
Abstract
Nonsteroidal anti-inflammatory drug derivatives (NSAIDs) are an important class of medications. Here we show a visible-light-promoted photoredox/nickel catalyzed approach to construct enantioenriched NSAIDs via a three-component alkyl arylation of acrylates. This reductive cross-electrophile coupling avoids preformed organometallic reagents and replaces stoichiometric metal reductants by an organic reductant (Hantzsch ester). A broad range of functional groups are well-tolerated under mild conditions with high enantioselectivities (up to 93% ee) and good yields (up to 90%). A study of the reaction mechanism, as well as literature precedence, enabled a working reaction mechanism to be presented. Key steps include a reduction of the alkyl bromide to the radical, Giese addition of the alkyl radical to the acrylate and capture of the α-carbonyl radical by the enantioenriched nickel catalyst. Reductive elimination from the proposed Ni(III) intermediate generates the product and forms Ni(I).
Similar content being viewed by others
Introduction
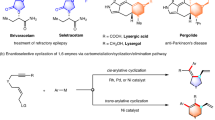
Enantioenriched α-aryl propionic acids are an important class of nonsteroidal anti-inflammatory medications (NSAIDs)1,2 and are also key building blocks for further elaboration. As a result, significant effort has been devoted to the asymmetric synthesis of α-aryl carboxylic acid derivatives. Among enantioselective methods to prepare these structural motifs, asymmetric hydrogenation3,4 and hydrocarboxylation5 stand out. A more efficient approach to access such tertiary stereocenters, however, would be the transition metal catalyzed asymmetric α-arylation of esters.
The asymmetric cross-coupling between α-halo esters and organometallic reagents (e.g., Grignard, organozinc, organoboron, and organosilicon) (Fig. 1a) was developed and driven mainly by G.C. Fu and coworkers6,7,8,9. A complementary approach was reported by Zhou10 and Gaunt11 involving asymmetric arylation of enol silane derivatives with aryl sulfonates or iodonium salts under enantioselective transition metal catalysis (Fig. 1b). Motivated by a desire to broaden the scope of coupling partners while also avoiding moisture- and air-sensitive organometallic reagents, Reisman12,13,14,15,16, Doyle17, Weix18, and their groups developed nickel catalyzed asymmetric reductive cross-electrophile coupling reactions using stoichiometric metal reductants (Zn or Mn). Inspired by their elegant studies, we disclosed an example of highly enantioselective nickel-photoredox catalyzed reductive cross-coupling of racemic α-chloro esters with aryl iodides (Fig. 1c) to construct enantioenriched NSAID derivatives19. Building on this work, we envisioned intercepting intermediates along the enantioselective cross-electrophile coupling reaction pathway with an olefin insertion step. Such a strategy could potentially lead to valuable enantioenriched olefin difunctionalization products.

a The classical asymmetric cross-coupling with organometallic reagents. b Transition metal catalyzed asymmetric arylation of enol silane derivatives. c Nickel-photoredox co-catalyzed asymmetric reductive arylation of racemic α-chloro esters. d A complementary photoredox/nickel catalyzed enantioselective carboarylation of alkenes. e This work: Nickel/photoredox catalyzed reductive asymmetric alky arylation of acrylates.
Recently, nickel-catalyzed reductive alkene difunctionalization reactions have gained notoriety as one of the most efficient strategies to install vicinal bonds in a single operation. Relevant examples were reported by Nevado20,21, Chu22,23, Martin24, Yuan25 and Molander26,27, among others. Impressive enantioselective three-component reactions were developed by Diao28, Chu29,30, Nevado31 as well as others32,33,34,35,36,37,38. In the final stages of preparing this manuscript, a complementary enantioselective three-component carboarylation of alkenes with alkyltrifluoroborates and aryl bromides was disclosed by Gutierrez and Chu29 (Fig. 1d). The redox-neutrality of this method is counterbalanced by the use of alkyltrifluoroborates, which must be prepared separately.
In this work, we present an example of enantioselective domino alky arylation of acrylates with alkyl- and aryl- bromides via cooperative nickel and photoredox catalysis (Fig. 1e). The organic photoredox 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) acts as “pseudoreductant” with Hantzsch ester (HEH) as the terminal electron donor. The products of this cascade process are NSAID derivatives of potential use in medicinal chemistry.
Results and discussion
Reaction development and optimization
We began our investigation into the three-component olefin difunctionalization reaction by choosing tert-butyl acrylate (1a), tert-butyl bromide (2a) and 1-(4-bromophenyl)ethan-1-one (3a) as model substrates (Table 1). After a systematic study of reaction conditions (see Table S1 in the Supporting Information for details), we were please to find that employing 4CzIPN (10 mol%), NiBr2 (10 mol%), L6 (11 mol%), Cy2NMe (3.0 equiv.) and HEH (3.0 equiv.) in N,N-dimethylacetamide (DMA, 0.33 M) under blue LED irradiation at room temperature for 24 h furnished the α-aryl ester (4a) in 82% assay yield (AY, determined by GC integration of the unpurified reaction mixture against an internal standard). Gratifyingly, the ee value of 4a was 92%. The (R) configuration of 4a was confirmed by comparison of its optical rotation with the literature value30 (see the Supporting Information for details). Based on our previous work19, enantioenriched bioxazoline (BiOX) frameworks were considered promising ligands for the three component coupling, giving up to 86% ee. (Table 1. entry 2–3). Further optimization, however, indicated the more electron-donating39 chiral biimidazoline (BiIM) ligands were better both in yield and enantioselectivity. Control experiments indicated that nickel salts, 4CzIPN, Cy2NMe and blue LEDs are all crucial for the success of this transformation (Table 1. entry 7–10). Additionally, HEH played a key role in the reaction yields. Only 32% AY of 4a was detected in the absence of HEH (Table 1. entry 11), although, the enantioselectivity remained 92%. In this case, it is possible that the Cy2NMe40,41,42 plays the role of reducing agent, albeit with much reduced efficiency. When other reductants, such as Mn, Zn and tetrakis(dimethylamino)ethylene (TDAE) were employed, however, only trace products were detected. The diminished reactivity with Mn, Zn and TDAE highlight the advantage of HEH under blue light in this process.
Substrate scope
With the optimized reaction conditions in hand, we next focused on the scope of the aryl bromide coupling partners using tert-butyl acrylate (1a) and 4-tert-butyl bromide 2a (Table 2). We were pleased to find that aryl bromides with functional groups in the 4-position, including electron-withdrawing groups (Ac, COOEt, CN, CHO, CF3, OCF3), halogens (F, Cl), neutral groups (H, Ph), and electron-donating groups (tBu, OMe, SMe, NMe2) all exhibited high to excellent ee values (83–93%) and moderate to good yields (45–90%). It is noteworthy that in prior studies, aryl bromides with electron-donating groups were poor substrates in reductive cross-coupling reactions43. Additionally, the 4-bromo phenyl BPin, which can be used for further transformations, was also well tolerated (71% yield, 86% ee).
Aryl bromides bearing functional groups in the 3-position (Ac and OMe) were fine substrates, furnishing the products 4p (60% yield, 88% ee) and 4q (77% yield, 88% ee), respectively. Disubstituted aryl bromides were good cross-coupling partners, affording products 4r with 89% ee and 75% yield. It is noteworthy that Flurbiprofen analog 4s and Naproxen analog 4t were obtained in good yields (83–84%) with excellent enantioselectivities (88–89%). Unfortunately, sterically hindered 2-substituted aryl bromides were not tolerated in this three-component asymmetric reductive cross-coupling (see Supporting Information for details).
Aryl bromides with extended π-systems, such as 2-bromonaphthylene, provided product 4u (80% yield, 90% ee). Heterocycles are important structural motifs in medicinal chemistry. Several aryl bromides containing heterocycles were, therefore, examined. 2-Bromodibenzo[b,d]furan, 5-bromo-2,3-dihydrobenzofuran, 5-bromobenzo[d][1,3]dioxole, 5-bromobenzofuran, 5-bromobenzo[b]thiophene, 5-bromo-1-methyl-1H-indole and 3-bromo-9-phenyl-9H-carbazole all exhibited good to excellent enantioselectivities (83–89%) with good yields (75–86%). To put the utility of this method to the test, ibuprofen and geraniol derivatives were subjected to the 3-component reaction. The desired products (4ac, 4ad) were generated in 79–82% yields and 87% de, 91% ee.
To further explore the applications of this three-component asymmetric reductive cross-coupling method, we next examined the scope of the alkyl bromides (Table 3). Initially, acyclic tertiary alkyl bromides were examined. We were pleased to find that tertiary alkyl centers containing primary alkyl bromides, aryl, esters and ketone functional groups were compatible with the reaction conditions and afforded their corresponding products (5a–5k) in good to excellent yields (60–84%) and good to excellent enantioselectivities (72–92%). Another sterically hindered 3o alkyl bromide, 1-bromo-1-methylcyclohexane, was also well-tolerated, furnishing the product l (42% yield, 88% ee). Use of 1o- or 2o-alkylbromides gave rise to two-component aryl-alkyl cross coupling under these conditions44, because reactions of radicals derived from these species at the nickel center are competitive with radical addition to the unsaturated ester.
To push the system beyond 3°-alkylhalides, additional optimization (see Table S2 in the Supporting Information for details) was conducted. Gratifyingly, moderate changes (aryl and alkyl iodides instead of their corresponding bromides, NiCl2·glyme replacing NiBr2 and a mix solvent acetone/DMA = 2:1 replacing DMA) under otherwise standard conditions enabled coupling of less substituted alkyl radicals (Table 4). Not only 1°-alkyl halides (1-iodobutane) but also 2°-alkyl halides (2-iodopropane, iodocyclopentane, iodocyclohexane, iodocycloheptane, iodocyclooctane) were tolerated, giving their corresponding products (6a–6f) in 15–75% yield and 76–92% ee. Other electron-deficient alkenes, such as N-phenylacrylamide, was tested under the standard conditions and afforded the product 6g (40% yield, 33% ee). It is noteworthy that the electron-rich vinyl benzoate was also tolerated, albeit with diminished yield and ee (6h, 45% yield, 50% ee).
Mechanistic Studies
We desired to gain insight into the reaction pathway. Based on our previous work19, a detail analysis of the model reaction was conducted (Fig. 2a). The product 4a was isolated in 79% yield with 92% ee. The fate of the HEH was the expected pyridine (isolated in 100% yield relative to HEH), derived from donation of 2 electrons and two protons. In the presence of HEH, Cy2NMe most likely acted as a base, as determined by the isolation of Cy2NMe·HBr (8, 56% yield). Only about 6% of the demethylated product, Cy2NH, was detected after workup. The demethylated amine (9) likely arises from oxidation of the amine by *4CzIPN to the amine radical cation, loss of H• to generate the iminium ion, and hydrolysis by advantageous water during the reaction or upon workup40.

a Detail analysis of the model reaction. b Radical trapping reaction via TEMPO. c Stoichiometric studies. d Intermediate verification reaction.
Additional experiments were then carried out to probe the reaction pathway. To explore the possibility of radical intermediates, the reaction was conducted under the standard conditions with the addition of the radical trap TEMPO. The presence of TEMPO shut down the formation of 4a, consistent with the involvement of radicals (Fig. 2b).
Stoichiometric studies, wherein NiBr2 was replaced by 1.0 equiv Ni(COD)2 in the presence of 1.1 equiv L6 but in the absence of 4CzIPN, HEH and Cy2NMe were carried out. This experiment was conducted both with and without blue light irradiation. With the light on 1 reacted with 2a and 3a to form cross-coupled product 4a (74% yield, 92% ee) (Fig. 2c). Likewise, when the reaction was conducted without irradiation, the cross-coupled product 4a was observed (73% yield, 92% ee). It is noteworthy that the ee of these stoichiometric reactions are identical to that observed under the standard catalytic conditions (Table 1, entry 1). These observations suggest that 1) the enantiodetermining step in the catalytic and stoichiometric reactions (Fig. 2b) are identical, and do not involve the photoredox cycle and 2) tert-butyl radical could be formed via SET reduction from the nickel catalyst (either Ni0 or NiI).
To probe the function of the photoredox cycle in this system, we performed the model reaction without Ni/L6 and bromobenzene (Fig. 2d). The radical addition/HAT quenching product (10) was obtained in 70% AY. This result indicates that in addition to reducing the Ni catalyst, the photoredox catalyst can undergo SET to the tert-butyl bromide to generate the tert-butyl radical. The radical then undergoes the Giese-type addition to the acrylate followed by abstraction of H• from HEH. Given that the olefin difunctionalization also proceeds via an α-carbonyl radical, it must be that the α-carbonyl radicals add to L*Ni(Ar)Br faster than they undergo HAT from HEH.
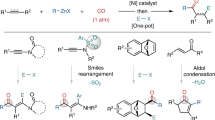
On the basis of related studies45, and the mechanistic experiments above, we present a proposed dual catalytic pathway for the enantioselective reductive three-component alkyl arylation reaction of alkenes with tertiary alkyl- and aryl-bromides (Fig. 3). The active (BiIM)Ni0 [E1/2red (NiII/Ni0) = −1.2 V vs. SCE] is generated in situ in two SET steps by the reduced photocatalyst 4CzIPN•− (E1/2red = −1.21 V vs. SCE)46,47,48,49. The resulting (BiIM)Ni0 catalyst oxidatively adds the aryl bromide to give the (BiIM)Ni(Ar)Br complex50. The alkyl bromide undergoes SET by either the Ni or reduced 4CzIPN•− (as described above45) to generate the alkyl halide radical anion, which looses bromide to generate the tertiary radical. Addition of the tertiary radical to the acrylate forms the α-carbonyl radical that is trapped by the (BiIM)Ni(Ar)Br complex to form the reactive NiIII species. The resulting NiIII intermediate51 then undergoes rapid reductive elimination to give the product 4a. In the photoredox cycle, photoexcited *4CzIPN (τ = 5.1 ± 0.5 μs)52 reacts with HEH to afford 4CzIPN•−. The resulting 4CzIPN•− is oxidized by the nickel catalyst (NiII/NiI) and/or 2a to regenerate 4CzIPN.

Key steps include oxidative addition of Ar–Br to Ni(0), radical generation by SET from nickel to the alkyl bromide, Giese addition of the alkyl radical to the acrylate, trapping of the α-carbonyl radical by Ni(II) to give Ni(III), which undergoes reductive elimination to give the product. The photocatalytic cycle involves reduction of 4CzIPN to 4CzIPN–, which is proposed to reduce the Ni catalyst to Ni(0).
In conclusion, we have developed an example of dual nickel/organic photoredox catalyzed asymmetric reductive difunctionalization of acrylates with alkyl- and aryl-bromides to generate NSAID derivatives. The advantages of this reductive protocol are that it uses two bench stable and commercially available electrophiles and it is broadly tolerant of functional groups, avoids preformed moisture- and/or air-sensitive organometallic reagents, and uses an organic reductant rather than the metal reductants used in most cross-electrophile coupling reactions.
Methods
General procedure 1
To an oven-dried vial equipped with a stir bar was added NiBr2 (2.2 mg, 0.01 mmol), L6 (5.4 mg, 0.011 mmol) and DMA (3.0 mL) under an argon atmosphere inside a glove box at RT to give a light green solution. After 30 min at RT, olefin (0.1 mmol, 1.0 equiv.), 3°-alkyl bromide (0.4 mmol, 4.0 equiv.) and aryl bromide (0.2 mmol, 2.0 equiv.) were added. Next, Cy2NMe (58.5 mg, 64 μL, 0.3 mmol, 3.0 equiv.), HEH (75.9 mg, 0.3 mmol, 3.0 equiv.) and 4CzIPN (8.0 mg, 0.01 mmol) were added. Once HEH and 4CzIPN were added, the solution turned from light green to yellow. The vial was sealed with a cap and removed from the glove box. The reaction mixture was stirred at RT under blue light. After 24 h, the color of the reaction mixture changed back to light green. The vial was opened to air and EtOAc (10 mL) was added. The resulting solution was then washed with brine (5 mL × 5) and the organic layer separated and dried over anhydrous Na2SO4, filtered and concentrated to give the crude product. The crude residue was purified by flash column chromatography to afford the corresponding product.
General procedure 2
To an oven-dried vial equipped with a stir bar was added NiCl2•glyme (2.2 mg, 0.01 mmol), L6 (5.4 mg, 0.011 mmol) and acetone/DMA (2.0/1.0, v/v, 3.0 mL) under an argon atmosphere inside a glove box at RT, giving a light green solution. After 30 min at RT, tert-butyl acrylate (0.1 mmol, 1.0 equiv.), 1° or 2°-alkyl iodides (0.4 mmol, 4.0 equiv.), 1-(4-iodophenyl)ethan-1-one (0.2 mmol, 2.0 equiv.), Cy2NMe (58.5 mg, 64 μL, 0.3 mmol, 3.0 equiv.), HEH (75.9 mg, 0.3 mmol, 3.0 equiv.) and 4CzIPN (8.0 mg, 0.01 mmol) were added. Once HEH and 4CzIPN were added, the solution turned from light green to yellow. The vial was sealed with a cap and removed from the glove box. The reaction mixture was stirred at RT under blue light. After 24 h, the color of the solution changed back to light green. The vial was opened to air and EtOAc (10 mL) was added to the solution, which was then washed with brine (5 mL × 5). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to give the crude product. The crude residue was purified by flash column chromatography to afford the corresponding product.
Data availability
The detailed experimental procedures and characterization of compounds data generated in this study are provided in Supplementary Information. The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon request.
References
Dhall, H., Kumar, A. & Mishra, A. K. Synthesis of some novel substituted phenyl β,β-diphenyl propanoate with analgesic and anti-inflammatory activity. Curr. Bioact. Compd. 14, 26–32 (2018).
Landoni, M. F. & Soraci, A. Pharmacology of chiral compounds: 2-arylpropionic acid derivatives. Curr. Drug Metab. 2, 37–51 (2001).
Pai, C.-C. et al. Highly effective chiral dipyridylphosphine ligands: synthesis, structural determination, and applications in the Ru-catalyzed asymmetric hydrogenation reactions. J. Am. Chem. Soc. 122, 11513–11514 (2000).
Li, J. et al. Asymmetric hydrogenation of α-substituted acrylic acids catalyzed by a ruthenocenyl phosphino-oxazoline-ruthenium complex. Org. Lett. 18, 2122–2125 (2016).
Alper, H. & Hamel, N. Asymmetric synthesis of acids by the palladium-catalyzed hydrocarboxylation of olefins in the presence of (R)-(-)- or (S)-(+)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate. J. Am. Chem. Soc. 112, 2803–2804 (1990).
Liu, F. et al. Cobalt-catalyzed enantioselective negishi cross-coupling of racemic α-bromo esters with arylzincs. Chem. Eur. J. 24, 2059–2064 (2018).
Jin, M., Adak, L. & Nakamura, M. Iron-catalyzed enantioselective cross-coupling reactions of α-Chloroesters with aryl grignard reagents. J. Am. Chem. Soc. 137, 7128–7134 (2015).
Mao, J. et al. Cobalt-bisoxazoline-catalyzed asymmetric kumada cross-coupling of racemic α-bromo esters with aryl grignard reagents. J. Am. Chem. Soc. 136, 17662–17668 (2014).
Dai, X., Strotman, N. A. & Fu, G. C. Catalytic asymmetric hiyama cross-couplings of racemic α-bromo esters. J. Am. Chem. Soc. 130, 3302–3303 (2008).
Huang, Z., Liu, Z. & Zhou Jianrong, S. An enantioselective, intermolecular α-arylation of ester enolates to form tertiary stereocenters. J. Am. Chem. Soc. 133, 15882–15885 (2011).
Bigot, A., Williamson, A. E. & Gaunt, M. J. Enantioselective α-Arylation of N-acyloxazolidinones with copper(II)-bisoxazoline catalysts and diaryliodonium salts. J. Am. Chem. Soc. 133, 13778–13781 (2011).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Catalytic asymmetric reductive acyl cross-coupling: synthesis of enantioenriched acyclic α,α-disubstituted ketones. J. Am. Chem. Soc. 135, 7442–7445 (2013).
Cherney, A. H. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling between vinyl and benzyl electrophiles. J. Am. Chem. Soc. 136, 14365–14368 (2014).
Hofstra, J. L., Cherney, A. H., Ordner, C. M. & Reisman, S. E. Synthesis of enantioenriched allylic silanes via nickel-catalyzed reductive cross-coupling. J. Am. Chem. Soc. 140, 139–142 (2018).
Kadunce, N. T. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling between heteroaryl iodides and α-chloronitriles. J. Am. Chem. Soc. 137, 10480–10483 (2015).
Poremba, K. E., Kadunce, N. T., Suzuki, N., Cherney, A. H. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling to access 1,1-diarylalkanes. J. Am. Chem. Soc. 139, 5684–5687 (2017).
Woods, B. P., Orlandi, M., Huang, C.-Y., Sigman, M. S. & Doyle, A. G. Nickel-catalyzed enantioselective reductive cross-coupling of styrenyl aziridines. J. Am. Chem. Soc. 139, 5688–5691 (2017).
Zhao, Y. & Weix, D. J. Enantioselective cross-coupling of meso-epoxides with aryl halides. J. Am. Chem. Soc. 137, 3237–3240 (2015).
Guan, H., Zhang, Q., Walsh, P. J. & Mao, J. Nickel/Photoredox-Catalyzed Asymmetric Reductive Cross-Coupling of Racemic α-chloro esters with aryl iodides. Angew. Chem. Int. Ed. 59, 5172–5177 (2020).
Garcia-Dominguez, A., Li, Z. & Nevado, C. Nickel-catalyzed reductive dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 139, 6835–6838 (2017).
Shu, W. et al. Ni-catalyzed reductive dicarbofunctionalization of nonactivated alkenes: scope and mechanistic insights. J. Am. Chem. Soc. 141, 13812–13821 (2019).
Zhao, X. et al. Intermolecular selective carboacylation of alkenes via nickel-catalyzed reductive radical relay. Nat. Commun. 9, 3488 (2018).
Guo, L., Tu, H.-Y., Zhu, S. & Chu, L. Selective, intermolecular alkylarylation of alkenes via photoredox/nickel dual catalysis. Org. Lett. 21, 4771–4776 (2019).
Sun, S.-Z., Duan, Y., Mega, R. S., Somerville, R. J. & Martin, R. Site-Selective 1,2-dicarbofunctionalization of vinyl boronates through dual catalysis. Angew. Chem. Int. Ed. 59, 4370–4374 (2020).
Zheng, S. et al. Selective 1,2-Aryl-aminoalkylation of alkenes enabled by metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 17910–17916 (2020).
Badir, S. O. & Molander, G. A. Developments in photoredox/nickel dual-catalyzed 1,2-Difunctionalizations. Chem 6, 1327–1339 (2020).
Campbell, M. W., Compton, J. S., Kelly, C. B. & Molander, G. A. Three-component olefin dicarbofunctionalization enabled by nickel/photoredox dual catalysis. J. Am. Chem. Soc. 141, 20069–20078 (2019).
Anthony, D., Lin, Q., Baudet, J. & Diao, T. Nickel-catalyzed asymmetric reductive diarylation of vinylarenes. Angew. Chem. Int. Ed. 58, 3198–3202 (2019).
Guo, L. et al. General method for enantioselective three-component carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. 142, 20390–20399 (2020).
Tu, H.-Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604–9611 (2020).
Wei, X., Shu, W., Garcia-Dominguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515–13522 (2020).
Wang, F., Chen, P. & Liu, G. Copper-catalyzed radical relay for asymmetric radical transformations. Acc. Chem. Res. 51, 2036–2046 (2018).
Gu, Q.-S., Li, Z.-L. & Liu, X.-Y. Copper(I)-catalyzed asymmetric reactions involving radicals. Acc. Chem. Res. 53, 170–18 (2020).
Jin, Y. & Wang, C. Nickel-catalyzed asymmetric reductive arylalkylation of unactivated alkenes. Angew. Chem. Int. Ed. 58, 6722–6726 (2019).
Tian, Z.-X. et al. Highly enantioselective cross-electrophile aryl-alkenylation of unactivated alkenes. J. Am. Chem. Soc. 141, 7637–7643 (2019).
Wang, K., Ding, Z., Zhou, Z. & Kong, W. Ni-catalyzed enantioselective reductive diarylation of activated alkenes by domino cyclization/cross-coupling. J. Am. Chem. Soc. 140, 12364–12368 (2018).
He, J. et al. Nickel-catalyzed asymmetric reductive 1,2-carboamination of unactivated alkenes. Angew. Chem. Int. Ed. 59, 2328–2332 (2020).
Wang, W., Ding, C. & Yin, G. Catalyst-controlled enantioselective 1,1-arylboration of unactivated olefins. Nat. Catal. 3, 951–958 (2020).
Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C-H arylation via photoredox and nickel dual catalysis. Nat. Commun. 10, 3549 (2019).
Fan, X., Gong, X., Ma, M., Wang, R. & Walsh, P. J. Visible light-promoted CO2 fixation with imines to synthesize diaryl α-amino acids. Nat. Commun. 9, 4936 (2018).
Wang, R., Ma, M., Gong, X., Fan, X. & Walsh, P. J. Reductive cross-coupling of aldehydes and imines mediated by visible light photoredox catalysis. Org. Lett. 21, 23–27 (2019).
Wang, R. et al. Visible-light-mediated umpolung reactivity of imines: ketimine reductions with Cy2NMe and water. Org. Lett. 20, 2433–2436 (2018).
Lucas, E. L. & Jarvo, E. R. Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem. 1, 0065 (2017).
Duan, Z., Li, W. & Lei, A. Nickel-catalyzed reductive cross-coupling of aryl bromides with alkyl bromides: Et3N as the terminal reductant. Org. Lett. 18, 4012–4015 (2016).
Neumann, M., Fueldner, S., Koenig, B. & Zeitler, K. Metal-free, cooperative asymmetric organophotoredox catalysis with visible light. Angew. Chem. Int. Ed. 50, 951–954 (2011).
Tellis, J. C., Primer, D. N. & Molander, G. A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 345, 433–436 (2014).
Zuo, Z. et al. Enantioselective decarboxylative arylation of α-amino acids via the merger of photoredox and nickel catalysis. J. Am. Chem. Soc. 138, 1832–1835 (2016).
Zuo, Z. et al. Merging photoredox with nickel catalysis: coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014).
Meng, Q.-Y., Wang, S., Huff, G. S. & Koenig, B. Ligand-controlled regioselective hydrocarboxylation of styrenes with CO2 by combining visible light and nickel catalysis. J. Am. Chem. Soc. 140, 3198–3201 (2018).
Weix, D. J. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 48, 1767–1775 (2015).
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692–700 (2017).
Uoyama, H., Goushi, K., Shizu, K., Nomura, H. & Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012).
Acknowledgements
We acknowledge the National Natural Science Foundation of China (22071107 and 21801128 to J.M.), Natural Science Foundation of Jiangsu Province, China (BK20170965 to J.M.), and Nanjing Tech University (BK20211588 and 39837112) for financial support. P.J.W. thanks the US National Science Foundation (CHE-1902509).
Author information
Authors and Affiliations
Contributions
P.Q. and H.G. are contributed equally to this work and mechanistic study with the help of Y.W., Q.L., F.Z. and D.X. The project was conceived by J.M. and P.Q. with help from P.J.W. The project was directed by J.M. and the manuscript was written by H.G., J.M. and P.J.W.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qian, P., Guan, H., Wang, YE. et al. Catalytic enantioselective reductive domino alkyl arylation of acrylates via nickel/photoredox catalysis. Nat Commun 12, 6613 (2021). https://doi.org/10.1038/s41467-021-26794-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26794-8
This article is cited by
-
Enantioselective reductive cross-couplings to forge C(sp2)–C(sp3) bonds by merging electrochemistry with nickel catalysis
Nature Communications (2025)
-
Photoredox/Cr-catalyzed enantioselective radical-polar crossover transformation via C-H functionalization
Nature Communications (2025)
-
Vinyl cyclopropanes as a unifying platform for enantioselective remote difunctionalization of alkenes
Nature Communications (2025)
-
Asymmetric paired oxidative and reductive catalysis enables enantioselective alkylarylation of olefins with C(sp3)−H bonds
Nature Communications (2024)
-
Nickel-catalysed enantioselective alkene dicarbofunctionalization enabled by photochemical aliphatic C–H bond activation
Nature Catalysis (2024)
