
Abstract
A base-assisted metal species modulation mechanism enables Ni-catalyzed stereodivergent transfer semihydrogenation of alkynes with water, delivering both olefinic isomers smoothly using cheap and nontoxic catalysts and additives. Different from most precedents, in which E-alkenes derive from the isomerization of Z-alkene products, the isomers were formed in orthogonal catalytic pathways. Mechanistic studies suggest base as a key early element in modulation of the reaction pathways: by adding different bases, nickel species with disparate valence states could be accessed to initiate two catalytic cycles toward different stereoisomers. The practicability of the method is showcased with nearly 70 examples, including internal and terminal triple bonds, enynes and diynes, affording semi-hydrogenated products in high yields and selectivity.
Similar content being viewed by others
Introduction
Divergent catalysis as a particularly appealing strategy from both academic and practical perspectives allows convenient control over selectivity towards different terminal products starting from the same material1,2,3,4,5,6,7. Predictably, it would be more beneficial for the distinction of reactivity and selectivity if the two target molecules are achieved in separate mechanistic pathways, which generally requires employment of different catalysts to initiate diverse catalytic cycles. Therefore, it would be mechanistically interesting and also operationally practical to develop novel strategies in which different catalytic species could be generated from the same catalyst precursor by simple adjustment of the reaction factors, leading to different products with high selectivity in two independent catalytic cycles.
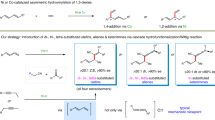
Transition metal-catalyzed stereodivergent hydrogen transfer of alkynes to produce both Z- and E-olefins have attracted remarkable interest in recent years8,9,10,11,12,13,14,15,16,17,18,19,20. Most pioneering examples actualize this transformation by regulation of catalytic systems to realize a Z to E isomerization process at the late stage (Fig. 1, above). For instance, Moran et al. showed that Ni-catalyzed transfer hydrogenation (TH) of alkynes with HCO2H selectively afforded Z-olefins, which isomerized to E-isomers by adding triphos ligand8. Another catalyst-modulated system was disclosed by Liu and coworkers in 2016, in which both isomers could be achieved using Co catalysts supported with specified bidentate ligands. The isomerization of Z-alkenes was suppressed by introducing bulky ligand due to the sterically unfavored coordination and insertion processes9. Recently Mei et al. reported that Pd-catalyzed semihydrogenation of alkynes with H2O delivered cis-olefins at room temperature in CH3CN, while isomerization of the double bond towards trans-olefins was facilitated at 80 °C in DMF10. Mechanistically, E-alkenes in the majority of reported strategies originate from the Z-isomer, requiring subtleness of the reaction conditions and the steric or electronic properties of the substrates. Therefore, mechanistically orthogonal stereodivergent semi-reduction of alkynes to both olefinic isomers, in which E-alkenes are generated directly from alkynes instead of the isomerization from Z-alkenes, is undoubtedly meaningful in both academic and practical perspectives. We envisioned that modulating the catalytic species at an early stage might initiate independent profiles to deliver both isomers in orthogonal manners (Fig. 1, bottom). Ideally, several issues should be addressed: (a) independent catalytic cycles should be initiated by simple adjustment of the reaction factor(s) to enable high yield and stereoselectivity8,9,10,11,12,13,14,21,22; (b) nonprecious metals and ligands without toxic additives would be more favorable23,24,25,26,27; (c) water is the first choice of the hydrogen donor for TH process10,28,29,30,31; (d) alkynes with various substituents should be hydrogenated in high yield and stereoselectivity in mild conditions, and over-reduction to saturated alkanes need to be avoided32,33. Pioneered by previous Ni-catalyzed alkyne hydrogenation8,34,35,36, we launched a project with nickel catalysts to address the above challenges. After laborious trials, we realize a Ni-catalyzed stereodivergent TH of alkynes with water in an innovative controlling mode, in which the key to the success of modulation is the judicious inclusion of the base. Notably, unlike most existing reports, formation of trans-olefins is unrelated to the isomerization of cis-olefin. Mechanistic investigations suggest that base modulated the valence state of active nickel species derived from the same simple pre-catalyst. Consequently, the isomers are achieved independently in completely disparate catalytic pathways: the in situ formed Ni(II) species delivered Z-alkenes, while the Ni(I) species selectively afforded E-alkenes as final products.

a Regulation of stereoselectivity in transition metal-catalyzed stereodivergent TH of alkynes. b Modulation of stereoselectivity in this work.
Results
Optimization of the reaction conditions
We initiated our exploration by evaluating the transfer hydrogenation of the model substrate 1a with simple nickel sources and 2,2′-bipyridine ligands (Table 1). The first obstacle to overcome is the activation of the inert H2O molecule in our nickel catalyst system37,38,39. Gratifyingly, boron reagents showed unique effect, and the alkenes were obtained in high yield and selectivity using Na2CO3 as base. B2pin2 turned out to be more efficient than other diboron compounds such as B2(OH)4, B2cat2 and B2neop2 (Supplementary Table 1)40,41,42. Although diboron compounds were found to be capable of activating water in Pd-catalyzed systems40,41,42,43,44, including hydrogenation of unsaturated C-C bonds to saturated alkanes40, it is, to our knowledge, the first case for such activation effect in Ni catalyst systems. Notably, E-alkene 3a was formed as the major isomer, and over-reduced alkane product was not observed. Solvents turned out to exert a profound influence on the reactivity (Supplementary Table 1), and 72% yield of alkenes were obtained with 11/89 isomeric ratio in DMF (entry 1). Decorating the bipyridine ligand with electron-withdrawing ester groups totally suppressed the reactivity (entry 2). Subsequent screening of other bipyridine derivatives as well as phenanthroline ligands L3-L6 provided comparatively inferior results to 2,2′-bipyridine (entries 3-6). Systematic screening of nickel catalyst, ligand, base, boron and water (Supplementary Tables 2 and 3) showed that base exerted an unexpected, yet decisive role in the control of selectivity. As shown in Table 1, the reaction was evidently inclined to E-selectivity by K2CO3, NaOH and CF3CO2Na, with the later showing the best result, affording 3a in 84% isolated yield and 6/94 Z/E ratio (entries 7–9). Interestingly, a slant to Z-selectivity was shown with CH3CO2Na, providing 2a with 69/31 Z/E ratio (entry 10). Organic bases such as DABCO and Et3N were also tested, and E-alkene 3a was delivered as the major product (entries 11 and 12). The catalyst loading could be lowered to 5 mol% with no erosion of the yield or selectivity (entry 13). The reactivity was almost totally shut down at a lower temperature of 60 °C (entry 14). The alkyne 1a was untouched at 40 °C, leaving all starting material recovered (entry 15). In contrast, comparable results were observed at higher temperatures (entries 16 and 17).
The above results inspired us to further proceed with other bases aiming at the optimization for Z-selective transfer semihydrogenation of 1a. As shown in Table 2, CH3CO2K and CH3CO2Cs acted similarly as CH3CO2Na, indicating that metal ions are not responsible for the selectivity reversal (entries 1 and 2). Only moderate selectivity was achieved when HCO2Na was added (entry 3). To our delight, PhCO2Na gave a promising result, providing the final olefins in 80/20 selectivity (entry 4). Again, dicarboxylate ligand L2 showed dramatically decreased reactivity (entry5). In contrast, 4,4′-dimethoxy-2,2′-bipyridine L3 improved the selectivity to 93/7 (entry 6). Ligands L4 and L5 bearing methyl groups at 3,3′- or 4,4′-positions both gave slightly reduced selectivity than L3 (entries 7 and 8). When the loading of the catalyst and base were reduced, alkenes were retrieved in slightly improved yield and selectivity (entries 9 and 10). Contrary to E-selective system (Table 1, entry 14), the reaction could still proceed smoothly at a lower temperature, albeit 1a was partially recovered (entry 11). Performing the reaction at higher temperatures resulted in poorer selectivities (entries 12 and 13).
Mechanistic investigations
Several questions deserve exploration to better understand this unprecedented system: (a) is water in the system indeed the hydrogen donor? (b) are alkenes generated from hydrometallation of in situ formed Ni-H species or hydrolysis of vinyl boron compounds? (c) does isomerization of Z-olefins take effect similarly as most precedents to afford E-olefins? (d) what are the roles of the bases in modulation of the reaction outcomes? To answer these questions, a series of mechanistic studies were carried out. Firstly, deuterium-labeled experiments were conducted (Fig. 2a). The deuterium was incorporated into both the 1,2-olefinic positions of 2a’ and 3a’ with D2O instead of H2O under both standard conditions (equations (1) and (2)). Similar results were also observed for unsymmetric alkynes 1bb and 1i, with the former leading to even higher deuterations (equations (7) and (8)). In contrast, there was no sign of deuteration on the products using DMF-d7 as solvent (equations (3) and (4)). When the reactions of 1a using D2O were placed in hydrogen atmosphere, comparative deuterium isotopic contents as in argon were observed (equations (5) and (6)), proving that releasing of H2 and consequent hydrogenation was not involved in the catalytic pathway. Control experimental studies of vinylboron reagents 4, 5 and diborylated vinyl derivatives 4′, 5′ were respectively performed under both reaction conditions with 0, 1.0, 2.0, and 3.0 equiv. of B2pin245,46,47,48. Olefin products 2g and 3g were not detected (Fig. 2b, (equations (9), (10), (11) and (12)). This, together with the reactions under H2 atmosphere, indicated that Ni-H species were formed between the nickel pre-catalyst and H2O assisted by B2pin2, which would deliver alkenyl nickel intermediates to accomplish the catalytic cycle.

a Deuterium labeling experiment. b Control experimental studies of vinylboron reagents. c Kinetic profiles of the reaction systems. d Competitive control experiments of the bases.
To deeper understand the process of selective semi-reduction, the kinetic behavior of the reaction system was monitored (Fig. 2c). The kinetic profile of Z-selective transfer semihydrogenation system showed that the concentration of 2a increased gradually throughout the reaction period, staying closely aligned with the conversion of 1a. After 5 h, 3a began to show up until the yield reached 6% (Fig. 2c, left). We postulate that the small amount of E-alkene in this system was generated from isomerization of the Z-product, which was suppressed in the initial period due to competitive coordination of alkyne 1a with the metal center. Consumption of most 1a after 5 h left space for the coordination of 2a for the subsequent isomerization process, which still need 1a as auxiliary since the selectivity remained unchanged after disappearance of 1a. The E-selective reaction profile with CF3CO2Na as base clearly indicated the nonexistence of Z/E isomerization (Fig. 2c, right). Approximately 6% of Z-alkene was already formed at the early stage of the reaction, which maintained in this level until 1a was completely converted. The concentration of 3a increased gradually, which was independent with the amount of 2a. The kinetic isotopic effect (kH/kD = 1.68) was observed when H2O was replaced by D2O in the Z-selective reactant stream (Supplementary Figure 1), and a kinetic isotopic effect of 1.08 was also obtained in the E-selective reduce system (Supplementary Figure 2), indicating that activation of H2O molecule delivering Ni-H species might not be involved in the rate-determining step in both selective hydrogenations.
To further verify the above inferences, a series of control experiments were conducted. When Z-alkene 2a was put in both standard conditions, only less than 5% of E-alkene was detected (Supplementary Fig. 3, equations (1) and (2)), demonstrating the reluctance of the Z/E isomerization in these conditions. Elevating the reaction temperature showed a beneficial effect for the isomerization, which was promoted to 13% by heating 2a at 120 °C under the Z-selective condition (Supplementary Fig. 3, equation (3)). Consistently, the reaction of 1a at 120 °C under this condition afforded the corresponding olefinic products in 86/14 selectivity (Supplementary Fig. 3, equation (4)), compared with 93/7 at 80 °C.
The color changes between the two reaction systems were significantly different. As shown in Supplementary Fig. 4, the Z-selective system seemed turbid and beige at the very beginning, which turned to light brown after several minutes and got darker later. The color changed to tan-yellow gradually in about one hour and became lighter to milk-white after another one hour, which remained till the end. A completely different visual appearance mutation was observed for the E-selective system, which looked transparent black and got darker quickly at the very early stage. Interestingly, as soon as the reaction was over as monitored of the crude mixture, the color changed to bright yellow immediately, which could be regarded as a simple hint for the complete of the reaction. We postulate that the dark color ascribes to the coordination of the triple bond to the metal center, which was terminated promptly once alkynes were exhausted33. The distinction in colors of the two systems indicates that different nickel species might be involved, leading to the corresponding olefinic products in totally unrelated pathways. The color variation of the control experiments on base was quite similar to the above observation (Supplementary Fig. 4, bottom): the initial pale green color changed to tint of turbidity yellow and clarify black color separately after addition of PhCO2Na and CF3CO2Na, respectively, indicating the formation of different nickel species was modulated with the choice of base.
Competitive control experiments of the bases were conducted to further illustrate their functions (Fig. 2d). After the standard Z-selective mixture using PhCO2Na was stirred for 1 h, another 2.0 equivalent of CF3CO2Na was added, and no apparent influence on the reaction outcomes was observed (equation (13)). By contrary, a worse selectivity was caused by addition of PhCO2Na into the E-selective system (28/72 vs 4/96) (equation (14)).
All the mechanistic insights and the visual phenomenon pointed to distinct catalytic pathways for the two reaction systems, inspiring us to further inquire whether different metal species were taking effect inherently. To detect whether nanoparticles were involved in our Ni-B-H2O system, general mercury drop experiments were performed41,43. The yield or selectivity was not affected in either system (Supplementary Fig. 5, equations (1) and (2)), excluding heterogeneous catalytic pathway. Despite the failure in capture of metallic intermediates, electron paramagnetic resonance (EPR) analyses provided clues on the active nickel species and the base effect. As shown in Fig. 3a (2), strong EPR signals were observed in the E-selective mixture, indicating the formation of Ni(I) or Ni(III) species49,50,51,52. The signals of such Ni species could not be found at ambient temperature, which is in accordance with our experimental observations that semihydrogenations of 1a were not permitted at rt (Supplementary Table 3, entry 25). In contrast, EPR active species was not observed in Z-selective system (Fig. 3a (1)) that features a Ni(0)/Ni(II) catalytic cycle. In agreement with the competitive experiments of bases (equation (14)), the EPR signals for the reactions using CF3CO2Na as base were markedly weakened after the addition of PhCO2Na (Fig. 3a (3)). In line with the fact that use of HCO2Na as base gave an almost 1:1 ratio of the Z- and E-alkenes (Table 2, entry 3), the EPR signal of the system with HCO2Na was less significant than that with CF3CO2Na (Fig. 3a (4)), but much more significant than that of PhCO2Na system (Fig. 3a (1)). EPR test results in the absence of alkyne and water are shown in Fig. 3a (5), which is in parallel to the studies in presence of starting materials: no signal was observed when PhCO2Na was employed, while a strong EPR signal showed up with CF3CO2Na as base, demonstrating that different Ni species are accessed at early stage without the participation of alkynes. In addition, we also analyze the mixture of NiBr2 and Ni(cod)2, which would generate Ni(I) species in situ (Fig. 3a (6))53,54. The similar signal compared with the system using CF3CO2Na further demonstrate the involvement of Ni(I) species in the E-selective protocol.

a EPR analyses. b Control experiment of Ni(I) species. c and d Control experimental studies of reductant in the Z-selective condition. e Proposed catalytic cycles.
Further control experiments were carried out to verify the key role of Ni(I) species in the E-selective hydrogenation process. As shown in Fig. 3b, in situ formed Ni(I) species by mixing NiBr2 and Ni(cod)2 resulted in olefin 3a with E-configuration as major product (40% yield, 18/82 Z/E). Noteworthy, the reaction was totally suppressed in this condition without base (Supplementary Table 3, entry 24), which is another evidence for the participation of CF3CO2Na to deliver Ni(I) species. On the other hand, the Z/E ratio dropped appreciably when Mn or Zn was added in the Z-selective condition (Fig. 3c), which might due to the generation and competitive act of Ni(I) species. Furthermore, the reactivity in condition A was suppressed when Zn or Mn was used instead of B2Pin2 (Fig. 3d), indicating that B2Pin2 not only interacts with bases to deliver active Ni species in this hydrogenation system, but also exists as an activator of water.
Although more experimental supports are awaited to uncover the detailed mechanism (initial NMR studies on the mechanism, see Supplementary Fig. 6), a general scenario could be delineated based on the above results and related literatures34,35,55,56,57,58,59,60,61,62,63 (Fig. 3e): NiBr2 would interact with the bases firstly, delivering carboxylates carrying different counter anions. The difference in electronic properties between the benzoate and the trifluoroacetate endows them with distinct reactivities towards B2pin2. Organic bases such as DABCO and Et3N inclined to deliver E-olefins (Table 1, entries 11 and 12), which is in consist of our proposal that counter anion from the base was not equipped to the metal center in the Z-selective catalytic cycle64. Consequently, Ni(II) species C is generated directly from the benzoate B and B2pin2 for Z-selective catalytic cycle. Activation of H2O molecule delivers Ni(II)-H species D, which undergo syn-addition to the triple bond to afford alkenyl Ni(II) intermediate E. Participation of another H2O molecule releases the cis-olefin and regenerate C with the assistance of B2pin2. Based on the kinetic experiments, coordination and insertion of the Z-alkene to the Ni-H species assisted by alkyne precursor would occur in the late stage of the reaction, followed by isomerization process resulting in slight stereo-impurity. We propose that isomerization of a vinyl Ni(I) species is responsible for the E-selectivity observed in this approach, the specific oxidation state at Ni could provide an opportunity for isomerization55,56. At the beginning of the cycle, Ni(II) species H might be generated firstly from nickel trifluoroacetate G and B2pin2, which furnishes Ni(0) species I in a reductive elimination step. Comproportionation between H and I occurs instantly, forging Ni(I) species J to initiate the catalytic cycle53,54. Activation of H2O molecule would deliver Ni(I)-H species L, followed by insertion of alkyne leading to vinyl Ni(I) intermediate M, which may undergo isomerization56 to E-alkenyl nickel intermediate N. Thermodynamically more stable product 3 is generated by hydrolysis of N, and the acquired nickel hydroxide O could be transformed back to Ni(I) species J in the aid of B2pin2.
Density functional theory (DFT) calculations were carried out to investigate the remarkably different impact of PhCO2Na and CF3CO2Na on Ni species65,66,67,68. The reaction free energy profiles are shown in Fig. 4. The Ni(II) precursor A reacted with PhCO2Na to afford nickel benzoate B, which is exergonic by 44.7 kcal/mol (Fig. 4a). The activation free energy barrier for one-ligand exchange of B towards PhCO2-Ni(II)-BPin C is 25.2 kcal/mol. The activation of C with H2O molecule requires a 14.3 kcal/mol of activation free energy en route to Ni(II)-H species D. Although the transition state of further ligand exchange from C to P could not be located, the process from P to Q was unfavored due to 29.9 kcal/mol of activation energy barrier, supporting our proposal that the catalytic cycle proceeds through benzoate D. As for the catalytic system using CF3CO2Na as base, LNiBr2 A firstly reacted with CF3CO2Na to give Ni(II) species G, which is exergonic by 48.4 kcal/mol. One-ligand exchange with B2Pin2 producing CF3CO2-[NiL]-BPin H is endothermic with 13.4 kcal/mol, and the energy barrier is 29.4 kcal/mol, which is in parallel with the experimental result that the reactivity was almost totally suppressed at a lower temperature of 60 °C (Table 1, Entry 14). Although we were unable to locate the transition state for further ligand exchange from H to P, the notably higher reaction ∆G of two-ligand exchange makes it more unlikely. For the one-ligand exchange pathway, subsequent reductive elimination and comproportionation process are exergonic, leading to Ni(I) species J and K to initiate the E-selective catalytic cycle. The activation free energy barrier for the reaction of H with H2O molecule en route to Ni(II)-H species R is nearly 10 kcal/mol higher than that of the reductive elimination. Besides, J + K is lower than R in the potential energy surfaces, basically excluding the participation of R in the catalytic cycle.

a The Free-energy reaction profile of Z-selective catalytic cycle. b The Free-energy reaction profile of E-selective reaction system.
Substrate scope
The synthetic practicability of this system was sufficiently embodied in the functional group compatibility investigations. In Fig. 5a, the Z-selective semi-reduction of various alkynes 1 using PhCO2Na as base is summarized. This reaction proceeded successfully toward substituted diarylethynes bearing a diverse set of substituents. Specifically, substrates bearing methyl or tert-butyl groups at p- or m-positions all worked smoothly under the standard conditions (2a–2d), as well as hindered isopropyl (2e) or phenyl (2f) groups located in the ortho-position of the aryl terminus, suggesting the insensitivity of the system to steric effect. Electron-donating methoxy substituent was well accommodated, and the diaryl alkenes were generated in high yields and selectivity (2h, 2i and 2j). Amino functional group 2k was no exception, well tolerated in this catalytic transfer semihydrogenation process. Z-olefins with electron-withdrawing trifluoromethyl (1l), cyano (1m, 1n), ester (1o) and acyl (1p) groups could also be achieved uneventfully. Fluoro- and chloro-containing products (2q-2t) were furnished from the corresponding alkynes, leaving space for further functionalization. Arylalkyne 1u bearing hydroxyl group provided the alkene product with 61/39 Z/E ratio. The relatively lower selectivity might be caused by isomerization of Z-olefin 2u, since the Z/E ratio of the olefins decreased from 86/14 to 53/47 by heating in DMF at 80 °C. The alkyne derivative containing Bpin-substituent was well tolerated, providing alkene 2v in 78% yield and 97/3 Z/E. The generality of the system was further showcased by the tolerance of naphthyl (2w) and heterocycles including thienyl (2x), benzofuryl (2y) and pyridyl motifs (2z), particularly the latter, considering pyridinyl ligands were used in our catalytic system. Moreover, running in a longer reaction time or higher temperature, alkynes carrying both naphthenic and linear alkyl terminuses could be reduced to the corresponding olefinic products efficiently (2bb–2hh). Notably, only Z-alkenes were formed specifically from the alkyl substrates, supporting our previous deduction that the E-alkenes in the Z-selective conditions might derive from the isomerization process, which was sluggish for alkyl alkenes due to their weak coordinating ability to the metallic species. The compatibility of the system was further underlined by successful involvement of unprotected primary OH group (2ff), which was unaffected under the catalytic conditions. Natural product derived alkyne with estrone skeleton proceeded smoothly, and the desired product 2gg was furnished in excellent yield and selectivity. Finally, internal alkyne 1ii bearing 1,2-dialkyl substituents also gave high yield and perfect stereoselectivity.

a Scope of TH of alkynes to Z-alkenes with water. b Exploration of substrate scope in E-selective condition. c Substrate scope with respect to terminal triple bonds, enynes, and diynes.
A survey on the substrate scope was performed next to demonstrate the robustness of the E-selective TH process using CF3CO2Na as base (Fig. 5b). Similar as the former system, diaromatic internal alkynes with a wide range of functional groups such as methyl (1a-1c), tert-butyl (1d), isopropyl (1e), methoxyl (1i, 1j), amino (1k), trifluoromethyl (1l), cyano (1m), ester (1o), acyl (1p) and halogen substituents (1q-1t) were all hydrogenated to the desired trans-alkenes uneventfully. Alkyne bearing a hydroxyl on the aromatic ring worked well to furnish the desired product 3u. Bpin group might interact competitively with the active species in this Ni-B hydrogenation system, thus olefin 3v was achieved in inferior selectivity (85/15 E/Z). Heteroaromatic rings including thienyl (1x), benzofuryl (1y) and pyridyl (1z, 1aa) substituents were compatible again, delivering the alkenyl heterocycles selectively. Trans-selective transfer semihydrogenation of alkyl acetylenes turned out to be challenging: monoaryl alkyne 1hh was reduced to 3hh in moderate yield and selectivity, while dialkyl alkyne 1ii delivered 3ii in much more inferior result. Propargylic esters were transformed to E-olefins (3jj–3ll) as single isomers in moderate yields and excellent selectivity. Consistent with the previous observation, for all the E-selective experiments, a mutation of color from black to bright yellow was observed as soon as the reaction finished.
Finally, we tested the reactivity of terminal alkynes, which are more inclined to over-reduction. As shown in Fig. 5c, alkene 6 was obtained in high yield in Z-selective conditions from 1mm, and saturated ethyl product was not observed. The condition could also be extended to diynes 1nn and 1oo, with both triple bonds being hydrogenated in high selectivity. Interestingly, the reaction of conjugated enyne 1pp in Z-selective conditions afforded diene 9 with E-configuration as the major product. On the contrary, Z-enyne 10 was obtained in high selectivity when 1,3-diyne 1qq was loaded in E-selective conditions.
Discussion
In conclusion, we have disclosed an unprecedented Ni-catalyzed stereodivergent transfer semihydrogenation of acetylenes with water. The configuration of the olefinic products was controlled by the choice of bases, which were demonstrated to influence the valence states of the catalytic nickel species. Consequently, E-alkenes were achieved independently from the direct reduction of alkyne precursors instead of isomerization of the Z-isomers. The strategy also features use of cheap catalysts and nontoxic reagents, and compatibility with an assortment of alkynyl substrates such as internal and terminal alkynes, 1,3-enynes and diynes. Besides its significance in semihydrogenation of alkynes, we believe that the mechanistic insights would lead to better understanding of the performance of nickel species, and also pave the way to further exploration of the other transition metal catalyst systems. Further pursuit including the development of the catalytic strategy and also detailed mechanistic studies are ongoing in our laboratory.
Methods
General procedure for Z-selective transfer semihydrogenation of alkynes 1
To a dry sealed tube were added alkyne 1 (0.3 mmol), NiBr2 (3.3 mg, 0.015 mmol, 5 mol%), L3 (7.1 mg, 0.033 mmol, 11 mol%), PhCO2Na (43.2 mg, 0.3 mmol, 1.0 equiv.) and B2pin2 (228.5 mg, 0.9 mmol, 3.0 equiv.). The flask was evacuated and refilled with argon, followed by the addition of H2O (16.2 µL, 0.9 mmol, 3.0 equiv.) and DMF (4 mL). The mixture was stirred at 80–100 °C for 8–30 h until the reaction was completed as monitored by TLC. The resultant solution was diluted with ethyl acetate, washed with HCl aqueous solution (1 M) and concentrated in vacuum. The mixture was detected by GC directly or after simple filtration in some cases to determine the Z/E ratio. The crude product was purified by chromatography on silica gel (300–400 mesh), eluted with petroleum ether with 0–20% of ethyl acetate to give alkene product. Careful column chromatography was able to partially deliver the major product in a pure form to provide precise NMR spectra of the major product. The overall isolated yield was calculated based on the combination of all parts.
General procedure for E-selective transfer semihydrogenation of alkynes 1
To a sealed tube were added alkyne 1 (0.3 mmol), NiBr2 (3.3 mg, 0.015 mmol, 5 mol%), L1 (5.2 mg, 0.033 mmol, 11 mol%), CF3CO2Na (81.6 mg, 0.6 mmol, 2.0 equiv.) and B2pin2 (228.5 mg, 0.9 mmol, 3.0 equiv.). The flask was evacuated and refilled with argon, followed by the addition of H2O (16.2 µL, 0.9 mmol, 3.0 equiv.) and DMF (4 mL). The mixture was stirred at 80 °C for 10–20 h until the reaction was completed as monitored by TLC. The resultant solution was diluted with ethyl acetate, washed with HCl aqueous solution (1 M) and concentrated in vacuum. The mixture was detected by GC directly or after simple filtration in some cases to determine the Z/E ratio. The crude product was purified by chromatography on silica gel (300–400 mesh), eluted with petroleum ether with 0–20% of ethyl acetate to give alkene product. Careful column chromatography was able to partially deliver the major product in a pure form to provide precise NMR spectra of the major product. The overall isolated yield was calculated based on the combination of all parts. All compounds were characterized (see the Supplementary Information).
Data availability
The data that support the findings of this study are available within the article, its Supplementary Information files. All data underlying the findings of this work are available from the corresponding author upon request. Supplementary Data 1 contains the data of the imaginary frequencies, free energies and coordinates of the optimized structures. Supplementary Data 2 contains the 1H, 19F, 13C NMR spectra.
References
Nájera, C., Beletskaya, I. P. & Yus, M. Metal-catalyzed regiodivergent organic reactions. Chem. Soc. Rev. 48, 4515–4618 (2019).
Irina P. Beletskaya, I. P., Nájera, C. & Yus, M. Chemodivergent reactions. Chem. Soc. Rev. 49, 7101–7166 (2020).
Ping, L., Chung, D. S., Bouffard, J. & Lee, S. Transition metal-catalyzed site- and Regio-divergent C–H bond functionalization. Chem. Soc. Rev. 46, 4299–4328 (2017).
Mei, L., Wei, Y., Tang, X. & Shi, M. Catalyst-dependent stereodivergent and regioselective synthesis of indole-fused heterocycles through formal cycloadditions of indolyl-allenes. J. Am. Chem. Soc. 137, 8131–8137 (2015).
Xu, S. et al. Enantioselective regiodivergent synthesis of chiral pyrrolidines with two quaternary stereocenters via ligand-controlled copper(I)- catalyzed asymmetric 1,3-dipolar cycloadditions. J. Am. Chem. Soc. 140, 2272–2283 (2018).
Yuen, O. Y. & So, C. M. Ligand control of palladium-catalyzed site-selective α- and γ-arylation of α,β-unsaturated ketones with (Hetero)aryl halides. Angew. Chem. Int. Ed. 59, 23438–23444 (2020).
Wang, J., Wu, P., Wu, J., Mei, G. & Shi, F. Chemodivergent tandem cyclizations of 2-indolylmethanols with tryptophols: C-N versus C-C bond formation. J. Org. Chem. 83, 5931–5946 (2018).
Richmond, E. & Moran, J. Ligand control of E/Z selectivity in nickel-catalyzed transfer hydrogenative alkyne semireduction. J. Org. Chem. 80, 6922–6929 (2015).
Fu, S. et al. Ligand-controlled cobalt-catalyzed transfer hydrogenation of alkynes: stereodivergent synthesis of Z- and E-alkenes. J. Am. Chem. Soc. 138, 8588–8594 (2016).
Zhao, C. Q. et al. Water as a hydrogenating agent: stereodivergent Pd-catalyzed semihydrogenation of alkynes. Org. Lett. 21, 1412–1416 (2019).
Shen, R. et al. Facile regio- and stereoselective hydrometalation of alkynes with a combination of carboxylic acids and group 10 transition metal complexes: selective hydrogenation of alkynes with formic acid. J. Am. Chem. Soc. 133, 17037–17044 (2011).
Kusy, R. & Grela, K. E- and Z‑selective transfer semihydrogenation of alkynes catalyzed by standard ruthenium olefin metathesis catalysts. Org. Lett. 18, 6196–6199 (2016).
Yang, J. et al. Ligand-controlled iridium-catalyzed semihydrogenation of alkynes with ethanol: highly stereoselective synthesis of E- and Z-alkenes. Chem. Commun. 55, 1903–1906 (2019).
Kusy, R., Lindner, M., Wagner, J. & Grela, K. Ligand-to-metal ratio controls stereoselectivity: highly functional-grouptolerant, iridium-based, (E)-selective alkyne transfer semihydrogenation. Chem. Catal. 2, 1346–1361 (2022).
Li, J. & Hua, R. Stereodivergent ruthenium-catalyzed transfer semihydrogenation of diaryl alkynes. Chem. Eur. J. 17, 8462–8465 (2011).
Chen, T., Xiao, J., Zhou, Y., Yin, S. & Han, L. B. Nickel-catalyzed (E)-selective semihydrogenation of internal alkynes with hypophosphorous acid. J. Organomet. Chem. 749, 51–54 (2014).
Li, K. et al. Cobalt catalyzed stereodivergent semi-hydrogenation of alkynes using H2O as the hydrogen source. Chem. Commun. 55, 5663–5666 (2019).
Oger, C., Balas, L., Durand, T. & Galano, J. Are alkyne reductions chemo-, regio-, and stereoselective enough to provide pure (Z)-olefins in polyfunctionalized bioactive molecules? Chem. Rev. 113, 1313–1350 (2013). 3.
Crespo-Quesada, M., Cárdenas-Lizana, F., Dessimoz, A. & Kiwi-Minsker, L. Modern trends in catalyst and process design for alkyne hydrogenations. ACS Catal. 2, 1773–1786 (2012).
Frihed, T. G. & Fürstner, A. Progress in the trans-reduction and trans-hydrometalation of internal alkynes. applications to natural product synthesis. Bull. Chem. Soc. Jpn. 89, 135–160 (2016).
Huang, Z., Wang, Y., Leng, X. & Huang, Z. An amine-assisted ionic monohydride mechanism enables selective alkyne cis-semihydrogenation with ethanol: from elementary steps to catalysis. J. Am. Chem. Soc. 143, 4824–4836 (2021).
Luo, J. et al. Controlled selectivity through reversible inhibition of the catalyst: stereodivergent semihydrogenation of alkynes. J. Am. Chem. Soc. 144, 13266–13275 (2022).
Srimani, D. et al. Iron pincer complex catalyzed, environmentally benign, e-selective semi-hydrogenation of alkynes. Angew. Chem. Int. Ed. 52, 14131–14134 (2013).
Cortese, N. A. & Heck, R. F. Palladium-catalyzed reductions of α,β-unsaturated carbonyl compounds, conjugated denes, and acetylenes with trialkylammonium formats. J. Org. Chem. 43, 3985–3987 (1978).
Semba, K., Fujihara, T., Xu, T. H., Terao, J. & Tsuji, Y. Copper-catalyzed highly selective semihydrogenation of non-polar carbon-carbon multiple bonds using a silane and an alcohol. Adv. Synth. Catal. 354, 1542–1550 (2012).
Whittaker, A. M. & Lalic, G. Monophasic catalytic system for the selective semireduction of alkynes. Org. Lett. 15, 1112–1115 (2013).
Wang, D. & Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 115, 6621–6686 (2015).
Richards, E. M. & Tebby, J. C. Reactions of phosphines with acetylenes. Part VIII. Synthesis of 1,2-dideuteriated olefins. J. Chem. Soc. (C) 1542-1544 (1969).
Chou, W., Clark, D. L. & White, J. B. The use of rieke zinc metal in the selective reduction of alkynes. Tetrahedron Lett. 32, 299–302 (1991).
Kataoka, Y., Takai, K., Oshima, K. & Utimoto, K. Selective reduction of alkynes to (Z)-alkenes via niobium- or tantalum-alkyne complexes. J. Org. Chem. 57, 1615–1618 (1992).
Chen, Z., Luo, M., Wen, Y., Luo, G. & Liu, L. Transition-metal-free semihydrogenation of diarylalkynes: highly stereoselective synthesis of trans-alkenes using Na2S·9H2O. Org. Lett. 16, 3020–3023 (2014).
van Laren, M. W. & Elsevier, C. J. Selective homogeneous palladium(0)-catalyzed hydrogenation of alkynes to (Z)-alkenes. Angew. Chem. Int. Ed. 38, 3715–3717 (1999).
Wang, Y., Huang, Z. & Huang, Z. Catalyst as colour indicator for endpoint detection to enable selective alkyne trans-hydrogenation with ethanol. Nat. Catal. 2, 529–536 (2019).
Thiel, N. O., Kaewmee, B., Ngoc, T. T. & Teichert, J. F. A simple nickel catalyst enabling an E-selective alkyne semihydrogenation. Chem. Eur. J. 26, 1597–1603 (2020).
Murugesan, K. et al. Nickel-catalyzed stereodivergent synthesis of E- and Z-alkenes by hydrogenation of alkynes. ChemSusChem 12, 3363–3369 (2019).
Li, K. et al. Anion controlled stereodivergent semi-hydrogenation of alkynes using water as hydrogen source. Asian J. Org. Chem. 10, 2143–2146 (2021).
Sato, T., Shoji Watanabe, S., Kiuchi, H., Oi, S. & Yoshio Inoue, Y. Hydrogenation of olefins using water and zinc metal catalyzed by a rhodium complex. Tetrahedron Lett. 47, 7703–7705 (2006).
Muhammad, O., Sonavane, S. U., Sasson, Y. & Chidambaram, M. Palladium/carbon catalyzed hydrogen transfer reactions using magnesium/water as hydrogen donor. Catal. Lett. 125, 46–51 (2008).
Yan, M. et al. Nanoporous gold catalyst for highly selective semihydrogenation of alkynes: remarkable effect of amine additives. J. Am. Chem. Soc. 134, 17536–17542 (2012).
Cummings, S. P., Le, T. N., Fernandez, G. E., Quiambao, L. G. & Stokes, B. J. Tetrahydroxydiboron-mediated palladium-catalyzed transfer hydrogenation and deuteriation of alkenes and alkynes using water as the stoichiometric H or D atom donor. J. Am. Chem. Soc. 138, 6107–6110 (2016).
Xuan, Q. & Song, Q. Diboron-assisted palladium-catalyzed transfer hydrogenation of n-heteroaromatics with water as hydrogen donor and solvent. Org. Lett. 18, 4250–4253 (2016).
Kong, W., Wang, Q. & Zhu, J. Water as a hydride source in palladium-catalyzed enantioselective reductive heck reactions. Angew. Chem. Int. Ed. 56, 3987–3991 (2017).
Ojha, D. P., Gadde, K. & Prabhu, K. R. Generation of hydrogen from water: a Pd-catalyzed reduction of water using diboron reagent at ambient conditions. Org. Lett. 18, 5062–5065 (2016).
Flinker, M. et al. Efficient water reduction with sp3-sp3 diboron(4) compounds: application to hydrogenations, H–D exchange reactions, and carbonyl reductions. Angew. Chem. Int. Ed. 56, 15910–15915 (2017).
He, G. et al. Copper(I)-catalyzed highly regio- and stereoselective boron addition–protonolysis of alkynamides to give alkenamides. Eur. J. Org. Chem. 2021, 6979–6989 (2013).
Bao, H., Zhou, B., Jin, H. & Liu, Y. Diboron-assisted copper-catalyzed Z-selective semihydrogenation of alkynes using ethanol as a hydrogen donor. J. Org. Chem. 84, 3579–3589 (2019).
Han, X., Hu, J., Chen, C., Yuanb, Y. & Shi, Z. Copper-catalysed, diboron-mediated cis-dideuterated semihydrogenation of alkynes with heavy water. Chem. Commun. 55, 6922–6925 (2019).
Huang, J. et al. Substrate-controlled Cu(OAc)2‑catalyzed stereoselective semireduction of alkynes with MeOH as the hydrogen source. ACS Omega 6, 11740–11749 (2021).
James, T. L., Smith, D. M. & Holm, R. H. Stereoelectronic preferences in electron transfer series of nickel with tridentate ligands containing hard-soft donor sets. Inorg. Chem. 33, 4869–4877 (1994).
Kim, J. S., Reibenspies, J. H. & Darensbourg, M. Y. Characteristics of nickel(0), nickel(I), and nickel(II) in phosphino thioether complexes: molecular structure and S-dealkylation of (Ph2P(o-C6H4)SCH3)2Ni0. J. Am. Chem. Soc. 118, 4115–4123 (1996).
Silver, S. C. et al. Protein delivery of a Ni catalyst to photosystem I for light-driven hydrogen production. J. Am. Chem. Soc. 135, 13246–13249 (2013).
Zhang, C. P. et al. A five-coordinate nickel(II) fluoroalkyl complex as a precursor to a spectroscopically detectable Ni(III) species. J. Am. Chem. Soc. 135, 8141–8144 (2013).
Day, C. S., Somerville, R. J. & Martin, R. Deciphering the dichotomy exerted by Zn(II) in the catalytic sp2 C–O bond functionalization of aryl esters at the molecular level. Nat. Catal. 4, 124–133 (2021).
Sheng, J. et al. Diversity-oriented synthesis of aliphatic fluorides via reductive C(sp3)−C(sp3) cross-coupling fluoroalkylation. Angew. Chem. Int. Ed. 60, 15020–15027 (2021).
Krasovskiy, A. & Lipshutz, B. H. Ligand effects on Negishi couplings of alkenyl halides. Org. Lett. 13, 3818–3821 (2011).
Barber, E. R. et al. Nickel-catalyzed hydroarylation of alkynes under reductive conditions with aryl bromides and water. J. Org. Chem. 84, 11612–11622 (2019).
Hashimoto, T., Shiota, K. & Yamaguchi, Y. Selective synthesis of secondary alkylboronates: markovnikov selective hydroboration of vinylarenes with bis(pinacolato)diboron catalyzed by a nickel pincer complex. Org. Lett. 22, 4033–4037 (2020).
King, A. E. et al. GordonNi(bpy)(cod): a convenient entryway into the efficient hydroboration of ketones, aldehydes, and imines. Eur. J. Inorg. Chem. 1635-1640 (2016).
Kumar, G. R., Kumar, R., Manda, R. M. & Reddy, M. S. A nickel-catalyzed anti-carbometallative cyclization of alkyne–azides with organoboronic acids: synthesis of 2,3-diarylquinolines. Chem. Commun. 54, 759–762 (2018).
Eberhardt, N. A. & Guan, H. Nickel hydride complexes. C. hem. Rev. 116, 8373–8426 (2016).
Wang, W., Chao Ding, C. & Yin, G. Catalyst-controlled Enantioselective 1,1-arylboration of unactivated olefins. Nat. Catal. 3, 951–958 (2020).
Nattmann, L., Saeb, R., Nöthling, N. & Cornella, J. An air-stable binary Ni(0)-olefin catalyst. Nat. Catal. 3, 6–13 (2020).
Clarke, C., Incerti-Pradillos, C. A. & Lam, H. W. Enantioselective nickel-catalyzed anti-carbometallative cyclizations of Alkynyl electrophiles enabled by reversible alkenylnickel E/Z isomerization. J. Am. Chem. Soc. 138, 8068–8071 (2016).
Harinath, A., Angaa, S. & Panda, T. K. Alkali metal catalyzed dehydro-coupling of boranes and amines leading to the formation of a B–N bond. RSC Adv. 6, 35648–35653 (2016). For similar interaction of nitrogen-containing organic bases with B2pin2.
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. A. Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 37, 785–789 (1988).
Frisch, M. J. et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT (2009).
Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21602015) (Y.L.) and Talent Development Fund of Jilin Province (Y.L.) are gratefully acknowledged. We thank Prof. Ruben Martin at Institute of Chemical Research of Catalonia (ICIQ) for his insightful discussion and generous help on the mechanistic studies.
Author information
Authors and Affiliations
Contributions
Y.W. developed the method and carried out most of the chemical reactions. Y.A., C.L. and J.Z. participated in the mechanistic studies. Z.L. conducted the DFT calculations. W.G., X.L., H.W. and Y.S.L. supported the synthesis of substrates. Y.L. designed and supervised the project. Y.L. and Y.W. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiao-Ning Guo, Wei-Hua Mu and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Ao, Y., Li, Z. et al. Modulation of metal species as control point for Ni-catalyzed stereodivergent semihydrogenation of alkynes with water. Nat Commun 14, 1655 (2023). https://doi.org/10.1038/s41467-023-37022-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37022-w
This article is cited by
-
Synthesis of 4-(het)aryl- and 4-(het)arylamino-substituted styrenes
Russian Chemical Bulletin (2025)
-
Scalable deoxygenative alkynylation of alcohols via flow photochemistry
Communications Chemistry (2024)
