
Abstract
The sluggish kinetics of oxygen evolution reaction (OER) and high iridium loading in catalyst coated membrane (CCM) are the key challenges for practical proton exchange membrane water electrolyzer (PEMWE). Herein, we demonstrate high-surface-area nano-metal diborides as promising supports of iridium-based OER nanocatalysts for realizing efficient, low-iridium-loading PEMWE. Nano-metal diborides are prepared by a novel disulphide-to-diboride transition route, in which the entropy contribution to the Gibbs free energy by generation of gaseous sulfur-containing products plays a crucial role. The nano-metal diborides, TaB2 in particular, are investigated as the support of IrO2 nanocatalysts, which finally forms a TaOx/IrO2 heterojunction catalytic layer on TaB2 surface. Multiple advantageous properties are achieved simultaneously by the resulting composite material (denoted as IrO2@TaB2), including high electrical conductivity, improved iridium mass activity and enhanced corrosion resistance. As a consequence, the IrO2@TaB2 can be used to fabricate the membrane electrode with a low iridium loading of 0.15 mg cm−2, and to give an excellent catalytic performance (3.06 A cm−2@2.0 V@80 oC) in PEMWE―the one that is usually inaccessible by unsupported Ir-based nanocatalysts and the vast majority of existing supported Ir-based catalysts at such a low iridium loading.
Similar content being viewed by others

Introduction
Proton exchange membrane water electrolyzer (PEMWE) is an advanced hydrogen production technology that can achieve large current density (>1 A cm−2), high hydrogen purity (>99.99%), and fast response (<5 s) towards dynamic electricity input1,2,3. The heart of PEMWE is a catalyst coated membrane (CCM) composed of solid electrolyte (i.e., perfluorosulfonic membrane), cathode catalyst layer, and anode catalyst layer (Supplementary Fig. 1). Industrially, Pt and Ir-based noble metals are required to catalyze the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode, respectively4,5,6,7,8. Due to the scarcity and expensiveness of Pt and Ir, strategies that can decrease noble metal loading in CCM and simultaneously maintain good catalytic performance deserve to be strongly pursued. The successful method is to anchor catalyst nanoparticles on a high-surface-area conductive support. An effective support material can enhance the dispersion of catalyst nanoparticles, stabilize them from agglomeration and thus benefit the exposure of large active surface areas9,10,11,12. In the approach, Pt/C as HER catalyst is commercially employed to construct a cathode catalyst layer of CCM with a low loading of <0.5 mgPt cm−2 13,14,15. But the carbonous supports are not suitable for Ir-based OER nanocatalysts because of their instability in highly oxidizing environments. Thanks to the lack of suitable support materials, the Ir loading in CCM is usually as high as a few mg cm−2 16,17,18. Therefore, it is highly desirable to find non-carbonous support materials possessing good electrical conductivity, large specific surface area, and high resistance from acid corrosion and oxidative decomposition for realizing low-iridium-loading PEMWE.
To this end, we turn our attention to boride ceramics, which are generally characterized by high melting points, high hardness, high chemical resistance, and high electrical conductivity. A number of various borides can be produced and used as, for example, high-temperature structural materials, anti-oxidation coating materials and wear resistant materials in industry19,20,21,22,23,24,25. Despite their great potential, boride ceramics have not been investigated as the support of Ir-based OER nanocatalysts for constructing anode catalyst layer of CCM in PEM electrolyzer. In part, this is because some potential borides (e.g., TaB2) prepared with some existing methods are usually composed of micrometer-sized particles with low surface areas, and sometimes contain large amounts of defects and impurities26,27,28,29,30. Thus, synthesizing high-quality borides with large surface areas is an essential prerequisite for meeting our goal of making efficient boride-supported anode catalysts and applying them in low-iridium-loading PEMWEs.
Herein, we present the entropy-driven synthesis of high-surface-area nano-metal diboride from the corresponding disulphide, and demonstrate the practical usefulness of boride ceramics, especially TaB2, as the effective support material of anode nanocatalyst IrO2 in PEMWE. The TaB2-supported IrO2 nanocatalyst (denoted as IrO2@TaB2) possesses a large electrical conductivity (0.1–0.2 S cm−1) similar to nano-IrO2 itself, shows about 10 times higher iridium mass activity than IrO2, and exhibits much lower iridium leaching than IrO2 during electrocatalysis. When integrated into a PEM single-cell electrolyzer, the IrO2@TaB2 can achieve 3.06 A cm−2 current density at 2.0 V and 80 °C (membrane: NafionTM N115), with a low iridium-loading of 0.15 mg cm−2. The performance of our PEMWE has reached that of US DOE 2023 target (1.9 V@2.5 A cm−2, platinum group metal (PGM) loading of 1 mg cm−2), under a much lower PGM loading (0.15 mgIr cm−2 and 0.27 mgPt cm−2).
Results
Fabrication and characterizations of nano-metal diborides
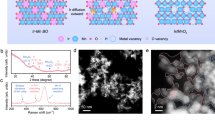
We achieve the structural transformation of metal disulphide into metal diboride (MS2 + B → MB2 + Sx) under a KCl–CsCl molten salt condition (Fig. 1a and Supplementary Table 1). The gaseous sulfur-containing products may consist of S2, S4, S8, and B2S3 at the reaction temperatures (1000–1100 °C), while S2 is considered to be the most abundant species31. We calculate the reaction enthalpy and entropic term to determine the Gibbs free enthalpy of the chemical reaction (ΔG = ΔH − TΔS). As exhibited in Fig. 1b and Supplementary Table 2, the entropic term TΔS of the gaseous sulfur-containing products is the dominant part of the total Gibbs energy. After reaching a certain temperature, the entropy gain would drive the reaction from a thermodynamically unfeasible process to a thermodynamically spontaneous process. According to the entropy-driven route, we can synthesize nine monometallic diborides, including MB2 (M = Ti, Zr, Hf, V, Nb, Ta, Mo, W, and Re). The powder X-ray diffraction (XRD) patterns in Fig. 1c confirm that the majority of as-synthesized samples are pure transition metal diboride phases. The morphologies of these borides are revealed by scanning electron microscopy (SEM, Supplementary Fig. 2). TaB2 and MoB2 possess a typical nanosheet structure, WB2 shows a nanowire morphology, and other MB2 samples (M = Ti, Zr, Hf, V, Nb, and Re) are composed of nanoparticles with sizes ranging from 50 to 200 nm. Most of these MB2 samples possess large Brunauer–Emmett–Teller (BET) surface areas within the range of 40–60 m2 g−1 (Fig. 1d and Supplementary Table 3), which are obviously higher than those of borides obtained by conventional high-temperature ceramic method32,33,34,35,36.

a A schematic presenting the synthesis and crystal structure of TaB2. b Temperature-dependent Gibbs free energy of the synthesis process of metal diborides. c XRD patterns of as-synthesized diborides samples. For comparison, the Joint Committee on Powder Diffraction Standard (JCPDS) cards of these metal diborides are included. d N2 adsorption-desorption isotherms of diborides samples. e TEM image and (f) HRTEM image of TaB2. The inset of Fig. 1f shows SAED pattern of TaB2.
We have compared the acid corrosion resistance and BET surface area of nine metal diborides synthesized above. Compared with TaB2, the TiB2, ZrB2, HfB2, VB2, CrB2, MoB2, and WB2 show more severe metal dissolution in acid (Supplementary Table 3). After the exposure to 0.5 M H2SO4 over a week (Supplementary Fig. 3), almost no Ta species are leached in the acidic solution, and TaB2 retains its pristine crystal structure well. These results imply the outstanding chemical stability of TaB2 in acid. In addition, TaB2 presents much higher BET surface area than HfB2, VB2, NbB2, and ReB2 (Supplementary Fig. 3). From the crystal structure perspective, the TaB2 comprises 3D metallic Ta–Ta framework and 2D graphene-like boron layers (also known as borophene subunits), suggesting a fast electron transport property37. Experiments present TaB2 has a high conductivity of 25.2 S cm−1 (Supplementary Fig. 4), which is significantly larger than those of previously-reported supports of IrO238,39,40,41, such as TiN (3.9 S cm−1), TaC (0.65 S cm−1) and TiO2 (1.8 × 10−4 S cm−1). Given its multi-advantages including large BET surface areas, great acid corrosion resistance, and high conductivity, the nano-TaB2 is specially selected to explore the potential as support of IrO2 catalysts. Moreover, the crust abundance of Ta is five orders of magnitude higher than that of Ir (Supplementary Table 4), and Ta costs around 2‰ of the Ir price ($367.5 for Ta vs. $164,662.0 for Ir per kilogram in 2023). Hence, the introduction of TaB2 supporting material is expected to reduce the cost of the anode catalyst layer and improve the feasibility of PEMWE.
We further characterized the TaB2 sample with SEM and transmission electron microscopy (TEM). The SEM (Supplementary Fig. 2f) and TEM images (Fig. 1e) present hat TaB2 nanosheets possess an edge length of 200–500 nm and a thickness of 5–20 nm. High-resolution TEM image (HRTEM) of TaB2 nanosheet (Fig. 1f) reveals its good crystallinity. In the HRTEM image and the corresponding selected-area electron diffraction (SAED) pattern (Fig. 1f, inset), interplanar spacing of 0.268 nm could be observed for three sets of lattice fringes, assigning to {001} planes of TaB2 phase. The angle of 60o between the three planes is consistent with the theoretical value. Moreover, N2 adsorption-desorption isotherms of TaB2 display typical type-II curves with H3 hysteresis loop (Fig. 1d), suggesting the presence of interlaminar pores among adjacent nanosheets. The BET surface area of TaB2 is calculated to be 54.6 m2 g−1. These results demonstrate the successful synthesis of high-quality TaB2 nanosheet with large surface areas.
Loading IrO2 nanocatalysts on boride supports
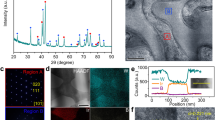
The loading of IrO2 nanocatalysts on TaB2 supports (IrO2@TaB2) is achieved through calcinating a mixture of K2IrCl6 and TaB2 in molten NaNO3 (i.e., 350 °C). The loading amounts of IrO2 can be controlled by changing the feeding of the iridium source. When TaB2 is not employed in the synthesis, pure IrO2 nanoparticles with a size of 1.4–2.0 nm are formed (Supplementary Fig. 5 and 6). A series of IrO2@TaB2 samples with Ir contents of 4 wt%, 10 wt%, 16 wt%, 25 wt%, and 40 wt% are synthesized, and their iridium contents are quantified by inductively coupled plasma atomic emission spectroscopy (ICP-OES). The XRD patterns of these IrO2@TaB2 samples are shown in Fig. 2a, with those of TaB2 and unsupported IrO2 as references. The diffraction peaks of TaB2 supports dominate the XRD patterns of IrO2@TaB2. In addition, a broadened peak at 30-40o can be attributed to IrO2, suggesting the very small size of IrO2. With the increase in IrO2 contents on TaB2 supports, the diffraction intensities of IrO2 increase gradually.

a XRD patterns, (b) Ta4f XPS spectra, (c) B1s XPS spectra, and (d) Ir4f XPS spectra of IrO2@TaB2 samples with different IrO2 loading. e Ir L1-edge XANES spectra of 16 wt% IrO2@TaB2 and IrO2. f Fourier transforms of the EXAFS spectra of 16 wt% IrO2@TaB2 and IrO2.
These IrO2@TaB2 samples are further characterized by X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS). Figure 2b shows Ta4f XPS spectra of IrO2@TaB2 samples and the reference TaB2. For TaB2, the surface comprises both metallic Ta elements and oxidized Ta elements, indicating slight surface oxidation. Unlike that of TaB2, the surface layers of IrO2@TaB2 samples are fully dominated by oxidized Ta elements, suggesting further surface oxidation of Ta species during the IrO2 loading process. With increasing IrO2 content in the samples, the signal intensities of Ta4f XPS decrease gradually, implying the decrease of surface Ta concentration. Similarly, the surface B concentration also decreases with increasing IrO2 content and is almost undetectable after the 16% Ir content (Fig. 2c). Based on the Ta, B and Ir XPS signal intensities (Fig. 2b–d), we conclude that the Ta and B species dominate the surfaces of IrO2@TaB2 samples with the Ir loading content ≤10%; when the Ir loading content >10%, the Ta and B signal intensities are very weak and the Ir species dominate the surfaces, indicating a dense coating of IrO2 formed at the support surface. In addition, the Ir4f XPS peaks of IrO2@TaB2 samples (Fig. 2d) present an obvious shift to larger binding energy relative to those of IrO2, and the deviation increases with the decrease in IrO2 contents on TaB2 supports. This indicates a slightly lower Ir oxidation in IrO2@TaB2 due to the electronic interaction between IrO2 and support. The electronic interaction and Ir oxidation state can be flexibly regulated by the Ir loading content.
The charge redistribution of IrO2 is also supported by X-ray absorption near-edge structure (XANES) spectra. We note that we chose 16 wt% IrO2@TaB2 as a representative sample, which exhibits an optimized iridium mass activity toward OER in a standard three-electrode cell. Unless otherwise specified, subsequent IrO2@TaB2 refers to the sample with 16 wt% Ir loading. We also note that we chose Ir L1-edge for the XANES measurements, considering that the Ir L3-edge overlaps with the Ta L2 and Ta L1-edges. Figure 2e shows the Ir L1-edge XANES spectra of IrO2@TaB2 and IrO2. Compared to that of IrO2, the white-line energy position of IrO2@TaB2 has a slightly weaker peak intensity, indicating an increased occupation in 5d electron orbital and a relatively lower Ir oxidation state in IrO2@TaB2. Moreover, extended X-ray absorption fine structure (EXAFS) exhibits similar structural information of IrO2@TaB2 and IrO2, and the corresponding fitting results (Fig. 2f and Supplementary Table 5) show that both the Ir-Ir and Ir-O bond distances in IrO2@TaB2 are close to those in IrO2. According to the above XPS and XAS results, we can reasonably conclude that: (i) the lattice of IrO2 has almost unchanged after loading on TaB2; (ii) however, the interfacial electronic interaction between IrO2 and support leads to the charge redistribution of IrO2.
The aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) was applied to investigate IrO2@TaB2 sample. As shown in Fig. 3a, the IrO2 nanoparticles are uniformly and tightly dispersed on the TaB2 sheets. The main sizes of IrO2 are about 1.5 nm (Supplementary Fig. 6). The energy dispersive X-ray spectrum (EDX) elemental mappings of IrO2@TaB2 sample in Fig. 3b and Supplementary Fig. 7 confirm that Ta and B elements show sheet morphology, while Ir and O elements are homogeneously distributed in supports. The side view of TEM image (Fig. 3c) reveals that an around 5 nm surface coating of IrO2 nanocrystallites is formed on TaB2 supports. The amorphous filler among IrO2 nanocrystallites can be attributed to TaOx layer on TaB2 surfaces. Both the side view and top view of TEM images (Fig. 3c, d) show that the IrO2 nanocrystallites are randomly but spatially interconnected. The HRTEM image (Fig. 3e) presents lattice fringes of the (11\(\bar{1}\)) plane (~0.224 nm) and (020) plane (~0.225 nm) of IrO2, respectively. The angle between the two planes is determined as 59.1o, equal to the theoretical value.

a The TEM image of IrO2@TaB2. b The elemental mapping images of IrO2@TaB2. c, d The side view and top view of aberration-corrected HAADF-STEM images of IrO2@TaB2. e High-resolution HAADF-STEM image of IrO2@TaB2. f Schematic illustration of microstructure of IrO2@TaB2.
Based on the above electron microscopy, electron diffraction, and spectroscopic evidence, the structural illustration of IrO2@TaB2 sample is shown in Fig. 3f. First, the surface of TaB2 support tends to form amorphous TaOx layer. Second, the IrO2 nanocrystallites are uniformly and tightly dispersed on the supports to form 5 nm thick IrO2 layers. Third, the IrO2 nanocrystallites with intrinsic conductive properties are spatially interconnected, and the IrO2 is electrically connected to the metallic TaB2 support, ensuring the formation of conductive networks. Fourth, the interfacial electronic coupling in TaOx/IrO2 catalytic layer results in the valence state reduction of Ir atoms.
Electrocatalytic performance in a three-electrode configuration
The electrocatalytic activities toward OER of IrO2@TaB2 were measured using a standard three-electrode configuration in 0.1 M HClO4 electrolyte. As the IrO2 content increases, the electrocatalytic activity for OER increased rapidly (Fig. 4a), and reaches its optimization when the Ir content is 16 wt%. However, a further increase in the Ir content of the IrO2@TaB2 results in a slight deterioration of the electrocatalytic activity. The required overpotential to generate a current density of 10 mA cm−2 (normalized over the electrode geometric area) for 16 wt% IrO2@TaB2 is 288 mV, lower than that of unsupported IrO2 (307 mV). The Tafel slope of 16 wt% IrO2@TaB2 is 42.6 mV dec−1 (Supplementary Fig. 8), close to that of unsupported IrO2 (45.1 mV dec−1), indicating that the the TaB2 support has no change to OER pathway of IrO2.

a The polarization curves toward OER in the presence of IrO2@TaB2 and IrO2 in 0.1 M HClO4. b Comparison of ECSA and current densities (jECSA) normalized by ECSAs for IrO2@TaB2 and IrO2. c Comparison of iridium mass activities (jIr) of Ir-based electrocatalysts at 1.53 V versus RHE40,42,43,44,45,46,47,48,49,50,51,52. d Chronopotentiometric curves of IrO2@TaB2 and IrO2 with a current density of 10 mA cm−2. e Contents of leached iridium in the electrolyte in the presence of IrO2@TaB2 and IrO2 during electrocatalysis. f The polarization curves for OER of IrO2@TaB2 and IrO2 in HClO4 electrolyte with different pH. g pH dependence of IrO2@TaB2 and IrO2 on the OER potential on the SHE scale. h DEMS signals of O2 products for 18O-labeled IrO2@TaB2 in 0.1 M HClO4 in H216O.
Electrochemical double layer capacitance (DLC) measurements were carried out to obtain the electrochemically active surface area (ECSA) of IrO2@TaB2 by estimating the accumulated charge amount at the electrode surface. Generally, the ECSA values become larger with increasing Ir content in IrO2@TaB2 (Fig. 4b, left and Supplementary Table 6). In order to compare the intrinsic activities of different catalysts, we normalized the measured currents by the ECSAs (Fig. 4b, right). The 16 wt% IrO2@TaB2 presents the highest current density (jECSA) at 1.53 V versus reversible hydrogen electrode (RHE) among these IrO2@TaB2 samples, which is 5.2 times larger than that of IrO2. We further compared the iridium mass activities of IrO2@TaB2 and IrO2 by normalizing the measured currents over the mass of iridium (jir). Although 16% IrO2@TaB2 has 70 wt% less iridium relative to IrO2, it displays a high iridium mass activity of 345 A g−1 at 1.53 V versus RHE (Fig. 4c and Supplementary Table 7), which is an order of magnitude higher than that of IrO2. The iridium mass activity of 16 wt% IrO2@TaB2 is superior to some recently-reported iridate electrocatalysts (e.g., 3C-SrIrO3, 6H-SrIrO3, Sr2IrO4, K0.25IrO2)42,43,44,45,46,47 and those representative supported catalysts (e.g., IrO2-TiO2, IrO2@Ir/TiN, IrO2/Nb0.2Ti0.8O2)40,48,49,50,51,52.
Besides high activity, IrO2@TaB2 displays great catalytic and structural stability for acidic OER. The galvanostatic measurements (Fig. 4d) demonstrate that IrO2@TaB2 remains steadily catalytic activity for more than 120 h of continuous operation at a current density of 10 mA cm−2, while IrO2 lost its catalytic activity after 40 h in the acidic electrolyte. Supplementary Fig. 9 exhibits the polarization curve for OER obtained from IrO2@TaB2 and IrO2 before and after 5000 cycles. IrO2@TaB2 presents no measurable loss of catalytic activity after the continuous polarization measurements, while IrO2 suffers from a severe deactivation. These results suggest that TaB2 support greatly enhances the catalytic stability of IrO2. Moreover, IrO2@TaB2 achieves a faradaic efficiency of nearly 100% during acidic OER (Supplementary Fig. 10), confirming that the observed current can be entirely attributed to the oxygen generation.
The leached amounts of cations during OER of IrO2@TaB2 and IrO2 were quantitatively determined by ICP-OES (Fig. 4e). There are no detectable leached Ta and B species for IrO2@TaB2 in the electrolyte, and the Ir dissolutions increase during the first two hours and then reaches a stable state. The constant Ir concentration for IrO2@TaB2 in the electrolyte is 0.25 mg L−1, which is much weaker than that for IrO2 (0.4–0.7 mg L−1), suggesting that the dispersion of IrO2 on TaB2 supports markedly improves the structural stability. The stability of IrO2@TaB2 is further evaluated by calculating the stability number (i.e., S-number proposed by Gieger et al.)53, which is a good metric that relates the amount of evolved oxygen to the dissolved iridium. As shown in Fig. 4e (inset), the S-number of IrO2@TaB2 during the acidic OER is 5.2 × 104, which is higher than that of IrO2 (2.9 × 104). In addition, IrO2@TaB2 still maintains the initial morphology and structure after OER, as supported by HRTEM, XRD, and XPS results (Supplementary Fig. 11–13). These results overall confirm the excellent electrocatalytic and structural stability of IrO2@TaB2 toward acidic OER.
Catalytic mechanism and origin of high activity
To investigate the catalytic mechanism, OER performance of IrO2@TaB2 and IrO2 were examined in HClO4 electrolytes with different pH. The catalytic activities of IrO2@TaB2 and IrO2 show strong pH dependence in the pH range of 0–1.5 on the standard hydrogen electrode (SHE) scale (Fig. 5a). The potential–pH dependence value of IrO2@TaB2 is −57.3 mV dec−1, which is near to that of IrO2 (−58.0 mV dec−1, Fig. 5b). Such a near −60 mV dec−1 Nernstian potential shift indicates a proton coupled electron transfer (CPET) process54, and the OER at Ir sites strictly follows the conventional adsorbates evolution mechanism (AEM) pathway. In addition, we applied in situ differential electrochemical mass spectrometry (DEMS) to study the reaction mechanisms. The O source of evolved oxygen product can be identified through labeling of catalysts with 18O isotope. When the 18O-labeled IrO2@TaB2 electrocatalyst works in H216O electrolyte, 34O2 (or 16O18O) to 32O2 ratio in gaseous product is 0.43% (Fig. 5c). It should be noted that the natural stable abundance of 18O isotope is about 0.2% in water55,56, indicating the minimum detected amount of 34O2 in OER products is about 0.4%. We further carried out DEMS measurement for 18O-labeled IrO2 catalyst under the same condition, the 34O2/32O2 intensity ratio from the reaction products is also 0.43% (Supplementary Fig. 14). These results overall demonstrate that like that of well-known IrO2 catalysts, the OER mechanism of IrO2@TaB2 catalyst strictly undergoes the adsorbate evolution mechanism (AEM), excluding the participation of lattice oxygen during the electrocatalysis.

a Stack structure and key materials of a PEMWE. b Cross-section SEM image of CCM employing 40% IrO2@TaB2 anode layer and 40% Pt/C cathode layer. c Polarization curves of PEMWEs using IrO2@TaB2 and IrO2 anodes at 80 °C with Nafion 115 membrane. d Comparison of current densities of PEM electrolyzers using different iridium-based catalysts at a cell potential of 1.9 V16,18,60,61,62,63,64,65,66,67,68,69. e Chronopotentiometry curve of PEMWEs using IrO2 and IrO2@TaB2 anodes operated at 1 A cm−2. f, g EIS curves of PEMWEs using IrO2@TaB2 and IrO2 anodes. h The comparison of ohmic resistance, activation resistance, and diffusion resistance for PEMWEs using IrO2@TaB2 and IrO2 anodes. The inset shows EEC model for EIS fitting.
After determining the catalytic mechanism, we sought to explain why IrO2@TaB2 possesses high activity for OER. Given that the crystal lattice and catalytic mechanism of IrO2 have almost no change after loading on TaB2, the enhanced performance of IrO2@TaB2 for OER is interpreted as follows. (i) The average particle sizes of IrO2@TaB2 and IrO2 are estimated to be 1.5 nm and 1.8 nm (Supplementary Fig. 6). Confinement of IrO2 nanoparticles on the TaB2 supports reduces IrO2 size, which provides more active sites to improve OER activity. (ii) The interfacial electronic coupling in TaOx/IrO2 catalytic layer is responsible for the high intrinsic activity of IrO2@TaB2. A metal−semiconductor heterojunction is constructed between TaOx and IrO2 on TaB2 surface, resulting in the formation of a surface electric field and strong electronic interaction (Supplementary Fig. 15a). The electrons will flow from the conduction band of TaOx to IrO2, driven by the work function differences, leading to electron-rich IrO2. The antibonding states of IrO2 are more fully occupied by electrons in the IrO2-TaOx heterojunction, which lowers the surface oxygen adsorption of IrO2 and consequently boosts the OER activity (Supplementary Fig. 15b, c). We note that the chemical and structural complexity of IrO2@TaB2 catalysts, including the uncertain surface structures of IrO2 nanoparticles (without specific exposed facets), amorphous TaOx layer, and their heterointerface, makes it unrealistic to establish a clear structural model of the catalyst. This limits us from accurately modeling the electrochemical processes of OER and quantitatively describing catalytic activity by advanced calculation methods (e.g., grand-canonical DFT)57.
Performance of PEMWE devices
The performance of as-prepared IrO2@TaB2 as an anode catalyst was finally evaluated on a real PEM electrolyzer (Fig. 5a). Unlike the three-electrode configuration to mainly reflect the performance of the catalyst itself, the PEMWE requires excellent electrical conductivity of catalyst (greater than 0.1 S cm−1) to deliver high current densities of several A cm−2 3,4. The electrical conductivity of IrO2@TaB2 becomes higher as the IrO2 content increases (Supplementary Fig. 16), and up to 0.17 S cm−1 for the 40% IrO2@TaB2 sample, which is similar to the conductivity of IrO2 itself (0.18 S cm−1). The structure and composition characterizations of the 40%IrO2@TaB2 sample by ICP-OES, XRD, XPS (Fig. 2a–c), and TEM (Supplementary Fig. 17) confirm that there is no essential difference between 40%IrO2@TaB2 and 16%IrO2@TaB2 sample, except for higher Ir loading of the former. Hence, the 40% IrO2@TaB2 sample is employed as the optimal anode catalyst to integrate into PEMWE. The Nafion 115 membrane is used as the PEM, which is a perfluorosulfonic polymer with a thickness of 125 μm. The CCM with a 5 cm2 working area (Supplementary Fig. 18) employing 40% IrO2@TaB2 anode layer and 40% Pt/C cathode layer is produced by a decal transfer method. SEM images in Fig. 5b and Supplementary Fig. 19 exhibit the cross-section and top view morphology of the CCM. Both the catalyst particles of IrO2@TaB2 anode and Pt/C cathode are composed of uniformly distributed agglomerates on the membrane surface. The thickness of both anode and cathode catalyst layers on the membrane is roughly 5 μm. As analyzed by ICP-OES, the CCM contains a low Ir loading of 0.15 mg cm−2 at the anode layer and a low Pt loading of 0.27 mg cm−2 at the cathode layer (Supplementary Fig. 20), respectively. The CCM shows lower total loadings of PGMs (0.42 mg cm−2) than the DOE 2023 target (1.0 mg cm−2) and even the DOE 2025 target (0.5 mg cm−2)58.
Even such low noble metals used in our CCM, the polarization curve of PEMWE reveals a current density of 3.06 A cm−2 with a cell potential of 2.0 V, operating at 80 °C and ambient pressure (Fig. 5c). It is surprising that the performance of our PEMWE has reached that of US DOE 2023 target (1.9 V@2.5 A cm−2), under such low PGM loadings (0.15 mgIr cm−2 and 0.27 mgPt cm−2). We note that the traditional CCMs in commercial PEMWE achieve reasonable activity and stability using a high Ir loading of 2–4 mgIr/cm2 to ensure sufficient in-plane conductivity and mechanical stability of catalyst layer59. In fact, the performance of PEMWE using IrO2@TaB2 as anode electrocatalyst exceeds the most recent reports of PEMWEs using novel anode electrocatalysts (e.g., Sr2CaIrO6, Ta0.1Tm0.1Ir0.8O2-δ, Ir@WOx)16,18,60,61,62,63,64,65,66,67, as shown in Fig. 5d and Supplementary Table 8. Even if several supported catalysts (e.g., Ir@B4C, Ir@Nb2O5-x) have reported to reach the DOE 2023 target, they require several times higher iridium loadings68,69.
We further compare the performance of PEMWEs using IrO2@TaB2 and commercial IrO2 as anode electrocatalysts (Fig. 5c), under the similar Ir loading in CCM. While IrO2 employing as anode layer, the PEMWE delivers a significantly lower current density of 1.9 A cm−2 relative to IrO2@TaB2 catalyst (3.06 A cm−2) at 2. 0 V cell potential. To evaluate catalyst stability, the IrO2 and IrO2@TaB2 anode cells were tested at a constant current density of 1 A cm−2. The PEMWE using IrO2 anode undergoes a severe deactivation under the low Ir loading of 0.2 mg cm−2, and the IrO2 anode cell exhibits reasonable stability when the Ir loading increases to 2 mg cm−2 (Fig. 5e and Supplementary Fig. 21–24). By comparison, the PEMWE using IrO2@TaB2 anode provides a steady operation for more than 120 h under a low Ir loading of 0.15 mg cm−2 (Fig. 5e), confirming excellent catalytic stability. We can speculate that the IrO2@TaB2 with larger material volume can forms a thicker catalyst layer relative to bare IrO2, so that the former possesses sufficient layer conductivity and stability at such a low Ir loading. The great activity and stability demonstrate the great potential of IrO2@TaB2 as a practical anode of real PEMWE for commercial application.
To deduce the origin of performance differences, we carry out chemical impedance spectroscopy (EIS) experiments on PEMWEs using IrO2@TaB2 and IrO2 as anode electrocatalysts, respectively (Fig. 5f, g). We further fit the EIS Nyquist plots by equivalent electrical circuit (EEC) model (Fig. 5h, inset), which shows good accordance with the experimental results. The total losses in a PEMWE mainly compose of ohmic resistance, activation resistance, and diffusion resistance70,71. (i) The Rcell denotes the cell ohmic resistance, which is related to ohmic losses of all components including membrane, catalyst layers, porous transport layers (PTL), bipolar plate, and their interfacial resistances. The RCell is almost unaffected at different current densities for IrO2@TaB2 anode cell, which can be reflected by high frequency resistance (HFR, Supplementary Fig. 25). The IrO2@TaB2 anode cell shows a slight decrease in ohmic resistance relative to the IrO2 anode cell. (ii) The Rac denotes the activation resistance to determine the reaction kinetics of anode and cathode electrocatalysts. Rac is sensitive to current density and is mainly contributed from the anode. The Rac of IrO2@TaB2 anode cell is significantly reduced compared with that of IrO2 anode cell, due to the much better catalytic performance IrO2@TaB2 relative to IrO2. (iii) The Rd denotes the diffusion resistance, as reflected by the Warburg diffusion element (Wd). Rd is negligible at a low current density and increases with the increase of current density. The IrO2@TaB2 anode cell presents much lower transport loss compared with the IrO2 anode cell. Taken overall, it can be concluded that the IrO2@TaB2 anode cell exhibits simultaneous decreases in both ohmic, activation, and diffusion losses relative to the IrO2 anode cell.
Discussion
In conclusion, we have demonstrated an entropy-driven disulphide-to-diboride transition route strategy that can be operated to synthesize large-surface-area metal diborides as the promising support of IrO2 nanocatalysts. As a demonstration, we have prepared TaB2-supported IrO2 nanocatalyst (i.e., IrO2@TaB2), which displays uniformly distributed IrO2 nanoparticles (∼1.5 nm), highly conductive networks, and great acidic corrosion resistance. Impressively, the as-prepared IrO2@TaB2 catalysts with an optimized IrO2 content exhibit a 10 times higher iridium mass activity than IrO2, while showing lower iridium leaching during acidic OER. When integrated into a PEMWE, the IrO2@TaB2 anode cell requires 2.0 V to attain 3.06 A cm−2 (reaching the US DOE 2023 target), under a much lower PGM loadings of 0.15 mgIr cm−2 and 0.27 mgPt cm−2 than DOE 2023 target (PGM loading of 1 mg cm−2). Our findings highlight the rational design of highly conductive, corrosion resistance, and large surface area materials as IrO2 supports for acidic OER, and stimulate practical applications of low-iridium-loading anode catalyst layers in industrial PEMWE.
Methods
Chemicals and reagents
Absolute ethanol (C2H6O), isopropyl alcohol ((CH3)2CHOH) and NaNO3 were purchased from Sinopharm Chemical Reagent Co., Ltd. TiS2, ZrS2, HfS2, VS2, NbS2, TaS2 and ReS2 were purchased from Nanjing MKNANO Tech. Co., Ltd. MoS2 and WS2 were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. HClO4, was purchased from Tianjin Xinyuan Chemical Co., Ltd. Nafion® perfluorinated resin solution was purchased from Sigma-Aldrich. K2IrCl6 and IrO2 were purchased from Shanghai Macklin Biochemical Co., Ltd. Pt/C (Pt 40 wt%) was purchased from Johnson Matthey Company. All the chemicals and reagents were used without further purification. Highly purified water (>18 MΩ cm resistivity) was obtained from a PALL PURELAB Plus system.
Materials synthesis
The metal diborides were synthesized by borothermal reduction in a molten salt medium (KCl-CsCl) with an equal molar ratio. The metal disulfide, boron powder, and molten salt are ground thoroughly in a mortar for 0.5 h. Then the mixture was transferred into a corundum boat and heated at 900–1100 °C under Ar atmosphere in a tubular furnace, and the heating rate was 10 °C/ min (Supplementary Table 1). After thorough cooling, the mixture was repeatedly washed with water and ethanol to remove soluble molten salts and byproducts. The final prepared metal diborides were dried at 80 °C for 12 h.
For the Preparation of IrO2@TaB2, K2IrCl6 was dissolved in isopropyl alcohol, then NaNO3 and prepared TaB2 support were added to the solution. The suspension was ultrasonically dispersed for 30 min and then dried at 90 °C for 5 h. Then the mixed powder was ground and heated in a muffle furnace at 5 °C/min to 350 °C for 1 h. After naturally cooling down, the product was washed fully with ethanol and water to remove residual salts, and the black catalyst powder was dried in an oven at 90 °C for 12 h to get IrO2@TaB2.
For the preparation of unsupported IrO2, the experimental process is the same as that for the preparation of IrO2@TaB2, except that TaB2 support was not added.
Characterizations
XRD patterns were conducted by a Rigaku D/Max 2550 X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). SEM images were obtained with field emission scanning electron microscopy (FESEM, JEOL 7800 F) at an accelerating voltage of 5 kV. EDX analysis was obtained with an EDX system attached to JEOL JSM7800F SEM. Low-magnification transmission electron microscope (TEM) images were obtained with a Philips-FEI Tecnai G2S-Twin microscope equipped with a field emission gun operating at 200 kV. High-resolution TEM images were recorded on a JEM-2100F electron microscope (JEOL, Japan). XPS measurements were carried out using a Thermo Fisher Scientific ESCALAB 250Xi with photoelectron spectroscopy system using a monochromatic Al Ka (1486.6 eV) X-ray source. ICP-OES was performed on a PerkinElmer Optima 3300DV ICP spectrometer. XAS (Ir L1-edge) were collected at BL14W beamline in Shanghai Synchrotron Radiation Facility (SSRF). The storage rings of SSRF were operated at 3.5 GeV with a stable current of 200 mA. Using Si (111) double-crystal monochromator, the data collection was carried out in fluorescence mode using Lytle detector. All spectra were collected in ambient conditions. Data reduction, data analysis, and EXAFS fitting were performed with the Athena and Artemis software packages. The energy calibration of the sample was conducted through a standard IrO2, which as a reference was simultaneously measured. For EXAFS modeling, EXAFS of the IrO2 is fitted and the obtained amplitude reduction factor S02 value (0.900) was set in the EXAFS analysis to determine the coordination numbers (CNs) in the Ir-O/Ir/Ta scattering path in the sample.
Electrochemical measurements in a three-electrode configuration
The performances of catalysts were studied in a standard three-electrode system by a CH Instrument (Model 660E) and the electrolyte was 0.1 M HClO4 solution bubbled with O2 gas. Linear sweep voltammetry (LSV) measurements were performed with the scan rate of 1 mV s−1 and compensated by 85% iR-drop. The chronopotentiometric curve was measured without iR-drop compensation. The counter electrode was a Pt wire and the reference electrode was a saturated calomel electrode (SCE). The potential was normalized with respect to the reversible hydrogen electrode (RHE), according to Eq. (1):
where 0.249 eV vs. the SCE electrode is the potential of zero net current. The SCE reference electrode was calibrated before testing according to the method proposed by Boettcher and co-workers72.
The working electrode was a glassy carbon electrode (GCE) loaded with catalysts. 4 mg catalyst powder was ultrasonically dispersed in 400 μL isopropyl alcohol containing 10 μL Nafion solution (5 wt.%). Then 2 μL catalyst ink was dropped on a GCE (Active area = 0.071 cm2) and dried in air. The catalyst loading on the working electrode was 0.28 mg cm−2. To avoid the passivation of GCE, a piece of carbon paper (Active area = 0.09 cm2) loaded with catalysts was used as the working electrode for long-term stability test, and the catalyst loading was 0.3 mg cm−2. In order to accurately evaluate the ion leaching and structural evolution behavior of the catalyst during the OER process, the catalyst ink was dropped onto the surface of a Ti plate (Active area = 1 cm2) with a catalyst loading of 10 mg cm−2. OER was catalyzed for 10 h at 10 mA cmgeo−2, and a portion of the electrolyte was taken out every 2 h for ICP-OES. After the catalysis, the catalyst sample was obtained by eluting the electrode with ethanol.
To calculate jgeo of catalysts, the measured current was normalized by the geometric area of GCE according to the Eq. (2):
where i is the measured current, and s is the geometric area of GCE.
To calculate jIr of catalysts, the measured current was normalized by the mass of iridium supported on GCE according to the Eq. (3):
where m is the loading mass of catalysts on GCE, and Ir (wt.%) is the mass fraction of iridium in catalysts.
To calculate ECSA of catalysts, we performed CV tests in the non-faradaic current region between 0.83 V and 0.93 V vs. RHE at different scan rates (10, 25, 50, 75, and 100 mV s−1). A linear trend could be obtained via plotting the currents difference (Δi) between the anodic and cathodic sweeps (ianodic − icathodic) at 0.88 V against the scan rate. The slope of the fitting line was equal to twice the geometric double layer capacitance (Cdl). The ECSA of the catalyst was estimated according to the Eq. (4):
where Cs represents the specific capacitance of the catalyst (0.06 mF cm−2).
To calculate jECSA of catalysts, we normalized the meatured current by the ECSA from Eq. (5):
For estimation of Faradaic efficiency, a 2 h-long measurement was performed to investigate the Faradaic efficiency of catalysis with the galvanostatic experiment of 10 mA cm−2. The oxygen gas released at every ten minutes was measured by the drainage collecting method. The mole of oxygen generated was calculated by the ideal gas law while the theoretical evolution of oxygen was calculated based on Faraday’s law, assuming that all the electron transfer at the anode was from OER. The ratio of the actual molar amount of oxygen released to the theoretical molar amount of oxygen in the OER process was identified as the Faraday efficiency of IrO2@TaB2 electrocatalyst.
In situ DEMS test was performed in a three-electrode cell with 0.1 M HClO4 solution as electrolyte. (i) The pristine IrO2@TaB2 was labeled with 18O isotope in 0.1 M HClO4 solution using H218O as solvent at 1.6 V for 10 min. (ii) The labeled electrode was washed with H216O to remove the residual H218O that was physically attached to the catalyst layer. Subsequently, CV tests were carried out at the rate of 50 mV s−1 in the potential range of 0.6 V and 1.2 V vs. RHE for removing the adsorbed 18O species on the surface of the catalyst. (iii) Three LSV cycles were applied for the labeled electrode in 0.1 M HClO4 solution using H216O as a solvent in the potential range of 1.2–1.7 V vs. RHE. The gaseous products including 36O2, 34O2, and 32O2 were monitored by the mass spectrometer. The fraction of 36O2, 34O2, and 32O2 can be estimated from the integral areas of corresponding mass signals of these gaseous products.
Electrochemical measurement of PEMWE
Before the construction of catalysis coated membrane (CCM), the N115 membrane was successively treated with 3 wt% H2O2, deionized water, and 0.5 M H2SO4 at 80 °C for 1 h. Then the treated N115 membrane was rinsed with deionized water. Commercial Pt/C (40 wt%) was used as a cathode electrocatalyst and commercial IrO2 or IrO2@TaB2 (Ir 40 wt%) was used as an anode electrocatalyst. In order to prepare the catalyst ink, the catalyst was dispersed into a mixed solution of isopropyl alcohol and distilled water (1:1, w/w). Subsequently, Nafion with an ionomer mass fraction of 10 wt% at the anode or 35 wt% at the cathode was added into the solution. The suspension was ultrasonically treated in an ice water bath for 1 h to obtain the catalyst ink. The anode catalyst ink and cathode catalyst ink were sprayed on polytetrafluoroethylene (PTFE) film respectively. Then the PTFE films supported with catalysts and a N115 membrane were hot pressed under 10 Mpa at 135 °C for 10 min. After cooling, the PTFE films were stripped to obtain CCM. The loading of the cathode was 0.27 mgpt cm−2 and the loading of the anode was 0.15 mgIr cm−2 for IrO2@TaB2 or 0.2 mgIr cm−2 for IrO2. The actual catalyst loading was determined by ICP-OES test. In order to construct a PEM electrolyzer, a well-defined pore Ti plate (provided by Hefei conservation of momentum green energy Co., Ltd) coated with Pt was used as the porous transport layer (PTL) of the anode, and a piece of carbon paper was used as the PTL of the cathode. The active area was 5 cm2. The PEM electrolyzer was operated at 80 °C and the reactant was deionized water, which was circulated through a peristaltic pump. The polarization curve of the PEM electrolyzer was collected at the cell voltage of 1.4–2.0 V, and the stability was tested by chronopotentiometry at 1 A cm−2.
Computation details
We performed all DFT calculations using the Vienna ab initio simulation package (VASP 5.4.4)73,74. The Generalized gradient approximation (GGA) with the Perdew−Burke−Ernzerhof (PBE) exchange correlation functional was employed with 500 eV cut-off energy75. All crystal structures were obtained from the ICSD (Inorganic Crystal Structure Database) database. During the structural optimization, we used k-point separation length of 0.04 2π A−1, while the k-point separation length was promoted to 0.03 2π A−1 for DOS calculation. For all calculations, we apply an energy convergence criterion of 10−5 eV with the force convergence criterion of 0.02 eV A−1.
For the Gibbs free energy change of the reaction, we use the Eq. (6) to calculate:
Where ΔE is the energy difference between product and reactant, ΔEzpe is the zero-point energy difference between product and reactant, T is the absolute temperature, and ΔS is the entropy difference between product and reactant. For the reaction:
Since only S2 is a gas, we only need to consider the zero-point energy and entropy of S2. The former can be calculated as 4.47 kJ mol−1, and the latter can be obtained as 228.2 J mol−1 K−1 from the database.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information. All other relevant data supporting the findings of this study are available from the corresponding authors upon request.
References
Wang, Y., Pang, Y., Xu, H., Martinez, A. & Chen, K. S. PEM Fuel cell and electrolysis cell technologies and hydrogen infrastructure development – a review. Energy Environ. Sci. 15, 2288–2328 (2022).
Chen, Y. et al. Key components and design strategy for a proton exchange membrane water electrolyzer. Small Struct. 4, 2200130 (2022).
Zhang, K. et al. Status and perspectives of key materials for PEM electrolyzer. Nano Res. Energy 1, e9120032 (2022).
Wu, Q. et al. Advances and status of anode catalysts for proton exchange membrane water electrolysis technology. Mater. Chem. Front. 7, 1025–1045 (2023).
Kibsgaard, J. & Chorkendorff, I. Considerations for the scaling-up of water splitting catalysts. Nat. Energy 4, 430–433 (2019).
Hansen, J. N. et al. Is there anything better than Pt for HER? ACS Energy Lett. 6, 1175–1180 (2021).
An, L. et al. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 33, 2006328 (2021).
Li, R. et al. IrW nanochannel support enabling ultrastable electrocatalytic oxygen evolution at 2 A cm−2 in acidic media. Nat. Commun. 12, 3540 (2021).
van Deelen, T. W., Hernández Mejía, C. & de Jong, K. P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2, 955–970 (2019).
Shan, J., Zheng, Y., Shi, B., Davey, K. & Qiao, S.-Z. Regulating electrocatalysts via surface and interface engineering for acidic water electrooxidation. ACS Energy Lett. 4, 2719–2730 (2019).
Chen, J. et al. Recent developments of nanocarbon based supports for PEMFCs electrocatalysts. Chin. J. Catal. 42, 1297–1326 (2021).
Shi, Y. et al. Electronic metal–support interaction modulates single-atom platinum catalysis for hydrogen evolution reaction. Nat. Commun. 12, 3021 (2021).
Liu, D. et al. Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution. Nat. Energy 4, 512–518 (2019).
Ye, S. et al. Highly stable single Pt atomic sites anchored on aniline-stacked graphene for hydrogen evolution reaction. Energy Environ. Sci. 12, 1000–1007 (2019).
Liang, X. et al. Developments and challenges of catalytic materials for green hydrogen production. Energy Lab 1, 220013 (2023).
Puthiyapura, V. K., Mamlouk, M., Pasupathi, S., Pollet, B. G. & Scott, K. Physical and electrochemical evaluation of ATO supported IrO2 catalyst for proton exchange membrane water electrolyser. J. Power Sources 269, 451–460 (2014).
Song, S. et al. Electrochemical investigation of electrocatalysts for the oxygen evolution reaction in PEM water electrolyzers. Int. J. Hydrog. Energy 33, 4955–4961 (2008).
Oh, H.-S., Nong, H. N., Reier, T., Gliech, M. & Strasser, P. Oxide-supported Ir nanodendrites with high activity and durability for the oxygen evolution reaction in acid PEM water electrolyzers. Chem. Sci. 6, 3321–3328 (2015).
Akopov, G., Yeung, M. T. & Kaner, R. B. Rediscovering the crystal chemistry of borides. Adv. Mater. 29, 1604506 (2017).
Carenco, S., Portehault, D., Boissière, C., Mézailles, N. & Sanchez, C. Nanoscaled metal borides and phosphides: recent developments and perspectives. Chem. Rev. 113, 7981–8065 (2013).
Zhang, J., Liu, J., Liao, H., Zeng, M. & Ma, S. A review on relationship between morphology of boride of Fe-B alloys and the wear/corrosion resistant properties and mechanisms. J. Mater. Res. Tech. 8, 6308–6320 (2019).
Martini, C., Palombarini, G., Poli, G. & Prandstraller, D. Sliding and abrasive wear behaviour of boride coatings. Wear 256, 608–613 (2004).
Zhang, S.-H. et al. Evolution of microstructure and anti-oxidation ability of ZrB2 improved by a unique inert glass phase. Ceram. Int. 45, 14291–14296 (2019).
Li, H. et al. Anti-oxidation and ablation properties of carbon/carbon composites infiltrated by hafnium boride. Carbon 52, 418–426 (2013).
Silvestroni, L. et al. A simple route to fabricate strong boride hierarchical composites for use at ultra-high temperature. Compos. Part B: Eng. 183, 107618 (2020).
Chen, H. & Zou, X. Intermetallic borides: structures, synthesis and applications in electrocatalysis. Inorg. Chem. Front. 7, 2248–2264 (2020).
Rao, L., Gillan, E. G. & Kaner, R. B. Rapid synthesis of transition-metal borides by solid-state metathesis. J. Mater. Res. 10, 353–361 (1995).
Guo, W.-M. et al. Synthesis of TaB2 powders by borothermal reduction. J. Am. Ceram. Soc. 100, 2368–2372 (2017).
Gürcan, K., Ayas, E. & Gaşan, H. Formation of TaB2 powders from high energy ball milling and borothermal reduction process. Mater. Chem. Phys. 235, 121732 (2019).
Jothi, P. R., Yubuta, K. & Fokwa, B. P. T. A simple, general synthetic route toward nanoscale transition metal borides. Adv. Mater. 30, 1704181 (2018).
Meyer, B. Elemental sulfur. Chem. Rev. 76, 367–388 (1976).
Jothi, P. R., Zhang, Y., Scheifers, J. P., Park, H. & Fokwa, B. P. T. Molybdenum diboride nanoparticles as a highly efficient electrocatalyst for the hydrogen evolution reaction. Sustain. Energy Fuels 1, 1928–1934 (2017).
Kamitaka, Y. et al. Platinum catalysts on niobium diboride microparticles for oxygen reduction reaction. Electrocatalysis 12, 188–198 (2021).
Shah, L. A. Low-temperature molten salt synthesis and characterization of nanowire-like TaB2 powder. JOM 73, 1023–1029 (2021).
Koc, R. & Hodge, D. B. Production of TiB2 from a precursor containing carbon coated TiO2 and B4C. J. Mater. Sci. Lett. 19, 667–669 (2000).
Wu, H. et al. Aqueous gelcasted ZrB2–SiC foams derived from composite poring mechanisms. Ceram. Int. 42, 1573–1580 (2016).
Li, Q. et al. Revealing activity trends of metal diborides toward ph-universal hydrogen evolution electrocatalysts with Pt-like activity. Adv. Energy Mater. 9, 1803369 (2019).
Daiane Ferreira da Silva, C. et al. Oxygen evolution reaction activity and stability benchmarks for supported and unsupported IrOx electrocatalysts. ACS Catal. 11, 4107–4116 (2021).
Karimi, F. & Peppley, B. A. Metal carbide and oxide supports for iridium-based oxygen evolution reaction electrocatalysts for polymer-electrolyte-membrane water electrolysis. Electrochim. Acta 246, 654–670 (2017).
Li, G. et al. Boosted performance of Ir species by employing TiN as the support toward oxygen evolution reaction. ACS Appl. Mater. Interfaces 10, 38117–38124 (2018).
Wang, S. et al. Defects tailoring IrO2@TiN1+x nano-heterojunctions for superior water oxidation activity and stability. Mater. Chem. Front. 5, 8047–8055 (2021).
Liang, X. et al. Activating inert, nonprecious perovskites with iridium dopants for efficient oxygen evolution reaction under acidic conditions. Angew. Chem. Int. Ed. 58, 7631–7635 (2019).
Yang, L. et al. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers. Nat. Commun. 9, 5236 (2018).
Sun, W., Song, Y., Gong, X. Q., Cao, L. M. & Yang, J. Hollandite structure Kx≈0.25IrO2 catalyst with highly efficient oxygen evolution reaction. ACS Appl. Mater. Interfaces 8, 820–826 (2016).
Zhang, R. H. et al. First example of protonation of ruddlesden-popper Sr2IrO4: a route to enhanced water oxidation catalysts. Chem. Mater. 32, 3499–3509 (2020).
Wu, Y. Y. et al. Highly efficient oxygen evolution activity of Ca2IrO4 in an acidic environment due to its crystal configuration. ACS Omega 3, 2902–2908 (2018).
Shang, C. Y. et al. Electron correlations engineer catalytic activity of pyrochlore iridates for acidic water oxidation. Adv. Mater. 31, 1805104 (2019).
Oakton, E. et al. IrO2-TiO2: a high-surface-area, active, and stable electrocatalyst for the oxygen evolution reaction. ACS Catal. 7, 2346–2352 (2017).
Karade, S. S., Sharma, R., Gyergyek, S., Morgen, P. & Andersen, S. M. IrO2/Ir composite nanoparticles (IrO2@Ir) supported on TiNxOy coated TiN: efficient and robust oxygen evolution reaction catalyst for water electrolysis. ChemCatChem 15, e202201470 (2023).
Hao, C. P., Lv, H., Mi, C. E., Song, Y. K. & Ma, J. X. Investigation of mesoporous Niobium-Doped TiO2 as an oxygen evolution catalyst support in an SPE water electrolyzer. ACS Sustain. Chem. Eng. 4, 746–756 (2016).
Oh, H. S. et al. Electrochemical catalyst-support effects and their stabilizing role for IrOx nanoparticle catalysts during the oxygen evolution reaction. J. Am. Chem. Soc. 138, 12552–12563 (2016).
Zhao, S., Stocks, A., Rasimick, B., More, K. & Xu, H. Highly active, durable dispersed iridium nanocatalysts for PEM water electrolyzers. J. Electrochem. Soc. 165, F82–F89 (2018).
Geiger, S. et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 1, 508–515 (2018).
Giordano, L. et al. pH dependence of OER activity of oxides: current and future perspectives. Catal. Today 262, 2–10 (2016).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: Toward boosted and sustained water oxidation catalysis. Joule 5, 2164–2176 (2021).
Curutchet, A., Colinet, P., Michel, C., Steinmann, S. N., & Bahers, T. L. Two-sites are better than one: revisiting the OER mechanism on CoOOH by DFT with electrode polarization. Phys. Chem. Chem. Phys. 22, 7031–7038 (2020).
Pivovar, B., Ruth, M. & Ahluwalia, R. H2NEW: Hydrogen (H2) from next-generation electrolyzers of water LTE Task 3c: system and techno-economic analysis. https://www.osti.gov/biblio/1900160 (2022).
Carmo, M., Fritz, D. L., Mergel, J. & Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrog. Energy 38, 4901–4934 (2013).
Retuerto, M. et al. Highly active and stable OER electrocatalysts derived from Sr2MIrO6 for proton exchange membrane water electrolyzers. Nat. Commun. 13, 7935 (2022).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotech. 16, 1371–1377 (2021).
Jiang, G. et al. Low-loading and highly stable membrane electrode based on an Ir@WOxNR ordered array for PEM water electrolysis. ACS Appl. Mater. Interfaces 13, 15073–15082 (2021).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Lv, H. et al. Synthesis and activities of IrO2/Ti1-xWxO2 electrocatalyst for oxygen evolution in solid polymer electrolyte water electrolyzer. J. Electroanal. Chem. 833, 471–479 (2019).
Yan, T. et al. IrO2 nanoparticle-decorated Ir-Doped W18O49 nanowires with high mass specific OER activity for proton exchange membrane electrolysis. ACS Appl. Mater. Interfaces 15, 6912–6922 (2023).
Hegge, F. et al. Efficient and stable low iridium loaded anodes for PEM water electrolysis made possible by nanofiber interlayers. ACS Appl. Energy Mater. 3, 8276–8284 (2020).
Faustini, M. et al. Hierarchically structured ultraporous iridium-based materials: a novel catalyst architecture for proton exchange membrane water electrolyzers. Adv. Energy Mater. 9, 1802136 (2019).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the Ir/Nb2O5−x catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Islam, J. et al. Enhancing the activity and durability of iridium electrocatalyst supported on boron carbide by tuning the chemical state of iridium for oxygen evolution reaction. J Power Sources 512, 230506 (2021).
Kang, Z. et al. Investigation of thin/well-tunable liquid/gas diffusion layers exhibiting superior multifunctional performance in low-temperature electrolytic water splitting. Energy Environ. Sci. 10, 166–175 (2017).
Kang, Z. et al. Performance improvement induced by membrane treatment in proton exchange membrane water electrolysis cells. Int. J. Hydrog. Energy 47, 5807–5816 (2022).
Stevens, M. B. et al. Measurement techniques for the study of thin film heterogeneous water oxidation electrocatalysts. Chem. Mater. 29, 120–140 (2017).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
X.Z. and H.C. acknowledge funding from the National Key R&D Program of China (no. 2021YFB4000200), the National Natural Science Foundation of China (NSFC) (no. 22179046 and 22279040), the State Grid Headquarter Science and Technology project (5419-202158490A-0-5-ZN), the Jilin Province Science and Technology Development Plan (no. 20220402006GH and 20210101403JC), and Interdisciplinary Integration and Innovation Project of JLU (JLUXKJC2021ZZ18).
Author information
Authors and Affiliations
Contributions
X.Z. and H.C. directed this research. Y.W. conducted most of the experiments. M.Z. and L.S. performed the theoretical calculations. Z.K. contributed to the performance measurement of PEMWE devices. Y.S., B.T., and Y.Z. contributed to data analysis. X.Z. and H.C. wrote the paper. X.Z. and H.C. supervised the project. All authors discussed and reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks David Sebastián and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Zhang, M., Kang, Z. et al. Nano-metal diborides-supported anode catalyst with strongly coupled TaOx/IrO2 catalytic layer for low-iridium-loading proton exchange membrane electrolyzer. Nat Commun 14, 5119 (2023). https://doi.org/10.1038/s41467-023-40912-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-023-40912-8
This article is cited by
-
Atomic-scale self-rearrangement of hetero-metastable phases into high-density single-atom catalysts for the oxygen evolution reaction
Nature Communications (2025)
-
Support-free iridium hydroxide for high-efficiency proton-exchange membrane water electrolysis
Nature Communications (2025)
-
Enhanced hydrogen peroxide photosynthesis via charge-complementary π-electron sites
Nature Communications (2025)
-
Tunnel-structured IrOx unlocks catalytic efficiency in proton exchange membrane water electrolyzers
Nature Communications (2025)
-
Atomically thin high-entropy oxides via naked metal ion self-assembly for proton exchange membrane electrolysis
Nature Communications (2025)
