
Abstract
Hydrogen peroxide (H2O2) has the wide range of applications in industry and living life. However, the development of the efficient heterogeneous catalyst in the direct H2O2 synthesis (DHS) from H2 and O2 remains a formidable challenge because of the low H2O2 producibility. Herein, we develop a two-step approach to prepare PdSn nanowire catalysts, which comprises Pd oxide layered on PdSn nanowires (PdL/PdSn-NW). The PdL/PdSn-NW displays superior reactivity in the DHS at zero Celcius, presenting the H2O2 producibility of 528 mol kgcat−1·h−1 and H2O2 selectivity of >95%. A layer of Pd oxide on the PdSn nanowire generates bi-coordinated Pd, leading to the different adsorption behaviors of O2, H2 and H2O2 on the PdL/PdSn-NW. Furthermore, the weak adsorption of H2O2 on the PdL/PdSn-NW contributes to the low activation energy and high H2O2 producibility. This surface engineering approach, depositing metal layer on metal nanowires, provides a new insight in the rational designing of efficient catalyst for DHS.
Similar content being viewed by others

Introduction
Hydrogen peroxide (H2O2) is widely used as oxidant in many fields because it is environmentally friendly and the byproducts only involve H2O and O2, as compared to other oxidants (e.g. Cl-containing oxidants and nitric acid)1,2. Currently, the anthraquinone oxidation process (AO process) is employed as the method to produce H2O2 in industry, whereas the AO process requires highly intense energy consumption and the addition of toxic organic solvents, such as alkylbenzene and so on. Therefore, it is desirable to develop sustainable process and more efficient strategy towards the production of H2O23,4,5,6,7.
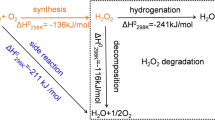
Direct H2O2 synthesis (DHS) from H2 and O2 is an effective alternative for small-scale and on-site H2O2 production, which is usually performed using Pd-based catalyst8,9,10,11. However, the catalytic activity was limited due to the high-side reactions such as hydrogenation and decomposition when employing a pure Pd-based catalyst, leading to a decreased net yield. Numerous efforts have been devoted to developing high-performance catalysts towards DHS, such as the use of bimetallic Pd-Au12,13,14,15, trimetallic Pd-Au-Pt16, acid and halide additives17,18,19,20,21. However, these Pd catalysts contain expensive gold and the H2O2 yield is still far from that achieved by the AO process22,23,24. The addition of Sn to Pd catalysts has attracted intensive attention due to the high stability and inert hydrogenation with Sn component for the H2O2 synthesis3. In particular, the synthesis of PdSn nanocatalyst involved a multi-step protocol of oxidation-reduction-oxidation (O-R-O), and a tin oxide surface layer that encapsulates small Pd-rich particles was formed while leaving larger PdSn alloy particles exposed. The PdSn nanocatalyst prepared via O-R-O presented the H2O2 production with high selectivity of >95%, while only showing the H2O2 producibility of ~70 mmol·gcat−1·h−1.
Metal nanowires (NWs) is one-dimensional (1D) structure material, which has been utilized in photonic25,26, electrical27,28, and plasmonic-related applications28,29. As compared to nanoparticles or bulk material, these 1D nanowire structures can expedite orientable electronic or ions transfer and diffusion to promote catalytic kinetics. Therefore, nanowires can provide a unique platform to study catalysis30,31. For example, in a nanowire-bacteria hybrid system, nanowires can capture electrons and deliver them to bacteria, allowing bacteria to ensure the conversion of CO226. On the other hand, cobalt oxide (CoO) nanorods/nanowires were reported to create oxygen vacancies on the nanofacets27. The catalyst exhibits excellent electrocatalytic ORR/OER performance due to the modulated electronic structure of cobalt oxide nanorods/nanowires revealed by simulation.
Herein, we develop an approach to prepare PdSn nanowires to directly produce H2O2 and found that a layer of Pd oxide on a Pd4Sn alloy nanowire (NW) prepared via two-step synthesis (Fig. 1) present efficient reactivity in the direct production of H2O2. This approach involves the synthesis of the surface-rough Pd4Sn nanowires (PdSn-NW, Sn:Pd molar ratio is 4) by a solvothermal method firstly (Supplementary Fig. 1)32. Then, metal Pd precursor was deposited onto the Pd4Sn nanowires, followed by dispersing on the TiO2 support. After rapid annealing of the material in air, an efficient catalyst for direct H2O2 synthesis was obtained and denoted as PdL/PdSn-NW catalyst (Supplementary Fig. 2). The unsupported PdL/PdSn-NW presents the morphology of worm-liked nanowires and some nanowires connect each other to form interconnected structures (Fig. 2a). The outmost surface of the structures contains Pd and Sn terminated with oxygen because both of the two metals were oxidized after the annealing in the air as shown in Fig. 1. We also synthesize and test the TiO2-supported PdSn nanowire catalyst prepared via one-step synthesis (denoted as PdSn-NW). As can be seen, the two unsupported materials show similar nanowire morphology (Fig. 2a and Supplementary Fig. 1). After loading onto TiO2, the nanowire morphologies do not change (see discussion below).

The supported PdSn-NW and PdL/PdSn-NW catalysts are supported on TiO2. The two-step protocol involving an annealing process was used for the synthesis of PdL/PdSn-NW. PdSn nanowire (NW) was prepared with a Pd: Sn molar ratio of 4 firstly and then, Pd precursor was deposited on the as-prepared PdSn nanowires again, followed by depositing on TiO2 and a rapid annealing in air. A PdSn-NW catalyst with Pd:Sn ratio of 4 was also synthesized for the purpose of comparison via one-step method by mixing Sn2+, Pd2+, PVP, EG, and NH4Br.

a Representative TEM image of the unsupported PdSn nanowire catalyst prepared by two-step (unsupported PdL/PdSn-NW). b H2O2 producibility of the supported PdL/PdSn-NW and Snx/PdSn-NW catalysts with different Pd/Sn ratios after annealing in air (350 °C, 8 min), demonstrating the addition of Sn has negative effect on the producibility of H2O2. c H2O2 producibility, hydrogenation, and decomposition of the supported PdL/PdSn-NW catalyst annealing at different temperatures in air, showing that the supported PdL/PdSn-NW annealing at 400 °C did not catalyze the hydrogenation and decomposition. d The comparison of the H2O2 producibility, hydrogenation, and decomposition of supported PdL/PdSn-NW catalyst with other Pd catalysts (Table 1). The error bars in b–d show the standard deviation in the measurements. The standard deviation was achieved from the repeated runs of three to five times using fresh catalyst each time. The error bars refers to the standard deviation of multiple times measurements of hydrogen peroxide productivity.
Results and discussion
Materials synthesis and catalytic performance
The catalytic reactivity of the TiO2-supported PdSn nanowire catalyst synthesized by the two-step (PdL/PdSn-NW, 4.1 wt.%Pd loading) was evaluated in the direct synthesis of H2O2 at zero Celcius. In comparison, we also synthesized PdSn-NW catalyst via one-step (PdSn-NW), PdSn nanoparticle (PdSn-NP), and SnOx supported on PdSn-NW (SnOx/PdSn-NW) via the two-step approach. The supports of all these reference catalysts are TiO2 and all the activity data reported in the work are achieved from catalysts supported on TiO2. Prior to dispersing on TiO2, the morphologies of these reference nanowire/nanoparticles are well characterized using TEM (Supplementary Figs. 3, 4). It should be mentioned that both TEM-EDS and X-ray photoelectron spectroscopy (XPS) analysis indicate that there is no residual Br ion detected on the Pd catalyst (Supplementary Fig. 5) and therefore, the effect of the Br ion in the H2O2 synthesis can be excluded. The supported PdL/PdSn-NW catalyst synthesized by two-step shows the high H2O2 producibility of ~680 mol/kg h−1 after annealing at 350 °C in the air (Fig. 2b). The addition of Sn onto the PdSn nanowires leads to the formation of SnOx/PdSn-NW catalyst, which is less active than the supported PdL/PdSn-NW catalyst after annealing at 350 °C in air. Moreover, with the increase of Sn loading (4.5–13 wt.%), the reactivity of the SnOx/PdSn-NW catalyst further decreases, indicating the addition of Sn on the PdSn nanowires has a negative effect on the reactivity of the PdSn-NW (Fig. 2b). However, the PdL/PdSn-NW catalyst annealing at 350 °C presents the high hydrogenation activity, which is undesirable for the DHS3. We investigated the effect of the annealing temperature on the reactivity of the PdL/PdSn-NW catalyst and found that annealing the PdL/PdSn-NW catalyst at 400 °C presented the H2O2 producibility of ~528 mol/kg h−1 with the complete absence of hydrogenation (Fig. 2c). Therefore, 400 °C was employed as the annealing temperature for all supported catalysts in the following discussion, unless otherwise noted. Moreover, the Pd/Sn ratios and metal loadings of all the catalysts were measured by EDS and ICP-OES analysis, and a good agreement was obtained using the two techniques (Supplementary Figs. 6 and 7).
When using the two-step approach to deposit Pd precursor onto the as-synthesized PdSn nanowires to form PdL/PdSn-NW catalyst, the Pd loading is precisely controlled to cover the PdSn nanowires as monolayer (Supplementary Note 1) and two layers, which are denoted as PdL/PdSn-NW and Pd2L/PdSn-NW, respectively. The annealed PdL/PdSn-NW catalyst shows superior reactivity than the PdSn-NW catalyst prepared via one-step (Fig. 2d and Supplementary Fig. 8). Although PdSn nanowire alone (PdSn-NW) shows no hydrogenation or decomposition activity (Fig. 2d), the H2O2 selectivity is only ~70% (Table 1). This is explained by the fact that the H2O2 selectivity is calculated from the first step in this two-step process (Supplementary Fig. 9). Furthermore, the annealed PdL/PdSn-NW catalyst presents outstanding catalytic activity for H2O2 production and higher H2O2 selectivity, as compared to Pd2L/PdSn-NW catalyst (Fig. 2d). This indicates that the addition of the extra Pd onto the PdL/PdSn-NW has a negative effect on the H2O2 production. Besides, both the H2O2 hydrogenation and decomposition are completely inhibited on the annealed PdL/PdSn-NW catalyst, as compared to other active Pd catalysts reported. Furthermore, the annealed PdL/PdSn-NW catalyst is very stable in the reaction and no deactivation is found in multiple runs (Supplementary Fig. 10). The reactivity of the supported PdL/PdSn-NW catalyst was also compared with other Pd-based catalysts (Fig. 2d and Table 1, Supplementary Tables 1 and 2), such as Pd nanoparticles, PdSn nanoparticles, and nice PdSn nanowires prepared by the approach reported in the literature32. None of them showed comparable H2O2 producibility and low decomposition/hydrogenation activity (Supplementary Fig. 11) as the supported PdL/PdSn-NW catalyst exhibiting the H2O2 selectivity of >95% under the same conditions. It should be mentioned that the H2O2 productivity slightly slows down over time (after 30 min) and it is partially ascribed to the decrease of the reactant gas concentration and the accumulation of H2O2 in the autoclave according to Le Chatelier’s principle (Supplementary Fig. 12). Besides, it possibly suggests that the catalyst changes in the presence of high concentration H2O2 and either become less active or begins to degrade H2O2 at high H2O2 concentrations.
Materials characterization
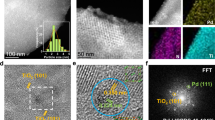
The composition and morphology of the PdL/PdSn-NW catalyst are first characterized by X-ray diffraction (XRD) and electron microscopy. XRD patterns of the unsupported PdSn nanowire prepared by two-step before and after annealing show the diffraction peaks located at lower diffraction angles (i.e., larger d spacings) as compared to that of metal Pd (Fig. 3a), indicating the crystal expansion of metal Pd due to the alloying of Sn atom. Annealing this unsupported PdSn nanowire at 350 °C or 400 °C does not make significant changes on the XRD patterns, indicating that the nanowire is thermally stable. When dispersing the nanowire on TiO2, XRD pattern of the supported PdL/PdSn-NW catalysts only presents the diffraction peaks of the support TiO2 (Fig. 3a and Supplementary Fig. 13), indicating the high dispersion of PdSn nanowire on the support. After annealing the supported PdL/PdSn-NW catalyst at different temperatures (350 °C and 400 °C, Supplementary Fig. 13a) in air, we do not find the presence of other diffraction peak or phase separation, indicating that the PdSn-NW and PdL/PdSn-NW catalysts are quite stable after the pretreatment. The representative high-resolution transmission electron microscopy (TEM)/scanning transmission electron microscopy (STEM) images of the PdL/PdSn-NW are shown in Fig. 3b, c. The nanowires have the diameter ranging from 5 to 10 nm and STEM-EDS line scanning shows the presence of both Pd and Sn in the nanowire (Supplementary Fig. 14). The high-resolution TEM of the nanowire is shown in Fig. 3b and the image exhibits the lattice fringes of 0.23 and 0.20 nm, corresponding to the (111) and (200) plane of Pd4Sn phase, respectively (more images are shown in Supplementary Fig. 15). STEM-EDS mapping demonstrated that both Pd and Sn signals are uniformly dispersed in the nanowires (Fig. 3c). Therefore, the electron microscopy characterization of the annealed PdL/PdSn-NW sample demonstrates that the nanowire morphology of PdSn-NW is well maintained after loading onto TiO2 and annealing in air.

a XRD patterns of the unsupported and supported PdL/PdSn-NW catalysts before and after annealing in air. b HRTEM image of the unsupported PdL/PdSn-NW sample showing the lattice fringes of the PdSn phase. c STEM image and STEM-EDS mapping of the supported PdL/PdSn-NW showing the close proximity of both Pd and Sn elements, indicating the successful synthesis of Pd4Sn alloy. d XPS spectra of Pd 3d core level of the supported PdL/PdSn-NW catalyst before and after annealing in air. e Surface valence band photoemission spectra of the supported PdL/PdSn-NW catalysts annealing at different temperatures in air. f HAADF-STEM image of the supported PdL/PdSn-NW catalyst after annealing showing a layer of Pd oxide on the PdSn nanowires.
XPS is used to investigate the surface species of the supported PdL/PdSn-NW catalyst during the annealing process (Fig. 3d, e, Supplementary Figs. 16–19). As shown in Fig. 3d, the XPS spectrum of the unannealed PdL/PdSn-NW catalyst shows that the main Pd species is Pd metal and the presence of a very small amount of Pd2+ prior to annealing. After annealing at 350 °C in air, the Pd2+ species content increases and Pd metal is still the dominant species on the surface. After annealing the PdL/PdSn-NW at 400 °C in air, we observe that the main Pd on the catalyst is Pd2+ species with a small amount of Pd metal, demonstrating the oxidation of metal Pd after annealing. Furthermore, the XPS analysis indicates that the Sn species on the supported PdL/PdSn-NW catalyst is SnOx (Supplementary Fig. 19) and the Pd/Sn ratio is ~2. Since Sn/PdSn-NW catalysts are less active and selective than PdSn-NW catalyst, the active site is not ascribed to the SnOx. Therefore, the rapid annealing process generated Pd oxide on the PdSn nanowires, which is observed in the HAADF-STEM image (Fig. 3f and Supplementary Fig. 20). The d-band model has been widely used in order to understand activity trends in metal-surface-catalyzed reactions33. Here, the d-band center obtained from XPS was used to study the interaction between gas adsorption/intermediates and metal surface during annealing in air. The surface valence band photoemission spectrum of the annealed PdL/PdSn-NW is more delocalized and widely distributed than that of the unannealed PdL/PdSn-NW (Fig. 3e), indicating the different activation ability of gas molecules or intermediates on the annealed PdL/PdSn-NW catalyst34. The annealed PdL/PdSn-NW catalyst, therefore, provides Pd oxide to modulate the adsorption/desorption behaviors of the reactants on the catalyst surface, which was confirmed by the improved H2O2 producibility of PdL/PdSn-NW (Table 1) and density functional theory (DFT) calculation below.
The chemical environment of the Pd atoms in the supported PdL/PdSn-NW catalyst after annealing in the air was examined via X-ray adsorption spectroscopy (Fig. 4a and Supplementary Figs. 21 and 22). We also performed the XAS measurements of the Pd foil, PdO, PdSn-NW, and PdSn-NP catalysts for comparison. For the reference Pd foil, a major peak attributing to the Pd-Pd scattering was observed. The EXAFS spectra of PdSn-NW and PdSn-NP catalysts show a similar or slightly expanded scattering distance as compared to that of the metal Pd foil (Fig. 4a), which is attributed to the Pd-Pd and PdSn scattering. The Pd-K edge EXAFS of the supported PdL/PdSn-NW catalyst after annealing (air, 400 °C) also have a major peak corresponding to Pd-Pd and PdSn scattering (Fig. 4a), indicating the main Pd phase on the PdL/PdSn-NW catalyst is metal Pd and the rapid annealing process does not change the bulk phase of the PdSn nanowires. EXAFS fit gave a coordination number of 4.2 at a bond distance of 2.76 Å (Supplementary Table 3). The EXAFS is fitted to Pd-Pd and PdSn scattering features because the XRD results clearly indicate that the bulk phase of the PdSn nanowire after annealing in air is metal Pd (Fig. 3a) and both XRD and XAS are bulk techniques. Furthermore, the high-resolution TEM (HRTEM) image shows the lattice fringes corresponding to metallic PdSn nanowire (Supplementary Fig. 15a). Since XPS is more surface sensors for observing the changes under different conditions than XRD and XAS, We performed near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) to in-situ investigate the surface properties of the PdSn catalysts (both PdSn nanoparticle and PdSn nanowire prepared via one-step and two-step approaches) in the flowing O2, H2 and O2/H2 mixture (Fig. 4b–d). Conventional PdSn nanoparticle catalyst showed the presence of Pd oxides in both vacumm and the flowing O2, whereas metal Pd was formed after switching to H2 or H2/O2 mixture (Fig. 4d), indicating that the conventional PdSn nanoparticles are not stable in H2 and H2/O2 atmospheres. For the PdSn nanowire catalysts, the PdL/PdSn-NW prior to annealing mainly shows the metal Pd phase on the catalyst surface (Fig. 4b) and there is a very small change for the Pd 3d XPS spectra of the catalyst after flushing in H2, O2, and H2/O2, indicating that the unannealing PdL/PdSn-NW maintain the metal Pd surface in the flowing H2/O2. Likewise, the Pd 3d XPS spectrum of the PdL/PdSn-NW after annealing in air at 400 °C indicated that Pd oxide is the main Pd species on the surface at vacuum (Fig. 4c). The introduction of a flow of O2 has no observed effect on the Pd 3d XPS spectrum of the annealed PdL/PdSn-NW. After introducing a flow of H2, we observed the presence of metal Pd, as well as the presence of Pd oxide. However, in the presence of H2/O2 mixture, the PdL/PdSn-NW surface after annealing was maintained as Pd oxide. Therefore, in-situ NAP-XPS evidenced that the surface species of the PdL/PdSn-NW after annealing is Pd oxide in the reaction feed of H2/O2, while the major Pd species on both PdSn-NP and PdSn-NW are metal Pd in the presence of H2/O2.

a EXAFS spectra of the PdK edge of PdSn catalysts after annealing in air. b In situ NAP-XPS of Pd 3d spectra for PdSn-NW in the presence of O2, H2, and O2/H2. c In situ NAP-XPS of Pd 3d spectra for PdL/PdSn-NW after annealing in air in the presence of O2, H2 and O2/H2. d In situ NAP-XPS of Pd 3d spectra for PdSn-NP after annealing in air in the presence of O2, H2 and O2/H2.
Theoretical studies on reaction mechanism
Based on the above reactivity and characterization results, we propose that the excellent reactivity of the PdSn nanowires prepared by the two-step approach (PdL/PdSn-NW) is associated with the Pd oxide layer on PdSn nanowire which was detected via AC-STEM and NAP-XPS (Figs. 3 and 4). The stable Pd oxide layer on PdSn nanowire in the presence of H2/O2 exhibited enhanced reactivity as compared to the conventional PdSn nanoparticle and PdSn nanowire prepared by a one-step approach. To better understand the unique properties of the Pd oxide layer on the PdL/PdSn-NW in direct H2O2 synthesis, we performed DFT calculations based on three models of PdO(101), Pd4Sn and PdO monolayer supported on Pd4Sn(111) (PdO@Pd4Sn) (Fig. 5a). The PdO(101) surface contains tetra-coordinated Pd (denoted as Pd4c) and tri-coordinated Pd (denoted as Pd3c) and the outmost surface atoms of the Pd4Sn model are Pd atom, while the PdO@Pd4Sn surface contains Pd4c and bi-coordinated Pd (denoted as Pd2c). Details for model construction can be found in the computational method section. Bader charge analysis shows that Pd4c on PdO@Pd4Sn and PdO(101) are charged similarly with +0.83|e| and +0.85|e| (Supplementary Fig. 23), respectively. Nevertheless, the charges of Pd2c on PdO@Pd4Sn and Pd3c on PdO(101) are different, with the former being charged +0.46|e| and the latter having a charge of +0.67|e|. The surface Pd atoms in Pd4Sn are slightly positive by +0.11|e|, which is therefore more metallic than those in PdO(101) and PdO@Pd4Sn. Thus, these three catalysts with different Pd chemical states may lead to different interactions between the adsorbate and the catalyst.

a DFT optimized structures of PdO(101), Pd4Sn, and PdO@Pd4Sn with b adsorption energies of H2, O2, H2 + O2, and H2O2, c free energy profiles for H2 activation, and d free energy profiles for O2 reduction by the surface hydrides on these three models. TS transition state. Color code: Pd, blue; O, red; H, white; Sn, yellow.
Subsequently, we investigated the adsorption behavior of the reaction molecules (O2, H2, and H2O2) on PdO(101), Pd4Sn, and PdO@Pd4Sn. The adsorption configurations and corresponding adsorption energies are shown in Supplementary Fig. 24 and Fig. 5b, respectively. The calculation results show that all three molecules exhibit strong adsorption on the PdO(101) surface. H2 prefers to adsorb on top of Pd3c and O2 tends to adsorb on the Pd3c-Pd3c bridge site, leading to adsorption energies of −0.62 and −1.56 eV, respectively. For H2O2, we found that it has a spontaneous dissociation to form two OHs on the two Pd3c sites on PdO(101), suggesting that PdO(101) has poor selectivity for H2O2 synthesis. For PdO@Pd4Sn, its binding to the three molecules is significantly weaker than that on PdO(101). H2 is physically adsorbed on the Pd2c top sites (2.76 Å away from Pd2c) with an adsorption energy of −0.11 eV, the adsorption of O2 on the Pd2c-Pd2c bridge sites leads to small adsorption energy of −0.42 eV. H2O2 adsorbs on Pd2c-Pd2c bridge sites, yielding moderate adsorption energy of −0.63 eV. Likewise, as for the Pd4Sn surface, H2 is adsorbed on the top of Pd while the O2 molecule connects with four surface Pd atoms, having the adsorption strengths as those located on between PdO(101) and PdO@Pd4Sn. To gain in-depth insight into the relation between electronic structure and catalytic behavior, the projected density of states (PDOSs) of the surface Pd- \({d}_{{z}^{2}}\) states on PdO(101) and PdO@Pd4Sn were calculated and compared since the \({d}_{{z}^{2}}\) states are considered to play an important role in molecule adsorption35,36,37. As can be seen in Supplementary Fig. 25, the distribution of \({d}_{{z}^{2}}\) of surface Pd3c on PdO is more localized than that of surface Pd2c on PdO@Pd4Sn. Furthermore, a sharp peak of \({d}_{{z}^{2}}\) states exists across the Fermi level on PdO(101), implying strong interaction between surface Pd and the reaction species via donating/accepting electrons. In contrast, on PdO@Pd4Sn, the distribution of Pd-\({d}_{{z}^{2}}\) state is less significant around the Fermi level, indicating the weaker binding of reaction species to Pd. It is therefore proposed that different adsorption behaviors of O2, H2, and H2O2 on the three models lead to different catalytic performances in the direct synthesis of H2O2.
In the following, we investigated the reaction mechanism of the O2 reduction on PdO(101), Pd4Sn, and PdO@Pd4Sn. First, we calculated the activation of the most stable adsorption state of H2, whose reaction pathways on these three models are shown in Fig. 5c. On PdO(101), H2 dissociation occurs readily with an energy barrier of 0.09 eV. In the final state, the two dissociated H atoms are adsorbed on the Pd3c-Pd3c bridge site and the Pd3c top site, respectively. In addition, it is found that the migration of H between two adjacent Pd3c sites is facile with a small energy barrier of 0.11 eV, while the migration of H from the Pd3c site to an adjacent oxygen site is difficult, requiring an energy barrier of 0.87 eV(Supplementary Fig. 26). It indicates that Pd-H hydride species are stable on PdO(101). On Pd4Sn, H2 activation on Pd4Sn is barrierless, similar to the previous DFT findings on Pd(111) surface38 (Supplementary Fig. 27). On PdO@Pd4Sn, the cleavage of H-H bond in the most stable H2 adsorbed has to overcome an energy barrier of 0.35 eV. Similar to the case on PdO(101), the Pd-H species is also stable because the energy barrier for H migration between Pd2c sites (0.69 eV) is lower than that (1.07 eV) of its migration to oxygen sites (Supplementary Fig. 28). Therefore, H2 readily dissociates on all of PdO(101), Pd4Sn, and PdO@Pd4Sn to produce stable Pd-H hydride, which provides a hydrogen source for the subsequent O2 reduction (Fig. 5d). We also calculated other possible H2 dissociation pathways but they possess higher energy barriers (Supplementary Figs. 29 and 30). Then, we investigate the O2 reduction to H2O2 (energy profile in Fig. 5d and configurations in Supplementary Figs. 31 and 32). On the Pd4Sn, the energy barriers of the first (*O2 + *H → *OOH) and second (*OOH + *H → *H2O2) hydrogenation of O2 is 0.55 eV and 0.65 eV with the reaction energies of 0.09 eV and −0.01 eV, respectively. On PdO@Pd4Sn, the *O2 + *H → *OOH reaction encounters an energy barrier of 0.53 eV, while the subsequent *OOH + *H → *H2O2 process has a lower energy barrier of 0.27 eV. This result indicates that the reaction of O2 reduction to produce H2O2 has a lower rate-determining energy barrier on PdO@Pd4Sn than that on Pd4Sn, in agreement with the experimental observations that PdO@Pd4Sn has better catalytic performance than Pd4Sn prepared by one-step. We did not infer the reaction pathway on PdO(101) because H2O2 was proved to be unstable on this surface (Fig. 5b and Supplementary Fig. 25), leading to low H2O2 production.
In addition, we simulated three main side reactions of *O2, *OOH, and *H2O2 dissociation on Pd4Sn and PdO@PdSn, and the corresponding structures of IS, TS, and FS are shown in Supplementary Figs. 33 and 34. On Pd4Sn(111), the *O2 and *OOH dissociations are barrierless while the *HOOH dissociation has a small barrier of 0.07 eV (Supplementary Fig. 33), suggesting the poor selectivity of H2O2 on Pd4Sn(111). On PdO@Pd4Sn, the energy barriers of the three side reactions are 1.74, 0.53, and 0.32 eV (Supplementary Fig. 34), respectively, which are larger than those of the corresponding competitive reactions (0.53, 0.27, and 0.14 eV for O2 hydrogenation, OOH hydrogenation, and H2O2 desorption, respectively, Supplementary Figs. 32, 34). Therefore, these results suggest the high selectivity of H2O2 on PdO@Pd4Sn, which is consistent with the experimental observations (Fig. 2d and Table 1).
Selective production of H2O2 is a challenge in the direct H2O2 synthesis (DHS) from H2 and O2. In this contribution, we develop a two-step approach to prepare PdSn nanowires to efficiently catalyze direct H2O2 synthesis (DHS). This approach involves the first synthesis of the surface-rough Pd4Sn nanowire (PdSn-NW) by a solvothermal method. Then Pd precursor was deposited onto the PdSn alloy nanowire (NW), followed by annealing in air. The as-prepared PdL/PdSn-NW via the two-step approach presents efficient reactivity with H2O2 producibility of >520 mol kgcat−1 h−1 and selectivity of >95% in the direct production of H2O2 at zero Celcius. For the PdSn nanowire prepared by one-step method and PdSn nanoparticle catalysts, the catalyst surfaces are prone to be reduced in the flowing H2/O2 and show low H2O2 reactivity.
The excellent H2O2 production over the PdL/PdSn-NW catalyst is attributed to the presence of Pd oxide layer on the PdSn nanowires. The Pd oxide layer is stable against reduction or oxidation in the flowing H2/O2, while other PdSn nanoparticles and PdSn nanowire catalysts undergo a reduction in the presence of H2/O2. The layered Pd oxide enables less adsorption of oxygen/hydrogen and decreases the rupture of both O-O and H-H bonds, as well as less adsorption of peroxide produced, leading to the complete inhibition of the H2O2 hydrogenation and decomposition. Therefore, engineering the surface of PdSn nanowires via two-step approach can generate a layer of Pd oxide on the PdSn nanowire, presenting excellent reactivity in the direct H2O2 synthesis, which provides a promising strategy to design and develop highly active DHS catalyst.
Methods
Chemicals
Bis (acetylacetonato) palladium (II) (Pd(acac)2, 99%), tin(II) acetate (Sn(Ac)2, 95%), methanol (CH3OH, HPLC grade), titanium dioxide (TiO2, P25, 99%) were purchased from Sigma-Aldrich. Palladium(II) nitrate dihydrate (Pd(NO3)2·2H2O, 99%), polyvinylpyrrolidone (PVP, MW = 58000), Tin(IV) chloride pentahydrate (SnCl4·5H2O, 99%) were purchased from J&K Scientific Ltd. Ethylene glycol (EG, analytical grade), N,N-Dimethylacetamide (DMAC, analytical grade), ammonium bromide (NH4Br, analytical grade) were purchase from Sinopharm Chemical Reagent Co. Ltd.(Shanghai, China). The de-ionized water (DI H2O,18 MΩ/cm) used in all experiments was obtained by passing through an ultra-pure purification system. All the chemicals were used without further purification.
Synthesis of unsupported PdSn nanowires via one-step
In a typical synthesis, the PdSn NWs (Pd4Sn, marked as PdSn) were synthesized according to our previous report32. Briefly, 7.6 mg of Pd(acac)2, 1.5 mg of Sn(Ac)2, 15 mg of NH4Br, 100 mg of PVP, 2 mL of DMAC, and 8 mL of EG were added into a vial (30 mL). And then, the mixture was sonicated for 15 min to ensure form a homogeneous solution. The solution was heated and maintained at 180 °C for 2 h in an oil bath. After that, the mixture was cooled down to room temperature. Finally, the precipitate was centrifuged and washed with ethanol/acetone mixture.
Synthesis of unsupported Pdx/PdSn-NW and Sny/PdSn-NW via two-step
In a typical synthesis, the Pdx/PdSn or Sny/PdSn NWs were synthesized by the following steps. Briefly, 7.6 mg of Pd(acac)2, 1.5 mg of Sn(Ac)2, 15 mg of NH4Br, 100 mg of PVP, 2 mL of DMAC, and 8 mL of EG were added into a vial (30 mL). Then, the mixture was sonicated for 15 min to ensure form a homogeneous solution. The solution was heated and maintained at 180 °C for 2 h in an oil bath. After that, the mixture was cooled down to room temperature to achieve the unsupported PdSn nanowires. The role of NH4Br in the synthesis of these worm structures is to induce the formation of the PdSn nanowires. Subsequently, the desired amount of Pd(acac)2 or Sn(Ac)2 solution (including 320 μL of EG and 80 μL of DMAC, e.g. 1.9 mg Pd(acac)2) was added into the above solution. Next, the mixture was continuously stirred and held for 30 min at room temperature, followed by heating to 150 °C (hold for 2 h) with vigorous stirring. Then, the mixture was cooled down to ambient temperature again. Finally, the precipitate was centrifuged and washed with ethanol/acetone mixture. The precipitate achieved with the addition of 1.9 mg Pd precursor in the second step is denoted as unsupported PdL/PdSn-NW and the sample achieved with the addition of Sn(Ac)2 solution in the second step is denoted as unsupported SnOx/PdSn-NW.
Synthesis of unsupported Pd nanoparticles
The unsupported Pd NPs were synthesized from a procedure similar to that of PdSn nanowires synthesized via one-step, but without the addition of Sn(Ac)2.
Synthesis of unsupported PdSn nanoparticles (PdSn-NP)
The bimetallic PdSn NP catalysts were synthesized according to a previous report3. In brief, 62.5 mg of Pd(NO3)2·2H2O, 2 mL of DI water were added into a vial (30 mL). The solution was heated to 80 °C with continuous stirring. Then, 1 mL of DI water solution (including 73.8 mg of SnCl4·5H2O) was added to the vial and held for 15 min under stirring. Next, 0.6 g of TiO2 and 1 mL of DI water were added to the solution, and the mixture was heated to 110 °C with continuous stirring in the air to evaporate the water. Subsequently, the solid sample was placed in a tube furnace and heated to 500 °C for 3 h under static air (heating rate of 10 °C/min). After cooling down, the solid sample was further heated to 200 °C for 2 h in a flow of 5% H2/Ar (heating rate of 10 °C/min). Then, the solid sample was annealed in static air at 400 °C for 8 min or 4 h, respectively (heating rate of 10 °C/min). The resultant was denoted as PdSn-NP.
Synthesis of catalysts supported on TiO2
Typically, the commercial TiO2 material (anatase) and 6 mL of CH3Cl were added into a 30 mL vial. The PdSn-NW or nanoparticle materials were firstly dispersed in 6 mL of CH3Cl and ultrasonicated for about 5 min. The resulting mixture was added to the TiO2 vial. Next, the mixture was continuously ultrasonicated for 30 min. Finally, the products were collected by centrifugation and dried at 60 °C overnight.
Annealing treatment of the supported catalysts
The as-synthesized catalysts were treated via a rapid annealing process. Firstly, the tube furnace was heated to 400 °C and held for 30 min under static air. Next, the appropriate amount of catalyst was placed in the porcelain boat and pushed into the tube furnace. The catalyst left for several minutes by annealing and was taken out (8 min) for cooling. The final products were denoted as PdL/PdSn-NW (supported on TiO2, the “NW” represents nanowires) and collected for further characterization.
Catalyst characterization
The crystallographic structure of all the samples was determined by Powder XRD (Rigaku Ultima IV, Japan) patterns using a Cu Kα X-ray source (λ = 1.54056 Å). The morphology of all the samples was imaged by transmission electron microscope (TEM, JEM-1400, JEOL Co., Japan). The HRTEM, line-scan analysis, and elemental mapping were carried out on FEI Tecnai F30 electron microscope with an acceleration voltage of 200 kV. STEM images were obtained on an FEI Titan Themis Z, operating at 60-300 kV to observe the surface morphology of the materials. Before microscopy examination, the catalyst powders were ultrasonically dispersed in ethanol and then a drop of the solution was put onto a carbon-coated copper grid. The elemental content of all the samples was examined by scanning electron microscopy energy dispersive spectrometer (SEM-EDS, ZEISS Sigma, Germany) and inductively coupled plasma-atomic emission spectrometry (ICP-AES, iCap 7000, USA). The chemical compositions and valence states of all the samples were analyzed by XPS (K-Alpha, USA). NAP-XPS (SPECS Surface Nano Analysis GmbH, Germany) measurements were carried out on a SPECS system equipped with a differentially pumped Phoibos hemispherical electron energy analyzer using monochromatic Al Kα radiation (1486.6 eV). The H2 or O2 (99.999%) flow was introduced into the NAP cell and the total pressure was kept constant at 0.2 mbar via the electronic back-pressure regulator. X-ray Absorption Fine Structure (XAFS) of all samples was analyzed by TLS-01C beamline of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan), and data were processed according to standard procedures using the Demeter program package (Version 0.9.24)39.
Direct H2O2 synthesis
All the catalysts supported on TiO2 were tested in the direct H2O2 synthesis. Direct H2O2 synthesis experiment was carried out in a stainless-steel semibatch autoclave with a volume of 50 mL. Briefly, 5 mg of supported catalyst, 5.54 g of MeOH, and 3.0 g of H2O (both HPLC grade) were added to the autoclave. Then purged three times with O2 (0.4 MPa) to the autoclave, and with filled with O2 (0.4 MPa) and 5% H2/Ar (3.6 MPa) until the total pressure of 4.0 MPa at room temperature. All experiments were performed in an ice-water bath and kept continuous stirring (1200 rpm) of reaction time for 15 min. The reaction was carried out at zero Celsius because it is much safer and easier to carry out in the lab. We also investigated the reactivity of the catalyst at room temperature for comparison. The producibility of the PdL/PdSn-NW catalyst at room temperature is slightly lower than that performed at zero Celcius in the direct H2O2 synthesis (Supplementary Fig. 35). Control experiments are also performed by varying the stirring rate from 100-1200 rpm, and varying the catalyst mass from 3 to 10 mg using autoclaves having different volumes (50 mL and 100 mL). All these experiments show similar H2O2 producibility under these conditions. Therefore, it indicates that there are no interphase mass transfer constraints in the reaction under the present conditions (Supplementary Fig. 36).
Quantification of gas product
The gas products were detected via a gas chromatograph (GC, 8890, Agilent) equipped with a thermal conductivity detector (TCD) and a MolSieve 5 A packing column (G3591-80022). Typically, after the direct H2O2 synthesis process, the autoclave was directly connected into the GC. The gas products were purged into GC by Ar.
The H2 conversion was calculated as follows:
H2O2 productivity evaluation
The H2O2 productivity was analyzed by titrating with acidified Ce(SO4)2 (0.05 M) and using ferroin (about 100 µL) as indicator3,40. The acidified Ce(SO4)2 solutions were standardized against (NH4)2Fe(SO4)2·6H2O using ferroin as an indicator. The H2O2 productivity and H2O2 selectivity were calculated based on the equations below:
H2O2 hydrogenation and decomposition
H2O2 hydrogenation and decomposition were carried out using similar procedures to direct H2O2 synthesis. Typically, for H2O2 hydrogenation test (H2O2 + H2 → 2H2O), 5 mg of supported catalyst (support is TiO2), 5.54 g of MeOH, 2.0 g of H2O and 1.1 g H2O2 (30%) were added in the autoclave. Then, 3.6 MPa of H2/Ar (5% H2) was filled into the autoclave. Afterward, the reaction time extended until 30 min. For the H2O2 decomposition test (H2O2 → H2O + O2), 5 mg of catalyst, 5.54 g of MeOH, 2.0 g of H2O, and 1.1 g H2O2 (30%) were added to the autoclave. Then, 3.6 MPa of N2 was filled into the autoclave. Subsequently, the reaction was carried out for 30 min. The H2O2 hydrogenation or decomposition rate was calculated based as follows:
Computational details
All spin-polarized DFT calculations were carried out by using the Vienna Ab initio Simulation Package. Interactions between ion cores and valence electrons were described by the projected augmented wave method and the exchange-correlation function was by the Perdew–Burke–Ernzerhof functional based on generalized gradient approximation41. Wavefunctions were expanded by the plane wave basis with a cutoff energy of 400 eV42. The van der Waals interactions were included via DFT-D2 in Grimme’s scheme43. Transition states were determined by the Climbing Image Nudged Elastic Band and Dimer method44,45, and charge states were obtained from Bader Charge analysis46. For all models (crystallographic information files of Pd4Sn, PdO@Pd4Sn, and PdO(101) were provided in supplementary materials as Supplementary Data 1–3, respectively), Monkhorst–Pack mesh with 2 × 2 × 1 grid was adopted to sample the Brillouin zone47. Convergence criteria for structural optimizations were set to 10−5 eV and 0.02 eV/Å for energy and force, respectively. And for transition states searching, the criterion for force was set to 0.05 eV/Å. The (2 × 3)-PdO(101) surface was cleaved from the optimized PdO unitcell (a = b = 3.056 Å, c = 5.381 Å, in agreement with experimental values (a = b = 3.043 Å and c = 5.335 Å)), containing 4 O-Pd-O tri-atomic layers. The bottom two layers were fixed while the other layers with surface species were fully relaxed in structural optimization. The lengths of a and b of supercell are 12.38 and 9.17 Å, respectively. Since the Pd unit is composed of 4 Pd atoms, uniform PdxSny can only be constructed as Pd2Sn2 and Pd3Sn. In addition, our experiments show that nanowires expose more Pd on the surface, we, therefore, simulate the slab with 4 layers of uniform Pd3Sn covered with one layer of Pd (Supplementary Fig. 37). Then, the total Pd/Sn ratio equals 4:1. For all slabs, a vacuum space of 15 Å is adopted along the z-direction to avoid the interaction between periodic images.
The Gibbs free energy (G) is obtained by VASPKIT code48 via the following formula:
in which E is the electronic energy of DFT calculations, EZPE and S are the zero-point energy and entropy computed from vibrational frequency analysis. T was the temperature adopted in our experiment, 273.15 K. ΔU0→T is the internal energy difference between 0 and T K.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Information. The raw data sets used for the presented analysis within the current study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Code availability
The code that supports the findings of this study is available from the corresponding author upon reasonable request.
References
Perry, S. C. et al. Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442–458 (2019).
Richards, T. et al. A residue-free approach to water disinfection using catalytic in situ generation of reactive oxygen species. Nat. Catal. 4, 575–585 (2021).
Freakley, S. J. et al. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 351, 965–968 (2016).
Xia, C. et al. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226–231 (2019).
Mehrotra, R. et al. Unassisted selective solar hydrogen peroxide production by an oxidised buckypaper-integrated perovskite photocathode. Nat. Commun. 12, 6644 (2021).
Wu, Q. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
Lin, Y.-J. et al. Thermocatalytic hydrogen peroxide generation and environmental disinfection by Bi2Te3 nanoplates. Nat. Commun. 12, 180 (2021).
Flaherty, D. W. Direct synthesis of H2O2 from H2 and O2 on Pd catalysts: current understanding, outstanding questions, and research needs. ACS Catal. 8, 1520–1527 (2018).
Yi, Y. et al. A review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 6, 1593–1610 (2016).
Xu, H. et al. Design of high-performance pd-based alloy nanocatalysts for direct synthesis of H2O2. ACS Catal. 7, 2164–2170 (2017).
Ricciardulli, T. et al. Effect of Pd coordination and isolation on the catalytic reduction of O2 to H2O2 over PdAu bimetallic nanoparticles. J. Am. Chem. Soc. 143, 5445–5464 (2021).
Landon, P. et al. Direct formation of hydrogen peroxide from H2/O2 using a gold catalyst. Chem. Commun. 2058–2059 (2002).
Richards, T. et al. The direct synthesis of hydrogen peroxide over supported pd-based catalysts: an investigation into the role of the support and secondary metal modifiers. Catal. Lett. (2022).
Wang, S. et al. Palladium-based bimetallic nanocrystal catalysts for the direct synthesis of hydrogen peroxide. ChemSusChem 13, 3243–3251 (2020).
Jin, Z. et al. Direct synthesis of pure aqueous H2O2 solution within aluminosilicate zeolite crystals. ACS Catal. 11, 1946–1951 (2021).
Edwards, J. K. et al. The direct synthesis of hydrogen peroxide using platinum-promoted gold–palladium catalysts. Angew. Chem. Int. Ed. 53, 2381–2384 (2014).
Chinta, S. et al. A mechanistic study of H2O2 and H2O formation from H2 and O2 catalyzed by palladium in an aqueous medium. J. Catal. 225, 249–255 (2004).
Liu, Q. et al. Direct synthesis of H2O2 from H2 and O2 over Pd–Pt/SiO2 bimetallic catalysts in a H2SO4/ethanol system. Appl. Catal., A 339, 130–136 (2008).
Choudhary, V. R. et al. Influence of nature/concentration of halide promoters and oxidation state on the direct oxidation of H2 to H2O2 over Pd/ZrO2 catalysts in aqueous acidic medium. Catal. Commun. 8, 1310–1316 (2007).
Edwards, J. K. et al. Switching off hydrogen peroxide hydrogenation in the direct synthesis process. Science 323, 1037–1041 (2009).
F. de L. e Freitas, L. et al. Tunable catalytic performance of palladium nanoparticles for h2o2 direct synthesis via surface-bound ligands. ACS Catal. 10, 5202–5207 (2020).
Wilson, N. M. et al. Mechanism for the direct synthesis of H2O2 on Pd clusters: heterolytic reaction pathways at the liquid–solid interface. J. Am. Chem. Soc. 138, 574–586 (2016).
Chen, L. et al. On the reaction mechanism of direct H2O2 formation over Pd catalysts. ACS Catal. 11, 2735–2745 (2021).
Selinsek, M. et al. Revealing the structure and mechanism of palladium during direct synthesis of hydrogen peroxide in continuous flow using operando spectroscopy. ACS Catal. 8, 2546–2557 (2018).
Dasgupta, N. P. et al. 25th anniversary article: semiconductor nanowires—synthesis, characterization, and applications. Adv. Mater. 26, 2137–2184 (2014).
Liu, C. et al. Nanowire–bacteria hybrids for unassisted solar carbon dioxide fixation to value-added chemicals. Nano Lett. 15, 3634–3639 (2015).
Ling, T. et al. Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis. Nat. Commun. 7, 12876 (2016).
Mai, L. et al. Nanowire electrodes for electrochemical energy storage devices. Chem. Rev. 114, 11828–11862 (2014).
Wei, H. et al. Plasmon waveguiding in nanowires. Chem. Rev. 118, 2882–2926 (2018).
Huo, D. et al. One-dimensional metal nanostructures: from colloidal syntheses to applications. Chem. Rev. 119, 8972–9073 (2019).
Garnett, E. et al. Introduction: 1D nanomaterials/nanowires. Chem. Rev. 119, 8955–8957 (2019).
Zhang, Y. et al. Defect engineering of palladium–tin nanowires enables efficient electrocatalysts for fuel cell reactions. Nano Lett. 19, 6894–6903 (2019).
Tong, W. et al. Crystal-phase-engineered PdCu electrocatalyst for enhanced ammonia synthesis. Angew. Chem. Int. Ed. 59, 2649–2653 (2020).
Nørskov, J. K. et al. The Electronic Factor in Heterogeneous Catalysis. In: Fundamental Concepts in Heterogeneous Catalysis) (2014).
Li, Z. et al. An adaptive machine learning strategy for accelerating discovery of perovskite electrocatalysts. ACS Catal. 10, 4377–4384 (2020).
Li, J. et al. Water adsorption on Pd {100} from first principles. Phys. Rev. B 76, 235433 (2007).
Filhol, J. S. et al. Surface phase stability diagram for Pd deposits on Ni(110): A first-principles theoretical study. Phys. Rev. B 64, 085412 (2001).
Todorovic, R. et al. A comparative density functional theory study of the direct synthesis of H2O2 on Pd, Pt and Au surfaces. Catal. Today 160, 242–248 (2011).
Ravel, B. et al. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Cao, K. et al. Efficient Direct H2O2 Synthesis Enabled by PdPb Nanorings via Inhibiting the O–O Bond Cleavage in O2 and H2O2. ACS Catal. 11, 1106–1118 (2021).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Xiao, P. et al. Solid-state dimer method for calculating solid-solid phase transitions. J. Chem. Phys. 140, 174104 (2014).
Henkelman, G. et al. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Yu, M. et al. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 134, 064111 (2011).
Monkhorst, H. J. et al. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Wang, V. et al. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Acknowledgements
Funds from the National High-Level Young Talents Program, National Natural Science Foundation of China (22072118 and 21973013), and National Natural Science Foundation of Fujian Province, China (2020J02025) are acknowledged. We also thank the financial support from State Key Laboratory for Physical Chemistry of Solid Surfaces and National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters of Xiamen University. Part fund was supported by Science and Technology Projects of Innovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM) (HRTP-[2022]-3) and the Fundamental Research Funds for the Central Universities (20720220008). S.L. thanks “Chuying Program” for the Top Young Talents of Fujian Province. J.H. would like to thank National Natural Science Foundation of China (Nos. 51772262, U20A20336, 21935009) and Natural Science Foundation of Hebei Province (No. B2020203037). Numerical computations were performed at Hefei advanced computing center. The authors thank Shanghai Synchrotron Radiation Facility (Shanghai Institute of Applied Physics) and beamline TLS-01C (Taiwan National Synchrotron Radiation Research Center) for providing the beam time.
Author information
Authors and Affiliations
Contributions
X.H., S.L., and H.X. conceived and supervised the research; H.-C.L. designed and performed the experiments; Q.W. performed and S.L. conducted the DFT calculations; C.D. and J.H. performed the HRTEM measurements; J.Z. performed the XAS analysis; Y.Zha. and F.L. help for the materials synthesis; M.C., Y.Zhe., K.Z., and G.F. helped with the data interpretations; H.-C.L., S.L., and H.X. wrote and edited the paper. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sang Soo Han and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Hc., Wan, Q., Du, C. et al. Layered Pd oxide on PdSn nanowires for boosting direct H2O2 synthesis. Nat Commun 13, 6072 (2022). https://doi.org/10.1038/s41467-022-33757-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-022-33757-0
This article is cited by
-
Keto-enol tautomerism as dynamic electron/hole traps promote charge carrier separation for hydrogen peroxide photosynthesis
Nature Communications (2025)
-
Palladium Bromide–Palladium Composite for Direct Synthesis of Hydrogen Peroxide
Topics in Catalysis (2025)
