plates (GF254) supplied by Yantai Chemicals (China). The TLC spots were visualized under ultraviolet light or by staining with an ethanolic solution of phosphomolybdic acid and cerium sulfate or iodine vapor. Flash column chromatography was performed using silica gel (200–300 mesh) from Qingdao Haiyang Chemicals. NMR spectra were recorded on Bruker AV III 400, Bruker AV III 500, Bruker AV III 850 instruments, and calibrated with tetramethylsilane (TMS) (δH = 0.00 ppm) and CDCl3 (δC = 77.00 ppm) as internal references. Multiplicities were designated as follows: s = singlet, d = doublet, t = triplet, q = quartet, br = broad, dd = double doublet, td = triple doublet, dt = double triplet, dq = double quartet, m = multiplet. Infrared spectra were measured on a Nicolet FT-380 spectrometer using film KBr pellet techniques. High-resolution mass spectra analyses were performed on a Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonics) with a 7-T magnet (Magnex) and an electrospray ionization source (Apollo II, Bruker Daltonics) under positive-ion mode. Optical rotations were measured on an Anton Paar MCP-500 polarimeter.
Experimental data
For detailed experimental procedures, see Supplementary Methods. NMR spectra were presented as Supplementary Figs. 11–66. For NMR comparison between the reference compounds and compounds that we synthetized in this study, including stemofoline (1), isostemofoline (2), stemoburkilline (7) and (11S,12R)-dihydrostemofoline (8), see Supplementary Tables 5–13. For ORTEP diagrams for compounds 30, see Supplementary Fig. 13.
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 1962403 for compound 30. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
References
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803 (2020).
Loiseleur, O. Natural products in the discovery of agrochemicals. Chimia 71, 810–822 (2017).
Hong, B., Dong, T. & Lei, X. Recent advances in target identification by natural product based chemical probes. Sci. China Chem. 59, 1088–1092 (2016).
Efficiency in Natural Product Total Synthesis (eds. Huang, P.-Q., Yao, Z.‐.J. & Hsung, R.-P.) (Wiley-VCH, Weinheim, 2018).
Classics in Total Synthesis III: Further Targets, Strategies, Methods (eds. Nicolaou, K.C. & Chen, J.S.) (Wiley-VCH, Weinheim, 2011).
Bisai, A., West, S. P. & Sarpong, R. Unified strategy for the synthesis of the “miscellaneous” lycopodium alkaloids: total synthesis of (±)-lyconadin A. J. Am. Chem. Soc. 130, 7222–7223 (2008).
Lee, A. S., Liau, B. B. & Shair, M. D. A unified strategy for the synthesis of 7-membered-ring-containing lycopodium alkaloids. J. Am. Chem. Soc. 136, 13442–13452 (2014).
Wagnières, O., Xu, Z., Wang, Q. & Zhu, J. Unified strategy to monoterpene indole alkaloids: total syntheses of (±)-goniomitine, (±)-1,2-dehydroaspidospermidine, (±)-aspidospermidine, (±)-vincadifformine, and (±)-kopsihainanine A. J. Am. Chem. Soc. 136, 15102–15108 (2014).
Xu, Z., Wang, Q. & Zhu, J.-P. Total syntheses of (−)-mersicarpine, (−)-scholarisine G, (+)-melodinine E, (−)-leuconoxine, (−)-leuconolam, (−)-leuconodine A, (+)-leuconodine F, and (−)-leuconodine C: self-induced diastereomeric anisochronism (SIDA) phenomenon for scholarisine G and leuconodines A and C. J. Am. Chem. Soc. 137, 6712–6724 (2015).
Jones, S. B., Simmons, B., Mastracchio, A. & MacMillan, D. W. C. Collective synthesis of natural products by means of organocascade catalysis. Nature 475, 183–188 (2011).
Zhao, G.-Y. et al. Bioinspired collective syntheses of iboga-type indole alkaloids. Org. Lett. 18, 2447–2450 (2016).
Zhang, J. et al. Diversity-oriented synthesis of lycopodium alkaloids inspired by the hidden functional group pairing pattern. Nat. Commun. 5, 4614–4623 (2014).
Hong, A. Y. & Vanderwal, C. D. A synthesis of alsmaphorazine B demonstrates the chemical feasibility of a new biogenetic hypothesis. J. Am. Chem. Soc. 137, 7306–7309 (2015).
Geng, H. & Huang, P.-Q. Rapid generation of molecular complexity by chemical synthesis: highly efficient total synthesis of hexacyclic alkaloid (−)-chaetominine and its biogenetic implications. Chem. Rec. 19, 523–533 (2019).
Greger, H. Structural classification and biological activities of Stemona alkaloids. Phytochem. Rev. 18, 463–493 (2019).
Pyne, S. G. et al. Phytochemical, synthetic and biological studies on Stemona and Stichoneuron plants and alkaloids: a personal perspective. Nat. Prod. Commun. 12, 1365–1369 (2017).
Wang, F.-P. & Chen, Q.-H. Stemona alkaloids: biosynthesis, classification, and biogenetic relationships. Nat. Prod. Commun. 9, 1809–1822 (2014).
Liu, X.-Y. & Wang, F.-P. Recent advances in the synthesis of Stemona alkaloids. Nat. Prod. Commun. 10, 1093–1102 (2015).
Pilli, R. A., Rosso, G. B. & De Oliveira, M. C. F. The chemistry of Stemona alkaloids: an update. Nat. Prod. Rep. 27, 1908–1937 (2010).
Pilli, R. A., Rosso, G. B. & De Oliveira, M. C. F. In The Alkaloids (eds Cordell, G. A.) Vol. 62, pp. 77–173 (Elsevier, San Diego, 2005).
Ma, K.-Q., Yin, X.-L. & Dai, M.-J. Total syntheses of bisdehydroneostemoninine and bisdehydrostemoninine by catalytic carbonylative spirolactonization. Angew. Chem. Int. Ed. 57, 15209–15212 (2018).
Yoritate, M. et al. Unified total synthesis of stemoamide-type alkaloids by chemoselective assembly of five-membered building blocks. J. Am. Chem. Soc. 139, 18386–18391 (2017).
Chen, J. B., Chen, J. C., Xie, Y. & Zhang, H. B. Enantioselective total synthesis of (−)-stenine. Angew. Chem. Int. Ed. 51, 1024–1027 (2012).
Chen, Z.-H., Tu, Y.-Q., Zhang, S.-Y. & Zhang, F.-M. Development of the intramolecular Prins cyclization/Schmidt reaction for the construction of the azaspiro[4,4]nonane: application to the formal synthesis of (±)-stemonamine. Org. Lett. 13, 724–727 (2011).
Irie, H. et al. The crystal atructure of a new alkaloid, stemofoline, from Stemona japonica. J. Chem. Soc. 1066–1066 (1970).
Tang, C.-P. et al. Alkaloids from stems and leaves of Stemona japonica and their insecticidal activities. J. Nat. Prod. 71, 112–116 (2008).
Jiwajinda, S. et al. Occurrence of insecticidal 16,17-didehydro-16(E)-stemofoline in Stemona collinsae. Phytochemistry 56, 693–695 (2001).
Seger, C. et al. Two pyrrolo[1,2–a]azepine type alkaloids from Stemona collinsae CRAIB: structure elucidations, relationship to asparagamine a, and a new biogenetic concept of their formation. Chem. Biodivers. 1, 265–279 (2004).
Lin, H. W. & Xu, R. S. Chemical studies on Stemona alkaloids II. studies on the minor alkaloids of Stemona parviflora Wright C.H. Acta Chim. Sin. 49, 1034–1037 (1991).
Sastraruji, T. et al. Phytochemical studies on Stemona plants: isolation of stemofoline alkaloids. J. Nat. Prod. 68, 1763–1767 (2005).
Mungkornasawakul, P. et al. Phytochemical studies on Stemona burkillii prain: two new dihydrostemofoline alkaloids. J. Nat. Prod. 67, 1740–1743 (2004).
Sastraruji, K. et al. Structural revision of stemoburkilline from an E-alkene to a Z-alkene. J. Nat. Prod. 72, 316–318 (2009).
Brem, B. et al. Feeding deterrence and contact toxicity of Stemona alkaloids: a source of potent natural insecticides. J. Agric. Food Chem. 50, 6383–6388 (2002).
Hosoya, T. et al. Inhibitors of nitric oxide production from Stemona javanica. Planta Med. 77, 256–258 (2011).
Chanmahasathien, W., Ampasavate, C., Greger, H. & Limtrakul, P. Stemona alkaloids, from traditional Thai medicine, increase chemosensitivity via P-glycoprotein-mediated multidrug resistance. Phytomedicine 18, 199–204 (2011).
Umsumarng, S. et al. Reversal of human multi-drug resistance leukaemic cells by stemofoline derivatives via inhibition of P-glycoprotein function. Basic Clin. Pharmacol. Toxicol. 116, 390–397 (2015).
Umsumarng, S. et al. A pharmacological strategy using stemofoline for more efficacious chemotherapeutic treatments against human multidrug resistant leukemic cells. Asian Pac. J. Cancer Prev. 19, 3533–3543 (2018).
Ye, Y. & Velten, R. F. Semi-syntheses of new stemofoline derivatives. Tetrahedron Lett. 44, 7171–7173 (2003).
Baird, M. C. et al. Semisynthesis and biological activity of stemofoline alkaloids. J. Nat. Prod. 72, 679–684 (2009).
Sastraruji, K. et al. Semisynthesis and acetylcholinesterase inhibitory activity of stemofoline alkaloids and analogues. J. Nat. Prod. 73, 935–941 (2010).
Beddoes, R. L., Davies, M. P. H. & Thomas, E. J. Synthesis of the tricyclic nucleus of the alkaloid stemofoline: X-ray crystal structure of (4RS,5RS,7SR,10RS)-10-butyl-5-hydroxy-l-azatricyclo [5.3.0.04,10]decan-2-one. J. Chem. Soc. 538–540 (1992).
Epperson, M. T. & Gin, D. Y. Enantiospecific synthesis of the bridged pyrrolizidine core of asparagamine A: dipolar cycloadditions of azomethine ylide derived from the sulfonylation of vinylogous amides. Angew. Chem. Int. Ed. 41, 1778–1780 (2002).
Baylis, A. M., Davies, M. P. H. & Thomas, E. J. Synthetic approaches to the polycyclic alkaloid stemofoline. Org. Biomol. Chem. 5, 3139–3155 (2007).
Baylis, A. M. & Thomas, E. J. Aspects of the chemistry of 8-azabicyclo[3.2.1]octanes. Tetrahedron 63, 11666–11671 (2007).
Burns, T., Helliwell, M. & Thomas, E. J. An asymmetric synthesis of the pentacyclic core of stemofoline. Tetrahedron Lett. 54, 2120–2123 (2013).
Anderson, B. K. & Livinghouse, T. Divergent stereocontrolled synthesis of the enantiopure tetracyclic cores of asparagamine A and stemofoline via an intramolecular 2‑propylidine-1,3-(bis)silane bicyclization. J. Org. Chem. 80, 9847–9855 (2015).
Ideue, E., Shimokawa, J. & Fukuyama, T. Synthesis of the common core structure of the stemofoline alkaloids. Org. Lett. 17, 4964–4967 (2015).
Kende, A. S., Smalley, T. & Huang, H. Total synthesis of (±)-isostemofoline. J. Am. Chem. Soc. 121, 7431–7432 (1999).
Brüggemann, M., McDonald, A. I., Overman, L. E. & Rosen, M. D. Total synthesis of (±)-didehydrostemofoline (asparagamine A) and (±)-isodidehydrostemofoline. J. Am. Chem. Soc. 125, 15284–15285 (2003).
Fang, C., Shanahan, C. S., Paull, D. H. & Martin, S. F. Enantioselective formal total syntheses of didehydrostemofoline and isodidehydrostemofoline through a catalytic dipolar cycloaddition cascade. Angew. Chem. Int. Ed. 51, 10596–10599 (2012).
Guo, L.-D. et al. Organocatalytic, asymmetric total synthesis of (−)-haliclonin A. Angew. Chem. Int. Ed. 55, 4064–4068 (2016).
Xiao, K.-J., Luo, J.-M., Ye, K.-Y., Wang, Y. & Huang, P.-Q. Direct, One-pot sequential reductive alkylation of lactams/amides with grignard and organolithium reagents through lactam/amide activation. Angew. Chem. Int. Ed. 49, 3037–3040 (2010).
Fan, T. et al. Versatile one-pot synthesis of polysubstituted cyclopent-2-enimines from α,β-unsaturated amides: imino-nazarov reaction. Angew. Chem. Int. Ed. 57, 10352–10356 (2018).
Huang, S.-Y. et al. Versatile construction of functionalized tropane ring systems based on lactam activation: enantioselective synthesis of (+)-pervilleine B. Chem. Commun. 49, 7088–7090 (2013).
Huang, P.-Q. et al. Enantioselective total synthesis of (+)-methoxystemofoline and (+)-isomethoxystemofoline. Chem. Commun. 51, 4576–4578 (2015).
Guo, L.-D. et al. Total synthesis of dapholdhamine B and dapholdhamine B lactone. J. Am. Chem. Soc. 141, 11713–11719 (2019).
Heathcock, C. H. The Enchanting Alkaloids of Yuzuriha. Angew. Chem. Int. Ed. Engl. 31, 665–804 (1992).
Krabbenhoft, H. O., Wiseman, J. R. & Quinn, C. B. 9-Methyl-9-azabicyclo[3.3.l]non-l-ene. J. Am. Chem. Soc. 96, 258–259 (1974).
Wauters, I. et al. Beyond the diketopiperazine family with alternatively bridged brevianamide F analogues. J. Org. Chem. 80, 8046–8054 (2015).
Yamazaki, N., Suzuki, H. & Kibayashi, C. Nucleophilic alkylation on anti-Bredt iminium ions. Facile entry to the synthesis of 1-alkylated 2-azabicyclo[3.3.1]nonanes (morphans) and 5-azatricyclo[6.3.1.01,5]dodecane. J. Org. Chem. 62, 8280–8281 (1997).
Suzuki, H., Yamazaki, N. & Kibayashi, C. Synthesis of the azatricyclic core of FR901483 by bridgehead vinylation via an anti-Bredt iminium ion. Tetrahedron Lett. 42, 3013–3015 (2001).
Seebach, D. & Locher, R. α,β-Unsaturated carbonyl compounds with sterically protected carbonyl groups-enforced a3 versus a1 reactivity. Angew. Chem. Int. Ed. Engl. 18, 957–958 (1979).
Thuring, J. J. F. et al. N-Phthaloylglycine-derived strigol analogues. Influence of the D-ting on seed germination activity of the parasitic weeds Striga hermonthica and Orobanche crenata. J. Agric. Food Chem. 45, 2284–2290 (1997).
Kuramochi, A., Usuda, H., Yamatsugu, K., Kanai, M. & Shibasaki, M. Total synthesis of (±)-Garsubellin A. J. Am. Chem. Soc. 127, 14200–14201 (2005).
Liao, X.-B., Zhou, H., Wearing, X. Z., Ma, J. & Cook, M. J. The first regiospecific, enantiospecific total synthesis of 6-oxoalstophylline and an improved total synthesis of astonerine and alstophylline as well as the bisindole alkaloid macralstonine. Org. Lett. 7, 3501–3504 (2005).
Auclair, S. X., Morris, M. & Sturgess, M. A. Rate enhancement in the Wacker oxidation of hydroxy-α,β-unsaturated esters: a fast neutral method for the preparation of masked β-ketoesters. Tetrahedron Lett. 33, 7739–7742 (1992).
Reiter, M., Ropp, S. & Gouverneur, V. Oxidative cyclization of β-hydroxyenones with palladium(II): a novel entry to 2,3-dihydro-4H-pyran-4-ones. Org. Lett. 6, 91–94 (2004).
Muniz, K. Palladium-carbene catalysts for aerobic, intramolecular Wacker-type cyclisation reactions. Adv. Synth. Catal. 346, 1425–1428 (2004).
Galán-Fernández, R., Clemente-Tejeda, D. & Bermejo, F. A. Oxidative cyclization of γ-alkylidene butenolides. Stereoselective preparation of spirolactones. Arkivoc ix, 171–184 (2012).
Chopin, N. et al. A rapid entry to diverse γ-ylidenetetronate derivatives through regioselective bromination of tetronic acid derived γ-lactones and metal-catalyzed postfunctionalization. Eur. J. Org. Chem. 146, 6259–6269 (2015).
Acknowledgements
We are grateful to the National Natural Science Foundation of China (22071204 and 21931010), the National Key R&D Program of China (2017YFA0207302), and the Program for Changjiang Scholars and Innovative Research Team in University of the Ministry of Education, China, for financial support. We thank Dr. J.L. Ye for assistance in the preparation of XRD documents and in checking the NMR data.
Author information
Authors and Affiliations
Contributions
P.-Q.H. conceived and directed the project and wrote the paper with assistance from X.-Z.H.; X.-Z.H. performed the experiments and analyzed the data; L.-H.G. improved the synthesis of 17. All authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, XZ., Gao, LH. & Huang, PQ. Enantioselective total syntheses of (+)-stemofoline and three congeners based on a biogenetic hypothesis. Nat Commun 11, 5314 (2020). https://doi.org/10.1038/s41467-020-19163-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19163-4
This article is cited by
-
Total syntheses of the parvistemoline alkaloids enabled by stereocontrolled Ir/Pd-catalyzed allylic alkylation
Nature Communications (2024)
-
Catalytic enantioselective reductive alkynylation of amides enables one-pot syntheses of pyrrolidine, piperidine and indolizidine alkaloids
Nature Communications (2023)
-
From the ethnomedicinal plants in northern Indochina to the development of novel anti-cancer therapeutic agents
Medicinal Chemistry Research (2023)
