
Abstract
There is tremendous interest in employing collective excitations of the lattice, spin, charge, and orbitals to tune strongly correlated electronic phenomena. We report such an effect in a ruthenate, Ca3Ru2O7, where two phonons with strong electron-phonon coupling modulate the electronic pseudogap as well as mediate charge and spin density wave fluctuations. Combining temperature-dependent Raman spectroscopy with density functional theory reveals two phonons, B2P and B2M, that are strongly coupled to electrons and whose scattering intensities respectively dominate in the pseudogap versus the metallic phases. The B2P squeezes the octahedra along the out of plane c-axis, while the B2M elongates it, thus modulating the Ru 4d orbital splitting and the bandwidth of the in-plane electron hopping; Thus, B2P opens the pseudogap, while B2M closes it. Moreover, the B2 phonons mediate incoherent charge and spin density wave fluctuations, as evidenced by changes in the background electronic Raman scattering that exhibit unique symmetry signatures. The polar order breaks inversion symmetry, enabling infrared activity of these phonons, paving the way for coherent light-driven control of electronic transport.
Similar content being viewed by others

Introduction
Mott Hubbard insulators are an example of strongly correlated electron system, where the energy scale for electron-electron repulsion is predominant relative to the kinetic energy of electrons1,2. On the other end, the energy scale for electron-phonon coupling (EPC) is predominant relative to the kinetic energy in a Peierls transition where charge density waves are observed3. In the intermediate regime of electronic correlation strength4 between that of weakly correlated Fermi-liquid metals and strongly correlated Mott insulators, a rich spectrum of competing electronic phenomena emerge such as unconventional superconductivity5,6, pseudogap phase7,8, and spin and orbital ordering9. In such systems, the energy scale for EPC can indeed be non-negligible in comparison with the correlation and exchange energy scales and could allow phonons to modulate emergent phenomena10,11,12, such as possible signatures of superconductivity in cuprates13,14,15,16, metal-insulator transitions in TaS217, VO218 and manganites19,20,21. Some of the ruthenates also lie in this intermediate correlation strength regime where correlation, exchange and EPC energy scales are all significant enough in influencing the physics underlying a rich spectrum of phenomena such as Mott insulators22, superconductivity23, pseudogap phase24,25, magnetic26, orbital27 and polar orders28. Ca3Ru2O7, the subject of this study exhibits an intermediate electron repulsion energy U ~ 1 eV, and onsite spin exchange coupling energy J ~ 0.4 eV27,29,30. However, despite two decades of research on this material, even the symmetry assignments of the phonons are currently incorrect; correcting these assignments in this study reveals new physics. Crucially, we show that the deformation potential (the square of which is proportional to the unitless EPC term \(\lambda\)31) for two of the correctly assigned B2 phonons in this system can reach up to ~10 eV/\({{{\text{\AA}}}}\), or ~0.1 eV per picometer of atomic displacement. Combining temperature dependent Raman spectroscopy with density functional theory (DFT), we show that these phonons (~52 meV) can modulate the electronic pseudogap (~10 meV) and mediate emergent density wave fluctuations that are absent in the ground state. The phonons are also infrared active, providing a pathway for potential optical control of the electronic transport instead of chemical pressure through doping as is conventionally explored32.
Ca3Ru2O7 is an n = 2 Ruddlesden-Popper phase of Can+1RunO3n+1, situated at the phase boundary between a Fermi liquid system, CaRuO3 (n = \(\infty\)), and a Mott insulator, Ca2RuO4 (n = 1). Slight impurity doping of 0.3% can induce a Mott insulating phase in Ca3Ru2O733. The ground state of Ca3Ru2O7 is a perplexing pseudogap electronic phase whose physical origin is still debated. The pseudogap is a partial gap that opens in their spectral function at specific k points. Recent density functional theory (DFT) calculations predict that a charge density wave with alternating \({{{{{\rm{Ru}}}}}}{{{{{{\rm{O}}}}}}}_{6}\) octahedral volumes can replicate the pseudogap band structure observed experimentally34. The presence of such density wave in the ground state has been postulated from the temperature dependent optical conductivity spectra24 and ARPES25 data. However, more recent ARPES35,36 and diffraction37,38,39 experiments have shown no evidence of density wave formation in the pseudogap phase\(.\) Although the ground state appears to lack the predicted density waves, we show that appropriate infrared optical phonons in Ca3Ru2O7 can indeed induce the predicted density wave fluctuations.
There are two central findings in this work. Firstly, correcting the previous erroneous symmetry assignments of phonons40 (Supplementary Note 1) reveals two dominant B2 phonons, labeled B2P and B2M, that strongly couple to the electronic and magnetic ground states: the scattering intensity of B2P and B2M are significantly enhanced respectively in the metallic and pseudogap phases. The magnitude of the deformation potential of these two phonons in Ca3Ru2O7 is comparable to known 2D systems with strong EPC, such as graphene41 (14.1 eV/Å) and MgB242 (6.5 eV/Å). First-principles frozen-phonon calculations with the important inclusion of spin-orbital coupling reveal that the B2P phonon mode opens the pseudogap, while the B2M mode closes it. Analysis of the eigen modes correlate the electronic pseudogap changes to phonon modulation of the Ru 4d orbital splitting and in-plane electron hopping. Secondly, a careful symmetry analysis of the background electronic Raman scattering reveals strong evidence for charge and spin density wave fluctuations mediated by the infrared active B2 phonons. Below 48 K, the electronic Raman scattering shows symmetry-dependent spectral weight transfer manifested by a distinctive rise above 550 cm−1 in the B2 spectra which is absent in the A1 spectra. This key symmetry signature in the electronic Raman scattering strongly points to the presence of charge and spin density wave like fluctuations mediated by optically excited B2 phonons (~52 meV) below the metal-pseudogap transition at 48 K (4 meV). The charge and spin density wave phase has been predicted34 in Ca3Ru2O7 but has been experimentally found to be absent in the ground state35,36,37,38,39. Our findings deepen the understanding of the role of electron-phonon coupling in modulating the pseudogap in correlated materials and pave the way for potential phononic control of electronic phases in ruthenates.
Results
Large anomalous phonon renormalization across the spin-reorientation transition
The polar metal Ca3Ru2O7 exhibits a rich range of ground states (Fig. 1b) realized by a subtle interplay between spin, charge, orbital and lattice degrees of freedom. Ca3Ru2O7 is a paramagnetic (PM) metal above \({{{{{{\rm{T}}}}}}}_{{{{{{\rm{N}}}}}}}\) = 56 K and is an antiferromagnetic metal with an easy axis along the a axis between 48-56 K (AFM-a phase, magnetic space group \({Bb}{2}_{1}^{{\prime} }{m}^{{\prime} }\)). It then transforms into an antiferromagnet with its easy axis along b axis below \({{{{{{\rm{T}}}}}}}_{{{{{{\rm{C}}}}}}}\) = 48 K (AFM-b phase, magnetic space group \(B{b}^{{\prime} }{2}_{1}{m}^{{\prime} }\)); the structures of two AFM phases are shown in Fig. 1a. The structural refinement from neutron diffraction indicates that the polar 2-fold axis is along the longer in-plane b-axis (b = 5.64 Å)43; this is contrary to the assignment in a previous Raman study on this material40, where the 2-fold axis was erroneously assigned to the shorter a-axis in-plane (a = 5.43 Å) (Supplementary Note 1). This error in the assignment of the 2-fold results in what are supposed to be the B2 Raman modes as A2 modes, and accordingly, the eigen displacements of these modes in the previous work40 are incorrect.

a Crystal structure of Ca3Ru2O7 with illustrated magnetic space group, symmetry operations and magnetic spins in the AFM-a and AFM-b phases. b Electronic and magnetic phase diagram of Ca3Ru2O7. c Schematically depicted Raman lab geometry with respect to the crystal lattice of Ca3Ru2O7. The \({E}_{i,s}\) and \({k}_{{is}}\) are polarization and wavevector or incident and scattered light respectively. d Representative polarized Raman intensities for Ca3Ru2O7 at AFM-b and AFM-a phases respectively.
The experimental geometry (Fig. 1c) involves 632.8 nm light propagates parallel to the c axis, while the light polarization is along the a or the b axes, and the lab coordinate system is defined such that x | |a, y | |b and z | |c. There are two non-degenerate configurations that produce non-zero experimentally observable Raman signal: \(\bar{z}\)(xx)z and \(\bar{z}\)(xy)z. The first and fourth letters in this notation respectively stand for the directions of propagation (wavevectors) of the incident (i) and scattered (s) light, whereas their polarizations are respectively denoted by the second and third letters. The character table in Table 1 correctly places the 2-fold symmetry parallel to the b axis44,45,46. \(\bar{z}\)(xx)z corresponds to the A1 phonons and \(\bar{z}\)(xy)z corresponds to the B2 phonons. The polarized spectra of Ca3Ru2O7 are measured at 50 K (AFM-a phase) and at 10 K (AFM-b phase) with 632.8 nm wavelength excitation (Fig. 1d). Similar spectra were observed in both the phases with 532 nm excitation (Supplementary Fig. 1), indicating that the Raman spectra presented are off-resonant. Self-consistent field calculations on electronic and phononic properties on Ca3Ru2O7 are performed with DFT LDA with on-site Coulomb repulsion of 1.2 eV applied on the 4d orbitals and spin-orbit interaction included. We fit the experimental spectra in Fig. 1e, and the obtained phonon peak positions are consistent with those identified by first and second derivatives of the spectra (see Supplementary Fig. 4). The converged fitting results of experimental phonon energy agrees well with the DFT predicted phonon energy (see Supplementary Note 3). The experimental fitting of phonon energy is robust against different background subtractions to account for the hybridization between phonon peaks and the electronic continuum (details in the Supplementary Note 4).
Reassignment of the B2 phonons reveal a crucial difference from the previous literature40,47: the broad feature between 380 \(c{m}^{-1}\) and 450 \(c{m}^{-1}\) in the B2 spectra (Fig. 2a) is composed of five phonon modes, indexed from B2(11) to B2(15), instead of one peak as identified in previous literature48,49,50. Further, the raw Raman spectra (Fig. 2b) shows that at 48 K near the phase transition, at least two peaks emerge as highlighted by the arrows. The experimental fitting of this composite peak is guided by frozen-phonon DFT calculations in the AFM-a and AFM-b phases. Comparison between the experimental and theoretical peak assignments (Fig. 2c, d) indicates a good agreement in both the AFM-b and AFM-a phases for these five phonons.

a Temperature dependent Raman spectra in \(\bar{z}\)(xy)z with B2(11) to B2(15) modes highlighted. The intensity is normalized to strongest Raman peak. b B2 spectrum at 48 K (Tc) showcase features of shoulder peaks highlighted with black arrows, compared with spectra at 18 K and 98 K. The DFT calculated B2(11) to B2(15) phonon energies compared to experimental observations in c AFM-a and d AFM-b phases.
The assignment of five peaks within the B2 Raman spectra from 350 to 450 cm−1 is further examined due to the congested peak features within this energy range. We can identify the features of five peaks from the taking first and second derivatives of the raw data of B2 Raman spectra as a function of temperature across Tc (Supplementary Fig. 4). The presence of five peaks in the congested B2 Raman spectra from 350 to 450 cm−1 can also be indirectly inferred by 3% Ti doping of the crystal without changing the point group of the crystal, where the Raman results shows a splitting of peaks from B2(12) to B2(15) peaks into two pairs of peak features with clear shoulders (Supplementary Figs. 5 and 6 and Supplementary Table 4). (Ti-doped sample is not discussed further in this study, since it requires a separate follow-up study). We also carefully compare our fitting model of five peaks against using a single Fano-line shape suggested by previous literature40 and conclude that our fitting model achieves better fits (Supplementary Fig. 7). For two phonon modes of interest, B2(13) and B2(15), the potential energy surface change is insensitive to the choice of DFT functionals (see Supplementary Fig. 8 in Supplementary Note 6). Thus, in this work, we present both experimental and computational evidence that the broad feature is composed of five phonon modes (instead of one) and are indexed from B2(11) to B2(15).
We present the region of interest in the B2 phonon spectra at two representative temperatures in Fig. 3a where clearly the B2(13) (henceforth labeled B2P) mode amplitude is suppressed while the B2(15) (henceforth labeled B2M) mode amplitude is enhanced upon heating above Tc (For fitting results of all observed modes, see Supplementary Figs. 9–12 in Supplementary Note 7). The overall temperature dependent fitting results (Fig. 3b) of B2P and B2M show a clear crossover of the Raman scattering intensity as well as that of the full-width-at-half-maximum (FWHM).

a Fitting of \(\bar{z}\)(xy)z Raman spectra between 340 cm–1 and 470 cm–1 at 50 K (top panel) and 10 K (bottom panel). the celeste/pink color-coded peak correspond to B2P/B2M. b The temperature dependent plots of amplitude (\(I\)) and FWHM (\(\varGamma\)) of B2P (celeste) and B2M (pink) modes. Error bars are standard deviations from fitting values. The celeste/pink lines are guides to the eye. c Raman colormap plot of the intensity difference between specified temperature and 13 K. The total area of region of interest highlighted in blue and red boxes in c are replotted in d to indicate an intensity anomaly observed at T* = 30 K. The pseudogap size (Δ) is extracted from ARPES data35 and is represented as grey data points. The broad grey line is a guide to the eye.
To understand these experimental trends, let us consider the origin of the Raman scattered intensity including EPC, which is given as follows51:
where \(\alpha\) is EPC term; \(F(\omega )\) is the phonon density of state at phonon frequency \(\omega\); \(N\left(0\right)\) is the electronic density of states at the Fermi surface, \({\varGamma }_{{{{{{\bf{q}}}}}},\nu }^{{ep}}\) is the electron-phonon-induced Raman peak width, \({\omega }_{q,\nu }\) is the phonon frequency of index \(\nu\) and wavevector \({{{{{\bf{q}}}}}}\). The Raman linewidth, \({\varGamma }_{{{{{{\bf{q}}}}}},\nu }^{{ep}}\), is given by the Fermi golden rule51,52 as follows:
Here, \(f\) is the Fermi-Dirac distribution function; \(\epsilon\) is the energy of the electron; \({g}_{\left({{{{{\bf{k}}}}}}{{{{{\boldsymbol{+}}}}}}{{{{{\bf{q}}}}}}\right)j,{{{{{\bf{k}}}}}}i,\nu }^{{ep}}\) is the EPC matrix for scattering from an electron state k located in band indexed i to k + q in band indexed j, via a phonon of wavevector q and mode index \(\nu\).
From the above equations, one can conclude that both the Raman intensity, I, and the FWHM are proportional to the determinant of the EPC matrix, \(|{g}_{\left({{{{{\bf{k}}}}}}{{{{{\boldsymbol{+}}}}}}{{{{{\bf{q}}}}}}\right)j,{{{{{\bf{k}}}}}}i,\nu }^{{ep}}|\), given that the joint DOS is not significantly modulated by the phonon perturbation (Supplementary Fig. 13 in Supplementary Note 8). An estimate for the deformation potential, which indicates EPC strength, can be obtained from \({\Gamma }_{{{{{{\boldsymbol{q}}}}}}={{{{{\boldsymbol{0}}}}}},\nu }^{{ep}}\) by excluding the combined contributions of the phonon-phonon scattering and the inhomogeneous broadening which is estimated to be ~6 \(c{m}^{-1}\) at Tc from lower energy \({B}_{2}\) phonons (Supplementary Fig. 9). Due to the quasi-2D nature of the ground state28,53, Ca3Ru2O7 exhibits a linear band dispersion near the Fermi level25,35, which justifies applying the 2D model41,52 based on the Fermi golden rule: \({\varGamma }_{{{{{{\bf{q}}}}}}={{{{{\boldsymbol{0}}}}}},\nu }^{{ep}}=\frac{{A}_{{uc}}}{8M{v}_{F}^{2}}{D}^{2}\), where \({A}_{{uc}}\) (=5.365\({{{\text{\AA}}}} \times\)5.535\({{{\text{\AA}}}}\)) is the area of the quasi-2D unit cell43 of Ca3Ru2O7, \(M\) (=434.374 g/6.02\(\times {10}^{23}\)) is the unit cell mass54, \({v}_{F}=5\times {10}^{4}{{{{{\rm{m}}}}}}/{{{{{\rm{s}}}}}}\) is the Fermi velocity25, and \(D\) is the deformation potential, which is related to EPC matrix by \({g}_{\left({{{{{\boldsymbol{k}}}}}}{{{{{\boldsymbol{+}}}}}}{{{{{\boldsymbol{q}}}}}}\right)j,{{{{{\boldsymbol{k}}}}}}i,\nu }^{{ep}}={D}_{\left({{{{{\boldsymbol{k}}}}}}{{{{{\boldsymbol{+}}}}}}{{{{{\boldsymbol{q}}}}}}\right)j,{{{{{\boldsymbol{k}}}}}}i,\nu }\sqrt{\hslash /(2M{\omega }_{{{{{{\boldsymbol{q}}}}}},\nu })}\). The measured \({\varGamma }_{{{{{{\bf{q}}}}}}={{{{{\boldsymbol{0}}}}}},\nu }^{{ep}}\) of \({B}_{2}^{{{{{{\rm{P}}}}}}}\) shows an increase of the estimated deformation potential from D ~ 6.7 \({{{{{\rm{eV}}}}}}/{{{\text{\AA}}}}\) in AFM-a phase to D ~ 9.7 \({{{{{\rm{eV}}}}}}/{{{\text{\AA}}}}\) in AFM-b phase. The \({B}_{2}^{{{{{{\rm{M}}}}}}}\) shows a decrease of the estimated deformation potential from D ~ 10.0 \({{{{{\rm{eV}}}}}}/{{{\text{\AA}}}}\) in AFM-a phase to D ~ 5.9 \({{{{{\rm{eV}}}}}}/{{{\text{\AA}}}}\) in AFM-b phase.
To further elaborate on the strong EPC of the two dominant B2 phonons, we zoom into the AFM-b phase where the pseudogap size gradually increases as the temperature decreases. As shown in Fig. 3c, the Raman intensity changes (I - I13K) exhibit two distinct regions, one where the change increases and the other where it decreases as the temperature decreases. These two regions are chosen according to the peak positions of the B2P (celeste) and B2M (pink) phonons due to their large Raman intensity changes across Tc. The area in these regions is integrated to represent the two respective phonons; the intensity of nearby phonon modes, B2(12) and B2(14), do not change significantly (Supplementary Fig. 10). The pseudogap size correlates well with Raman intensity as a function of temperature across the metal-to-pseudogap phase transition (Fig. 3d). Further, the Raman intensity is close to saturation at T* but there is no abrupt anomaly in the phonons at T* indicating that T* is not a distinct phase transition; this is consistent with the experimental evidence seen in the recent optical pump probe study55. On the other hand, \({T}^{*}\) likely corresponds to an anisotropic change in the electronic band structure near the Fermi-Surface35.
Microscopic Mechanism for the coupling of the two B 2 phonons to the Pseudogap
We investigate the coupling of the B2P and B2M phonons to the electronic structure via the frozen-phonon approach in DFT. The eigenmode of B2P in the AFM-b phase and that of the B2M in the AFM-a phase are shown in Fig. 4a–c. The B2P mainly modulates in-plane Ru-O bonding lengths, \({d}_{\parallel }\), as a “scissoring” mode in which the two nearest oxygen atoms move toward and away from each other. On the other hand, the B2M mainly modulates the apical Ru-O bonding lengths, \({d}_{\perp }\), between two perovskite layers (for numerical phonon eigenvectors of B2M and B2P and further analysis of the two modes, see Supplementary Tables 5, 6 and Supplementary Figs. 14–16 in Supplementary Note 9).

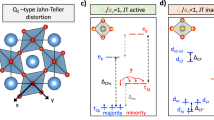
a Illustration of the Ru and O atoms, bond lengths between oxygens and the bond angle of O-Ru-O. b The illustrations of the eigen mode of B2P, and c that of B2M. d The averaged ratio of RuO6 octahedra cage apical Ru-O bond length (\({d}_{\perp }\)) over in-plane bond length (\({d}_{\parallel }\)) modulated by two phonons. e Averaged in-plane bond angle (\(\theta\)) modulated by the two phonons. (see Supplementary Figs. 15 for phonon modulated bond length and angles in two types of RuO6) f The density of states plots of the AFM-b ground state (middle) and the modulated AFM-b state by B2M (top) and B2P (bottom). g Correlation between octahedra distortion (\({d}_{\perp }\)/\({d}_{\parallel }\)), in plane bond angle (\(\theta\)) and the density of states near the Fermi surface. The lines are guides to the eye.
In Ca3Ru2O7, the octahedral cage without any phonon distortions is slightly compressed in its equilibrium phase43, since on the average, \(\frac{{d}_{\perp }}{{d}_{\parallel }}\, < \) 1. The B2P modulation further compresses the RuO6 octahedron along \({d}_{\perp }\) direction (Fig. 4d), while the B2M mode elongates it irrespective of the sign of the Eigen vectors. Such octahedral distortions are linked to the occupation of the Fermi level DOS in strongly correlated systems32. Due to the quasi-2D nature of the ground state28,53, only the in-plane Ru-O hopping parameter affects the electronic ground state. The averaged in-plane Ru-O-Ru bond angle \(\theta\) plotted in Fig. 4e is largely suppressed by the B2P mode, while the angle is slightly increased by the B2M mode. The decreasing bond angle leads to less electron cloud overlap between Ru 4d and O 2p orbitals which reduces the electronic bandwidth W. The DFT calculation results in Fig. 4f shows that the pseudogap opens/closes as the ground state is modulated by the B2P/B2M phonons, respectively. A clear correlation is seen (Fig. 4g) between the \(\frac{{d}_{\perp }}{{d}_{\parallel }}\), \(\theta\) and N(0) from the DFT calculations using the frozen phonon modulated DOS: B2P/B2M phonons respectively decrease/increase the \(\frac{{d}_{\perp }}{{d}_{\parallel }}\), the \(\theta\), and hence the N(0) in the metallic and pseudogap phase. Although the observed phonons are not thermally excited to induce the pseudogap opening at Tc, the modulation of the pseudogap by B2M and B2P phonons revealed by first principles calculations highlights the key role of structure distortion in the Mott transitions which is in agreement with the structural changes underlying the insulating phases in Ti-doped Ca3Ru2O756,57 and Ca2RuO458.
Density wave fluctuations from phonon-assisted structural distortions
Next, we present evidence for density wave fluctuations from the Raman electronic background. The imaginary part of the electronic Raman response function (\({\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\)) of Ca3Ru2O7 is obtained after subtracting the phonon and magnon peaks to get electronic Raman scattering intensity (\({I}_{u}(\omega,\, T)\)) and correcting for the Bose factor (\(n\)): \({\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\propto \frac{1}{(1+n\left(\omega,T\right))}{I}_{u}(\omega,\, T)\). In turn, \({\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) probes the weighted charge correlation function: \({\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)=\left\langle {\rho }_{u}(\omega ){\rho }_{u}(-\omega )\right\rangle\), where \({\rho }_{u}\left(\omega \right)={\sum }_{{{{{{\boldsymbol{k}}}}}}}{\gamma }_{u}\left({{{{{\boldsymbol{k}}}}}}\right)N({{{{{\boldsymbol{k}}}}}})\) is related to charge density operator \(N({{{{{\boldsymbol{k}}}}}})\) and Raman vertex \({\gamma }_{u}({{{{{\boldsymbol{k}}}}}})\), which denotes the scattering amplitude governed by the Raman selection rule (Supplementary Note 10). Thus, Raman response function has sensitivity to electron transitions in reciprocal space where \({\left|{\gamma }_{u}\left({{{{{\boldsymbol{k}}}}}}\right)\right|}\neq0\).
The experimental Raman response function \({\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) as well as the Raman vertex, \({\gamma }_{u}\left({{{{{\boldsymbol{k}}}}}}\right)\), of the B2 (\({\gamma }_{{xy}}\)) and the A1 (linear combination of \({\gamma }_{{x}^{2}+{y}^{2}}\) and \({\gamma }_{{x}^{2}-{y}^{2}}\)) phonons computed from group theory are depicted in Fig. 5a and c, respectively. The Fermi surface information25,35,36 combined with \({\gamma }_{u}({{{{{\boldsymbol{k}}}}}})\) (insets of Fig. 5a, c) indicates that the B2 spectra are mostly sensitive to the pseudogap feature centered around (\(\pm \frac{\pi }{2a}\), \(\pm \frac{\pi }{2a}\)), and the A1 spectra are mostly sensitive to metallic response at the M and M1 points, and are less sensitive (albeit with non-zero response) to the pseudogap features around the \(\Gamma\) and the quasi-1D bands close to M and M1. Indeed, the pseudogap features shows up as a dip in differential Raman response \(\Delta {\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)=\Delta {\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)-\Delta {\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, 49K\right)\) below ~400 cm-1 in both B2 and A1 spectra (Fig. 5b, d). From Gaussian fitting of the pseudogap dip feature, the probed pseudogap size is \(2{\Delta }_{{pseudogap}}=22.5\pm 1.4{meV}\) from B2 Raman response function and \(2{\Delta }_{{pseudogap}}=21.4\pm 1.3{meV}\) from the A1 Raman function at 28 K. This agrees well with the average pseudogap size extracted from optical conductivity measurements24, which was reported to be ~20 meV. By integrating the dip area between 120 cm-1 and 400 cm-1 in both the B2 and the A1, the differential Raman responses avoid the Drude response at lower frequency; with this analysis, we observe that the area of the dip feature can be well described by a BCS type gap (\(\Delta \left(T\right)\propto \tanh (2.2\sqrt{{T}_{c}/T-1})\)), which is consistent with ultrafast optical study on Ca3Ru2O755 (Fig. 5e). Thus, the electronic Raman scattering response is a sensitive probe of the changes in the pseudogap.

Temperature dependence of a B2 and b A1 Raman response function (\({\chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,T\right)\)) of Ca3Ru2O7 across Tc. The corresponding momentum-space structure of the form factor \({\gamma }_{{{{{{\boldsymbol{k}}}}}}}^{\mu }\) are depicted in the insets (Supplementary Note 10) overlayed with a schematic of momentum resolved electronic band features in the low-energy excitation near the Fermi surface from ARPES studies25, 35, 36. The background responses of c B2 and d A1, after subtracting the background response at 49 K to highlight the dip feature below 400 cm-1 arising from the pseudogap opening, and the hump feature seen above 550 cm-1. e Temperature dependence of the pseudogap Raman signal obtained by summing between 120 cm-1 and 400 cm-1 in \(\Delta {\chi }_{{B}_{2}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,T\right)\) (red circles) and \(\Delta {\chi }_{{A}_{1}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,T\right)\) (black circles). The signal is normalized to the maximum signal at 28 K and the solid line is a BCS gap model. f Correlation between B2P spectral weight and charge transfer Raman response at 300\(\mu\)W (black circles) and 600\(\mu\)W (red circles) incident laser power. g A schematic diagram of the proposed 3 step mechanism for the hump feature in B2: the orbital flip excitation is mediated by B2 phonons. The orbital flip excitation transiently differentiates the neighboring RuO6 octahedra and induces dynamical charge and spin density fluctuations. Errorbars of integrated area is estimated from variance of featureless section in \({\Delta \chi }_{u}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,T\right)\), and the errorbars of phonon spectral weight are standard deviations from fitting results.
In addition to the pseudogap feature, a hump in the differential Raman response is seen in \(\Delta {\chi }_{{B}_{2}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) above ~550 cm−1 but is conspicuously absent in \(\Delta {\chi }_{{A}_{1}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) (Fig. 5b, d). In literature, we can an find excellent agreement of this hump feature with the optical conductivity spectra24. The feature was assigned to electron-hole quasiparticle excitation across the neighboring Ru atoms from dxy to dxz/yz that benefits from the energy gain through Hund’s coupling (Supplementary Note 11). However, the absence of the hump features in \(\Delta {\chi }_{{A}_{1}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) in our present study is rather surprising and suggests new insights into its origin.
Both \({\chi }_{{A}_{1}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) and \({\chi }_{{B}_{2}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) have sensitivity to dxz/yz quasi-1D band near the Fermi-surface25,35 where proposed Fermi nesting feature is at. The pseudogap dip area in \(\Delta {\chi }_{{B}_{2}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,\, T\right)\) is not sensitive to the laser line intensity, while the increasing intensity of incident laser gives rise to an increase in the B2P phonon amplitude below Tc (Supplementary Fig. 17). Furthermore, the intersite quasiparticle excitation hump area, \(\Delta {\chi }_{{B}_{2}}^{{{{\hbox{'}}}{{\hbox{'}}}}}\left(\omega,T\right)\), above ~550 cm-1 is linearly proportional to the B2P phonons amplitude (Fig. 5e). It is highly likely that since B2P mode shows an anomalously large EPC (order of ~10 \({{{{{\rm{eV}}}}}}/{{{\text{\AA}}}}\)) below Tc, which is not observed in the FWHM of other B2 modes (Supplementary Figs. 9 and 10), the intersite excitation appears to be mainly mediated by the B2P mode. However, it is worth noting that Raman cannot uniquely single out the B2P mode (Supplementary Fig. 18). A further look into the selection rule for intersite dxy to dxz/yz orbital electron-hole quasiparticle excitation suggests that in-plane excitation of electrons from dxy to O 2pz is forbidden via the electric dipole transition between electronic states, unless the excitation is mediated by the B2 phonons between vibronic states.
Using group theory analysis (Supplementary Note 13), we propose a 3-step mechanism (Fig. 5g) to explain the electronic background above ~550 cm−1 using symmetry resolved quasiparticle information from the electronic Raman. The electron hopping is initiated in Step I by two simultaneous steps: (1) charge excitation from O \({{{{{{\rm{p}}}}}}}_{{{{{{\rm{z}}}}}}}\) orbital to Ru \({{{{{{\rm{d}}}}}}}_{{{{{{\rm{xz}}}}}}/{{{{{\rm{yz}}}}}}}\) orbital (octahedra 1) and (2) the electron from neighboring Ru \({{{{{{\rm{d}}}}}}}_{{{{{{\rm{xy}}}}}}}\) (octahedra 2) hopping to the O \({{{{{{\rm{p}}}}}}}_{{{{{{\rm{z}}}}}}}\) orbital, assisted by the B2 phonons. Then in Step II, the charge imbalance between the two RuO6 cages would promote the electron hopping from (1) O \({{{{{{\rm{p}}}}}}}_{{{{{{\rm{z}}}}}}}\) to Ru \({{{{{{\rm{d}}}}}}}_{{{{{{\rm{xz}}}}}}/{{{{{\rm{yz}}}}}}}\) (octahedra 2) and followed by electron hopping from (2) Ru \({{{{{{\rm{d}}}}}}}_{{{{{{\rm{xz}}}}}}/{{{{{\rm{yz}}}}}}}\) (octahedra 1) to O \({{{{{{\rm{p}}}}}}}_{{{{{{\rm{z}}}}}}}\). Eventually, the electron configuration would return to the ground state (Step III). This provides a unique case where symmetry-breaking phonons drive charge and spin fluctuation like a density wave modulation by exciting electrons across neighboring RuO6. The energy cost for this hopping is estimated as following: Hund’s coupling energy benefits the intersite charge transfer and the energy cost is \(U-3J+D-{\omega }_{B2}\) (Supplementary Note 11), where U is the electron correlation energy, J is the Hund’s coupling energy, D is the gap splitting between \({{{{{{\rm{d}}}}}}}_{{{{{{\rm{xy}}}}}}}\) and \({{{{{{\rm{d}}}}}}}_{{{{{{\rm{xz}}}}}}/{{{{{\rm{yz}}}}}}}\) and \({\omega }_{B2}\) is the gain of phonon energy via vibronic mixing. With reported values of U ~ 1 eV, J ~ 0.4 eV, D ~ 0.3 eV27,29,30 and considering the phonon energy of B2P (0.05 eV), this would result in an energy barrier of ~0.05 eV for the \({{{{{{\rm{d}}}}}}}^{4}+{{{{{{\rm{d}}}}}}}^{4}\to {{{{{{\rm{d}}}}}}}^{3}+{{{{{{\rm{d}}}}}}}^{5}\) electron transition; this agrees with our experimental result that shows an increase in the electronic Raman scattering above 550 cm−1 ~ 0.07 eV.
Based on this clear symmetry-based evidence for the density wave fluctuations mediated by the B2 phonons (but not the A1 phonons), a revisiting of the ground state of Ca3Ru2O7 is merited. The formation of static charge density waves requires structural distortion. With the most recent high energy X-ray pair distribution function study on Ca3Ru2O759, which has larger precision on Ru-O bond length than required to resolve the proposed static density wave phase, the space group remains Bb21m, which indicates that the static density wave distortion is not observed in the low temperature, in which case the predicted space group would change34. Below the first order transition temperature Tc = 48 K, the bond angles and bond length below Tc undergo a continuous transition as a function of temperature but the structure distortion does not promote electron hopping between intersite Ru atoms39,43(see detailed analysis in Supplementary Note 14). This indicates that synchronized modulation of the in-plane electron hopping integral by a phonon such as B2P is a viable means to drive the structure towards a charge density wave distortion (see Supplementary Figs. 16a, 20). Indeed, temperature dependent electronic Raman suggests that the B2P phonon assists in the formation of the density wave fluctuations in Ca3Ru2O7. While the density wave fluctuation is largely incoherent and short-lived as indicated by the broad nature of the hump feature (for the discussion of the lifetime of the density wave fluctuation, see Supplementary Note 15), resonantly driving the infrared (IR) active B2P mode with mid IR pulses is a promising avenue to create long-lived charge order states in Ca3Ru2O7.
Using Raman spectroscopy combined with DFT calculations, this work presents a significant revision of the symmetry assignment of previously reported phonon spectra40 in Ca3Ru2O7, which alters the interpretation of underlying physics. In particular, two phonon modes labeled B2P and B2M show a strong cross-over effect in their mode specific deformation potential across the metal-pseudogap transition temperature Tc = 48 K. Employing DFT calculations with spin-obit coupling, we discover the microscopic mechanism through which the two phonon modes close and open the pseudogap, respectively. Two phonons modulate the bandwidth near the Fermi surface and the RuO6 octahedral compression ratio, which in turn are coupled to the pseudogap modulation. Temperature-dependent electronic Raman scattering presents evidence for the formation of the phonon-mediated charge and spin density fluctuation that has been predicted by theory but absent from ground state34. Our study indicates that this density wave phase can be potentially stabilized by resonant light excitation of IR active B2P phonon mode. Our finding in Ca3Ru2O7 motivates a potential pathway for modulating competing phases by phonon excitations using light in correlated systems with strong electron-phonon coupling.
Methods
Sample preparation
High quality calcium ruthenate Ca3Ru2O7 single crystals were grown by the floating zone techniques using a mirror-focused furnace60, and aligned ex situ by X-ray Laue diffraction for polarized Raman measurement. The composition of the sample was determined via X-ray diffraction.
Raman spectroscopy measurements
The Raman experiments were carried out in a 4 K helium-flow Oxford microstat He cryostat connected to an Agilent TPS-compact pump station with base pressures of \(5\times {10}^{-6}\) torr. Temperature-dependent Raman spectra were collected using a Horiba LabRam HR Evolution with a freespace Olympus BX51 confocal microscope. A 632.8 nm linearly polarized HeNe laser beam was focused at normal incidence using a LWD 50x objective with a numerical aperture of 0.5, with the confocal hole set to 100 μm. A Si back-illuminated deep depleted array detector and an ultra-low-frequency volume Bragg filter were used to collect the spectra, dispersed by a grating (1800 gr/mm or 600 gr/mm) with an 800 mm focal length spectrometer.
The laser power was maintained below 300 \(\mu\)W in all the temperature-dependent measurements using 600 gr/mm (unless stated otherwise), to minimize laser heating and maintain the power well below the damage threshold. The 1800 gr/mm data was collected using 600 \(\mu\)W 632.8 nm laser and 400 \(\mu\)W 532 nm laser. The temperature presented in this paper is after correcting for the laser heating. Laser heating was calibrated as described in the Supplementary Note 16 with thermal conductivity values from reference55. Polarized spectra were obtained using a half-waveplate to rotate the polarization of the incident beam, with a fixed analyzer. Spectra were averaged over 60 minutes in the case of temperature-dependent measurements, with a temperature stability of \(\pm\)0.1 K. The temperature dependent spectra are shown in Supplementary Fig. 2. The intensity of colormap in Supplementary Fig. 2 is normalized to the Raleigh line intensity.
Raman peak fitting
The A1, and B2 peaks were fit with a standard Lorentz lineshape, except the B2(13) and B2(15) peaks that were fit with a Pseudo Voigt lineshape. A nonlinear least-squares fitting procedure was used. To ensure robustness of the temperature-dependent fits, the same initial fit values and constraints were used for each set of temperature-dependent spectra. The B2 peaks were fit after subtracting the background with Savitzky-Golay filter to account for potential hybridization between background and phonon peaks. The experimental fitting is robust against different background subtractions. Detailed description of background subtraction method and robustness of fitting demonstration can be found in the Supplementary Note 4 and 5.
Electronic structure and phonon calculations
Self-consistent field calculations are performed with the Vienna Ab-initio Simulation Package (VASP) using projected augmented wave method61,62. To correct for the self-interaction issue, the on-site Coulomb repulsion of 1.2 eV is applied on the 4d orbitals using Dudarev’s approach34,55,63. The spin-orbit interaction is included for the atomic position optimizations. The lattice parameters in all the DFT studies are from the reported experimental structures with a space group of Bb21m43. Our results show that AFM-b is more stable than AFM-a (with an energy difference of 17.3 meV). For low and high-temperature phases, the bi-layer antiferromagnetic orderings are respectively along the [001] and [010] axes, which correspond respectively to the AFM-a and AFM-b phases. Previous literature64 has shown that LDA performs better than PBEsol for describing the phononic properties in Ca3Ru2O7. Therefore, LDA functional65 is utilized to study the electronic and phononic properties in this work. The optimization of the atomic positions is carried out with a plane-wave cutoff of 650 eV and the Brillouin zone is sampled using a Gaussian smearing with a 20 meV width on a 10×10×6 Γ-centered k-mesh. The energy and forces are converged to be within 10–8 eV and 0.1 meV/Å. The phononic properties are calculated using frozen phonon approach that is implemented in phonopy66. We represent phonons using mass-weighted atomic displacements QIα≡\(\sqrt{{m}_{I}}\)uIα, where \(I\) is the atom index, \(\alpha\) is the space direction, \({m}_{I}\) is the mass of atom \(I\) (in atomic units of mass, a.m.u = 1.6605 · 10−24 g), and \({u}_{I\alpha }\) is the displacement of atom \(I\) along direction \(\alpha\) (in Å). The dynamical matrix \({{{{{\boldsymbol{D}}}}}}({{{{{\boldsymbol{q}}}}}})\) (the mass-rescaled interatomic force constant matrix for monochromatic perturbations of atomic positions of wave vector \({{{{{\boldsymbol{q}}}}}}\)) can then be expressed as
where ΦIα,Jβ(R) = ∂2E/∂uIα(0)∂uJβ(R) is the (un-rescaled) interatomic force constant matrix between atoms Iα and Jβ residing in unit cells that are separated by the translation vector R. Since the Raman response is related to the zone-center phonons (q ≈ 0), we consider the dynamical matrix at the \(\Gamma\)-point:
By diagonalizing the dynamical matrix, one obtains the resonant frequencies \({\omega }_{n}\) and the normal modes \({{{{{{\boldsymbol{e}}}}}}}_{n}\):
Therefore, the atomic displacements can be decomposed along the normal modes as
where \({Q}_{n}\equiv {\sum }_{I\alpha }{Q}_{I\alpha }^{*}{e}_{I\alpha,n}\) denotes the coordinate of the \(n\)th normal mode [in (a.m.u)1/2 Å].
The electronic band structures at \({k}_{z}\) = 0 for the AFM-a and AFM-b phases are shown in Supplementary Fig. 22. LDA + U + SOI predicts a suppressed band crossing of the low-temperature AFM-b compared to the AFM-a phase with a prominent effect near \({M}_{1}\), which is in good agreement with the previous studies55. With the electronic structure established, we applied finite difference method to calculate the A1 and B2 vibrational frequencies and modes which corresponds to the predominant peaks in the low-temperature Raman measurements.
To perform the Raman assignment and make meaningful comparisons between DFT and experiments, we carefully compare the B2 vibrational modes of both AFM-b and AFM-a phases by ordering them in the ascending order of their vibrational frequencies. As shown in Supplementary Fig. 23, the vibrational eigen modes of 1 Q are in good correspondence between the AFM-b and AFM-a phases.
Data availability
Supplementary Information is available for this paper. The Raman data generated in this study have been deposited in the Figshare database at https://doi.org/10.6084/m9.figshare.24022497. Additional data are available from the corresponding author upon request.
References
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Basov, D. N., Averitt, R. D., van der Marel, D., Dressel, M. & Haule, K. Electrodynamics of correlated electron materials. Rev. Mod. Phys. 83, 471–541 (2011).
Gruner, G. Density waves in solids. (CRC press, 2018).
Baldini, E. Nonequilibrium dynamics of collective excitations in quantum materials. (Springer, 2018).
Sigrist, M. & Ueda, K. Phenomenological Theory of Unconventional Superconductivity. Rev. Mod. Phys. 63, 239–311 (1991).
Stewart, G. R. Unconventional superconductivity. Adv. Phys. 66, 75–196 (2017).
Timusk, T. & Statt, B. The pseudogap in high-temperature superconductors: an experimental survey. Rep. Prog. Phys. 62, 61–122 (1999).
Rice, T. M., Yang, K. Y. & Zhang, F. C. A phenomenological theory of the anomalous pseudogap phase in underdoped cuprates. Rep. Prog. Phys. 75 (2012).
Hotta, T. Orbital ordering phenomena in d- and f-electron systems. Rep. Prog. Phys. 69, 2061–2155 (2006).
Subedi, A., Cavalleri, A. & Georges, A. Theory of nonlinear phononics for coherent light control of solids. Phys. Rev. B 89, 220301 (2014).
Forst, M. et al. Nonlinear phononics as an ultrafast route to lattice control. Nat. Phys. 7, 854–856 (2011).
Zhang, J. & Averitt, R. D. Dynamics and Control in Complex Transition Metal Oxides. Annu. Rev. Mater. Res. 44, 19 (2014).
Mankowsky, R. et al. Nonlinear lattice dynamics as a basis for enhanced superconductivity in YBa2Cu3O6.5. Nature 516, 71–73 (2014).
Hu, W. et al. Optically enhanced coherent transport in YBa2Cu3O6.5 by ultrafast redistribution of interlayer coupling. Nat. Mater 13, 705–711 (2014).
Fausti, D. et al. Light-Induced Superconductivity in a Stripe-Ordered Cuprate. Science 331, 189–191 (2011).
Liu, B. et al. Pump Frequency Resonances for Light-Induced Incipient Superconductivity in YBa2Cu3O6.5. Phys. Rev. X 10, 011053 (2020).
Stojchevska, L. et al. Ultrafast switching to a stable hidden quantum state in an electronic crystal. Science 344, 177–180 (2014).
Liu, M. K. et al. Terahertz-field-induced insulator-to-metal transition in vanadium dioxide metamaterial. Nature 487, 345–348 (2012).
Zhang, J. D. et al. Cooperative photoinduced metastable phase control in strained manganite films. Nat. Mater 15, 956–960 (2016).
Polli, D. et al. Coherent orbital waves in the photo-induced insulator–metal dynamics of a magnetoresistive manganite. Nat. Mater 6, 643–647 (2007).
Rini, M. et al. Control of the electronic phase of a manganite by mode-selective vibrational excitation. Nature 449, 72–74 (2007).
Nakatsuji, S., Ikeda, S. I. & Maeno, Y. Ca2RuO4: New Mott insulators of layered ruthenate. J. Phys. Soc. Jpn. 66, 1868–1871 (1997).
Maeno, Y. et al. Superconductivity in a Layered Perovskite without Copper. Nature 372, 532–534 (1994).
Lee, J. S. et al. Pseudogap dependence of the optical conductivity spectra of Ca3Ru2O7: A possible contribution of the orbital flip excitation. Phys. Rev. Lett. 98, 097403 (2007).
Baumberger, F. et al. Nested Fermi surface and electronic instability in Ca3Ru2O7. Phys. Rev. Lett. 96, 107601 (2006).
Suzuki, H. et al. Spin waves and spin-state transitions in a ruthenate high-temperature antiferromagnet. Nat. Mater 18, 563 (2019).
Das, L. et al. Spin-Orbital Excitations in Ca2RuO4 Revealed by Resonant Inelastic X-Ray Scattering. Phys. Rev. X 8, 011048 (2018).
Lei, S. M. et al. Observation of Quasi-Two-Dimensional Polar Domains and Ferroelastic Switching in a Metal, Ca3Ru2O7. Nano Lett. 18, 3088–3095 (2018).
Fatuzzo, C. G. et al. Spin-orbit-induced orbital excitations in Sr2RuO4 and Ca2RuO4: A resonant inelastic x-ray scattering study. Phys. Rev. B 91, 155104 (2015).
Mizokawa, T. et al. Spin-orbit coupling in the Mott insulator Ca2RuO4. Phys. Rev. Lett. 87, 077202 (2001).
An, J. M. & Pickett, W. E. Superconductivity of MgB2: Covalent bonds driven metallic. Phys. Rev. Lett. 86, 4366–4369 (2001).
Peng, J. et al. Mott transition controlled by lattice-orbital coupling in 3d-metal-doped double-layer ruthenates. Phys. Rev. B 96, 205105 (2017).
Tsuda, S. et al. Mott transition extremely sensitive to impurities in Ca3Ru2O7 revealed by hard x-ray photoemission studies. Phys. Rev. B 87, 241107 (2013).
Puggioni, D., Horio, M., Chang, J. & Rondinelli, J. M. Cooperative interactions govern the fermiology of the polar metal Ca3Ru2O7. Phys. Rev. Res. 2, 023141 (2020).
Horio, M. et al. Electronic reconstruction forming a C-2-symmetric Dirac semimetal in Ca3Ru2O7. Npj Quantum Mater. 6, 29 (2021).
Markovic, I. et al. Electronically driven spin-reorientation transition of the correlated polar metal Ca3Ru2O7. P Natl Acad. Sci. USA 117, 15524–15529 (2020).
Sokolov, D. A. et al. Metamagnetic texture in a polar antiferromagnet. Nat. Phys. 15, 671 (2019).
Dashwood, C. D. et al. Spontaneous cycloidal order mediating a spin-reorientation transition in a polar metal. Phys. Rev. B 102, 180410 (2020).
Petkov, V. et al. Lattice distortions and the metal-insulator transition in pure and Ti-substituted Ca3Ru2O7. J. Phys.-Condens Mat. 51, 015402 (2023).
Iliev, M. N. et al. Raman spectroscopy of Ca3Ru2O7: Phonon line assignment and electron scattering. Phys. Rev. B 71, 214305 (2005).
Yan, J., Zhang, Y. B., Kim, P. & Pinczuk, A. Electric field effect tuning of electron-phonon coupling in graphene. Phys. Rev. Lett. 98, 166802 (2007).
Yildirim, T. et al. Giant anharmonicity and nonlinear electron-phonon coupling in MgB2: A combined first-principles calculation and neutron scattering study. Phys. Rev. Lett. 87, 037001 (2001).
Yoshida, Y. et al. Crystal and magnetic structure of Ca3Ru2O7. Phys. Rev. B 72 (2005).
Aroyo, M. I. et al. Crystallography online: Bilbao Crystallographic Server. Bulg Chem. Commun. 43, 183–197 (2011).
Aroyo, M. I. et al. Bilbao crystallographic server: I. Databases and crystallographic computing programs. Z Kristallogr 221, 15–27 (2006).
Aroyo, M. I., Kirov, A., Capillas, C., Perez-Mato, J. M. & Wondratschek, H. Bilbao crystallographic server. II. Representations of crystallographic point groups and space groups. Acta Crystallogr A 62, 115–128 (2006).
Liu, H. L., Yoon, S., Cooper, S. L., Cao, G. & Crow, J. E. Raman-scattering study of the charge and spin dynamics of the layered ruthenium oxide Ca3Ru2O7. Phys. Rev. B 60, R6980–R6983 (1999).
Karpus, J. F., Gupta, R., Barath, H., Cooper, S. L. & Cao, G. Field-induced orbital and magnetic phases in Ca3Ru2O7. Phys. Rev. Lett. 93, 167205 (2004).
Snow, C. S. et al. Pressure-tuned collapse of the Mott-like state in Can+1RunO3n+1 (n=1,2): Raman spectroscopic studies. Phys. Rev. Lett. 89, 226401 (2002).
Karpus, J. F. et al. Spectroscopic study of the field- and pressure-induced phases of the bilayered ruthenate Ca3Ru2O7. Phys. Rev. B 73, 134407 (2006).
Allen, P. B. Neutron Spectroscopy of Superconductors. Phys. Rev. B 6, 2577 (1972).
Lazzeri, M., Piscanec, S., Mauri, F., Ferrari, A. C. & Robertson, J. Phonon linewidths and electron-phonon coupling in graphite and nanotubes. Phys. Rev. B 73, 155426 (2006).
Yoshida, Y. et al. Quasi-two-dimensional metallic ground state of Ca3Ru2O7. Phys. Rev. B 69, 220411 (2004).
Prohaska, T. et al. Standard atomic weights of the elements 2021 (IUPAC Technical Report). Pure Appl. Chem. 94, 573–600 (2022).
Yuan, Y. K. et al. Ultrafast quasiparticle dynamics in the correlated semimetal Ca3Ru2O7. Phys. Rev. B 99, 155111 (2019).
Ke, X. et al. Emergent electronic and magnetic state in Ca3Ru2O7 induced by Ti doping. Phys. Rev. B 84, 201102 (2011).
Peng, J. et al. From quasi-two-dimensional metal with ferromagnetic bilayers to Mott insulator with G-type antiferromagnetic order in Ca3(Ru1-xTix)2O7. Phys. Rev. B 87, 085125 (2013).
Souliou, S. M. et al. Raman Scattering from Higgs Mode Oscillations in the Two-Dimensional Antiferromagnet Ca2RuO4. Phys. Rev. Lett. 119, 067201 (2017).
Petkov, V. et al. Lattice distortions and the metal-insulator transition in pure and Ti-substituted Ca3Ru2O7. J. Phys-Condens Mat. 35, 015402 (2023).
Bao, W., Mao, Z. Q., Qu, Z. & Lynn, J. W. Spin valve effect and magnetoresistivity in single crystalline Ca3Ru2O7. Phys. Rev. Lett. 100, 247203 (2008).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Wang, Y. et al. Thermodynamic and electron transport properties of Ca3Ru2O7 from first-principles phonon calculations and Boltzmann transport theory. Phys. Rev. B 107, 035118 (2023).
Perdew, J. P. & Zunger, A. Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems. Phys. Rev. B 23, 5048–5079 (1981).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scripta Mater 108, 1–5 (2015).
Acknowledgements
H.W., Y.X., Y.W., Z.W., L-Q. C, I.D. and V.G. acknowledge the primary support from US department of Energy, Office of Science, Basic Energy Sciences, under Award Number DE-SC0020145 as part of the Computational Materials Science Program. H.P. and V.G. acknowledge support from the Department of Energy grant DE-SC0012375 for contributions to Raman analysis. R.Z., L.M., Z.M., and V.G. acknowledge support from the NSF MRSEC Center for Nanoscale Science grant number DMR-2011839 for partial contributions to crystal growth and performing the Raman experiments in the Materials Characterization Laboratory. Major support for crystal growth and characterization was provided by the National Science Foundation through the Penn State 2D Crystal Consortium-Materials Innovation Platform (2DCC-MIP) under NSF cooperative agreement DMR-1539916 and 2039351. G.H., G.B. and R.C. acknowledge support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, under Contract No. DE-AC02-05CH11231: Materials Project program KC23MP as well as from the Communauté Française de Belgique, grant ARC 18/23-093 for the phonon and electron-phonon theoretical work. First-principles calculations were carried out partially on the resources of NERSC supported by the Office of Science of the US Department of Energy under contract No. DE-AC02-05CH11231 (Y.W.).
Author information
Authors and Affiliations
Contributions
H.W. and V.G. conceived the project. Raman spectroscopy measurements and analysis were carried out by H.W., H.P., Z.R., M.W. and V.G. Density functional calculations were done by Y.X., Yi.W., Z.W., R.C., G.B., G.H., I.D., and L-Q. C. Crystal Growth and characterization were done by L.M., Yu.W. and Z.Q.M. The paper was written by H.W. and V.G. with inputs from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Edoardo Baldini, Hanghui Chen and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H.(., Xiong, Y., Padma, H. et al. Strong electron-phonon coupling driven pseudogap modulation and density-wave fluctuations in a correlated polar metal. Nat Commun 14, 5769 (2023). https://doi.org/10.1038/s41467-023-41460-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-023-41460-x
